

Abstract
A consistently high level of stallion fertility plays an economically important role in modern horse breeding. We performed a genome-wide association study for estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in Hanoverian stallions. A total of 228 Hanoverian stallions were genotyped using the Equine SNP50 Beadchip. The most significant association was found on horse chromosome 6 for a single nucleotide polymorphism (SNP) within phospholipase C zeta 1 (PLCz1). In the close neighbourhood to PLCz1 is located CAPZA3 (capping protein (actin filament) muscle Z-line, alpha 3). The gene PLCz1 encodes a protein essential for spermatogenesis and oocyte activation through sperm induced Ca2+-oscillation during fertilization. We derived equine gene models for PLCz1 and CAPZA3 based on cDNA and genomic DNA sequences. The equine PLCz1 had four different transcripts of which two contained a premature termination codon. Sequencing all exons and their flanking sequences using genomic DNA samples from 19 Hanoverian stallions revealed 47 polymorphisms within PLCz1 and one SNP within CAPZA3. Validation of these 48 polymorphisms in 237 Hanoverian stallions identified three intronic SNPs within PLCz1 as significantly associated with EBV-PAT. Bioinformatic analysis suggested regulatory effects for these SNPs via transcription factor binding sites or microRNAs. In conclusion, non-coding polymorphisms within PLCz1 were identified as conferring stallion fertility and PLCz1 as candidate locus for male fertility in Hanoverian warmblood. CAPZA3 could be eliminated as candidate gene for fertility in Hanoverian stallions.
Introduction
Stallion fertility is of increasing importance due to the widespread use of artificial insemination in horse breeding and the strong seasonal breeding restricted to six months of the year. Studies in human and mice discovered a large number of genes with influence on male fertility [1]–[5], whereas there are only few reports about genes with impact on stallion fertility [6]–[10]. To date, genome-wide association studies (GWAS) with dense genotyping arrays greatly enhance the possibilities to clarify the role of known candidates and to identify new promising candidate genes for mammalian fertility [11]–[13].
Several studies implicated candidate genes for stallion fertility that have been shown to play a substantial role in equine male reproduction. In Hanoverian stallions, single nucleotide polymorphisms (SNPs) within the genes prolactin receptor (PRLR) [6], inhibin beta A (INHBA) [7], follicle stimulating hormone (FSHB) [8], angiotensin converting enzyme (ACE) [8], spermatogenesis associated 1 (SPATA1) [9] and cysteine-rich secretory protein 3 (CRISP3) [10] were shown significantly associated with stallion fertility. Recently, a GWAS employed in seven Thoroughbred stallions with impaired acrosome reaction (IAR) and 37 controls suggested equine FK506 binding protein (FKBP6) as an IAR-susceptibility locus in Thoroughbred stallions [14].
The objectives of the present study were to perform a GWAS for stallion fertility in Hanoverian warmblood horses. Estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) were employed as the target trait for stallion fertility in Hanoverian warmblood horses. The most significant association for stallion fertility was found for a single nucleotide polymorphism (SNP) within the gene phospholipase c zeta (PLCz1) on horse chromosome (ECA) 6. Close to PLCz1, capping protein (actin filament) muscle Z-line, alpha 3 (CAPZA3) was identified as a second potential candidate gene. We sequenced PLCz1 and CAPZA3 using cDNA and genomic DNA samples to construct the gene models and to perform mutation detection. Validation of the 48 polymorphisms identified within PLCz1 and CAPZA3 was performed in 237 Hanoverian stallions.
Results
Genome-wide association
In 228 Hanoverian warmblood stallions, the genome-wide association study (GWAS) for stallion fertility using a mixed linear model (MLM) analysis revealed the highest association for the genomic region at 45,571,963–45,612,102 base pairs (bp) on ECA6 for EBV-PAT (Figure 1A). Within this region, the highest associated SNP BIEC2-952439 (g.45586821C>T) reached a −log10P-value of 4.14 and explained 6.72% of the variance of EBV-PAT. Within this region, seven protein-coding genes, eight pseudogenes und two non-coding RNAs are annotated using the horse genome reference assembly EquCab2.0 (http://www.ensembl.org/Equus-caballus/) (Figure S1). We located the SNP BIEC2-952439 within intron 8 of the phospholipase c zeta 1 (PLCz1) gene, annotated at 45.571–45.612 Mb. Downstream to PLCz1, the gene capping protein (actin filament) muscle Z-line, alpha 3 (CAPZA3) is annotated at 45.612–45.614 Mb. The observed −log10P-values were plotted against the expected −log10P-values and the quantile-quantile (Q-Q) plots illustrated that the population stratification was eliminated through the model employed (Figure 1B). Analysis of the haplotype structure of the SNP BIEC2-952439 flanking genomic region identified 15 haplotype blocks. Haplotype block 9 at 45,402,588–45,586,821 bp extends over 194 kb and includes the genome-wide significantly associated SNP BIEC2-952439 (Figure S1).
Figure 1. Manhattan plot of −log10P-values of the genome-wide association study for estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in Hanoverian stallions using a mixed linear model analysis (A) and Q-Q plots of observed versus expected −log10P-values (B) from the mixed linear model analysis for EBV-PAT.

(A) On the X-axis, the SNPs are given by horse chromosome number. The −log10P-values for each SNP effect are plotted against the SNP position on each chromosome. Chromosomes are differentiated by colors. The color keys are given below the plot. The blue line indicates the threshold of the −log10P-values >4.0. The peak value (BIEC2-952439) is located on horse chromosome 6 at 45.556 Mb. (B) The expected −log10P-values (solid line) are plotted against the observed −log10P-values (black dots).
Sequencing of PLCz1 and CAPZA3
We selected PLCz1 and CAPZA3, alias LOC100067825, for re-sequencing because intron 8 of PLCz1 contained the EBV-PAT-associated SNP BIEC2-952439 and CAPZA3 was the second candidate gene in this region, approximately 27 kb downstream to BIEC2-952439. We detected four transcript variants of the equine PLCz1 in cDNA amplicons from testis tissue of each of the six stallions (Table 1, Figure S2). Each two transcripts differed in the size of the first untranslated exon. Transcripts JX545317 and JX545318) cover a smaller exon 1 (exon 1a) with 114 bp, whereas transcripts JX545319 and JX545320 show an extended exon 1 (exon 1b) containing 174 bp (Figure S3). Two transcripts (JX545318 and JX545320) contained a premature termination codon (PTC). The PTC resulted from the use of alternative acceptor splice sites at the boundary between intron 3 and exon 4 (Figure 2). Bioinformatic analyses using NetGene2 [15] and HSF 2.4.1 [16] for splice site prediction supported the existence of two adjacent acceptor splice sites at this intron-exon-boundary. For the non-PTC-containing transcripts (JX545317 and JX545319), the acceptor site was predicted with a reliability of 86% (HSF 2.4.1) and 97% (NetGene2). The acceptor site for the PTC-containing transcripts reached a reliability of 82% (HSF 2.4.1) and 63% (NetGene2). The PTC position at the exon-exon-junction (136 bp downstream to the translation start) identified in the equine PLCz1 is identical to the PTC position in human (Figure 3).
Table 1. Transcript variants detected in the equine PLCz1.
| Transcript variant ID | Exon 1 | PTC | mRNA | CDS | Amino acids |
| JX545317 | 114 | - | 2220 | 1917 | 638 |
| JX545319 | 174 | - | 2259 | 1917 | 638 |
| JX545320 | 174 | 5′ exon 4 | 2262 | 1605 | 534 |
| JX545318 | 114 | 5′ exon 4 | 2223 | 1605 | 534 |
| ENSECAT00000012304 | not annotated | - | 2088 | 1917 | 638 |
| XM_001497766 | not annotated | - | 2092 | 2049 | 682 |
Variants were detected in cDNA sequences of testis tissues from six Hanoverian stallions. The size of the untranslated exon 1, mRNA and coding sequence (CDS) in base pairs, the location of the premature termination codon (PTC) and the predicted number of amino acids are given.
Figure 2. Chromatograms of transcripts detected in equine PLCz1.

(A) The sequences of all animals tested show both adjacent splice sites of the cDNA. Both, forward and reverse sequences show ambiguous double traces when elongation passes exon boundary. (B, C) Visualization of single trace sequence is used to portray each single transcript. (B) For the transcripts without a premature termination codon (PTC), exon 4a starts with the residues GAT, (C) whereas PTC-containing transcript variants harbor an extended exon 4b and start with the residues TAG. (D) Genomic sequence was used to illustrate usage of adjacent acceptor splice sites AG-TA for PTC-containing transcripts and AG-GA for non-PTC-containing transcripts. Vertical lines mark the exon/exon boundary or intron/exon boundary, respectively. The black open boxes indicate specific nucleotide residues used.
Figure 3. Comparison of equine PLCz1 transcript variants with and human PLCz1.

Transcript variants (1) ENSECAT00000012304, (2) ENSECAT00000012372, (3) XM_001497766.3, (4) JX545317, (5) JX545319, (6) ENST00000318197, (7) NR_073075.1, (8) JX545318, and (9) JX545320 are presented. (1–4) Human and equine variants without a premature termination codon (PTC) in sequences are indicated. (5–9) A PTC is activated within human and equine variants at similar positions (c.136). The PTC region is highlighted by a red box.
The non-PTC-containing transcripts (JX545317 and JX545319) showed an open reading frame (ORF) of 1917 bp. For PTC-containing transcripts, we performed an in-silico analysis using ATGPR for prediction of ORFs. Based on this analysis, two possible ORFs were distinguished. The first ORF (ORF1) predicted with a very low probability (0.06%) encodes a 45 amino acid (aa) containing protein. In human, at a similar position a truncated protein of 45 aa is predicted to elicit nonsense-mediated mRNA decay (NMD) (Figure S4). Due to the very low probability for the predicted ORF1 according to the in-silico analysis, we assumed that a protein resulting from ORF1 deems to be non-existent in horses. The second ORF (ORF2) was predicted with a probability of 25% to contain 1605 bp and to encode a protein of 534 aa. The usage of the alternative splice site extended exon 4 (exon 4b) by three base pairs (Figure S1). Translation start for ORF2 is located at the 3′end of exon 4 and translation termination is consistent with the non-PTC-containing transcripts.
Mutation detection
Several mutations within PLCz1 were reported to affect human male fertility [17]. PLCz1 is involved in male fertility by triggering [Ca2+] oscillation during fertilization [18]. CAPZA3 localized predominantly in the neck region of ejaculated sperm may play a role in sperm architecture and male fertility in man [19] and mice [20], [21]. Polymorphism detection was performed using genomic DNA of 19 fertile Hanoverian stallions with low (n = 6), moderate (n = 6), high (n = 3) and very high (n = 4) EBV-PAT (Table S1). Re-sequencing of all exons and exon-intron boundaries of PLCz1 and CAPZA3 revealed a total of 46 SNPs, whereof 45 were located within PLCz1 and one within CPAZA3 (Table S2). In addition, two intronic indels were detected (g.45610678delA and g.45581388delTTAA) within PLCz1. Validation of exonic mutations was done using the cDNA sequences of the six Hanoverian stallions employed for the analysis of the gene models. All exonic mutations could be identified in these six animals. In addition, to preclude effects of the indels on cDNA sequence, we verified the two intronic indels in three out of six of these animals.
Within PLCz1, we identified 40 novel SNPs and confirmed five SNPs previously annotated by the Broad Institute (http://www.broadinstitute.org/ftp/distribution/horse_snp_release/v2/). Out of these 45 SNPs, eight were exonic, 33 intronic, one in the 5′UTR and three in the 5′ promoter region (Figure 4). In total, four exonic SNPs were missense mutations. A missense mutation within exon 9 (g.45586134G>C) caused an exchange of glutamine with glutamic acid (p.Gln400Glu) and had been classified as possibly damaging using PolyPhen-2 [22]. All other missense mutations were classified as benign. Within CAPZA3, a missense mutation (p.Leu259Pro) was detected. This mutation had been classified as probably damaging using PolyPhen-2 [22].
Figure 4. Equine gene model of PLCz1 and polymorphisms detected.
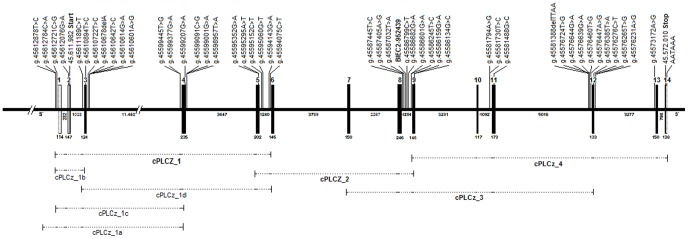
Sequence analyses of PLCz 1 revealed four transcripts of which the full-length variant (JX545317) is shown. Translated exons are shown as solid black boxes, untranslated exons as open boxes. Numbers above indicate the exon number. Continuous lines represent introns, numbers below the exons and introns indicate the sizes in base pairs. The predicted translation start and stop and the polyadenylation site is indicated. The highly associated single nucleotide polymorphism (SNP) BIEC2-952439 identified in a genome-wide association study (GWAS) is given in bold. The dashed lines below represent the coverage of the complementary DNA (cDNA) primers.
Association analysis of candidate gene-associated polymorphisms
Association analysis in the detection sample (n = 19) revealed six SNPS and one indel significantly associated with EBV-PAT within PLCz1 (Table S3). No EBV-PAT-associated SNP was found within CAPZA3. The minor allele frequencies (MAF) of the polymorphisms were at 0.05–0.37. Nominal P-values were at 0.015–0.05.
Validation of candidate gene-associated polymorphisms
In total, we genotyped 16 polymorphisms and imputed the remaining 32 SNPs in 237 Hanoverian stallions. In order to genotype all polymorphisms possibly associated with EBV-PAT, we performed three rounds of genotyping and imputation. First, we genotyped the seven polymorphisms significantly associated in the detection sample and in addition, two synonymous and two non-synonymous exonic SNPs including the CAPZA3-related SNP. Then, we imputed the genotypes of the remaining 37 SNPs and performed an association analysis for all 48 polymorphisms. In the second round, we genotyped 13 polymorphisms and imputed 35 SNPs and in the third step, genotypes of 16 SNPs were available and imputed the genotypes of 31 SNPs. After each round, the SNPs with imputed genotypes and highest associations with EBV-PAT were genotyped. In the final association analysis, 14 polymorphisms were significantly associated with EBV-PAT (Table 2). After accounting for multiple testing using a Bonferroni correction, two genotyped SNPs (g.45599001G>A, PBonf = 0.047 and BIEC2-952439 (g.45586821C>T), PBonf = 0.047) and one imputed SNP (g.45595060G>T, PBonf = 0.037) reached significant associations with EBV-PAT.
Table 2. Significant association for 14 PLCz1-polymorphisms with estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in 237 Hanoverian stallions.
| Polymorphism | MAF | PIC | HET | P-HWE | R2 | P-Allele | P-Genotype | P-Bonf |
| g.45595060G>T | 0.148 | 0.368 | 0.397 | 0.005 | 5.31 | 0.001 | 0.003 | 0.047 |
| g.45599001G>A | 0.238 | 0.297 | 0.401 | 0.110 | 6.40 | 0.002 | 0.001 | 0.047 |
| g.45581730T>C | 0.046 | 0.085 | 0.093 | 0.454 | 3.95 | 0.004 | 0.004 | |
| g.45586682G>A | 0.439 | 0.371 | 0.481 | 0.719 | 3.03 | 0.011 | 0.040 | |
| g.45576724T>G | 0.435 | 0.371 | 0.473 | 0.554 | 2.83 | 0.013 | 0.049 | |
| g.45599377G>A | 0.205 | 0.273 | 0.367 | 0.049 | 5.03 | 0.018 | 0.005 | |
| g.45594075C>T | 0.217 | 0.282 | 0.367 | 0.223 | 5.03 | 0.021 | 0.005 | |
| g.45586821C>T | 0.247 | 0.303 | 0.376 | 0.878 | 6.45 | 0.027 | 0.001 | 0.047 |
| g.45581794A>G | 0.232 | 0.293 | 0.380 | 0.314 | 3.84 | 0.031 | 0.016 | |
| g.45581388delTTAA | 0.232 | 0.293 | 0.329 | 0.238 | 2.47 | 0.032 | 0.072 | |
| g.45586245T>C | 0.458 | 0.373 | 0.511 | 0.662 | 3.99 | 0.033 | 0.014 | |
| g.45576460T>G | 0.006 | 0.013 | 0.013 | 0.922 | 1.88 | 0.047 | 0.046 | |
| g.45586601G>A | 0.460 | 0.373 | 0.515 | 0.577 | 4.80 | 0.069 | 0.006 | |
| g.45612878T>C | 0.030 | 0.056 | 0.042 | 0.0001 | 4.60 | 0.067 | 0.008 |
For each polymorphism, minor allele frequency (MAF), polymorphism information content (PIC), heterozygosity (HET), test for Hardy Weinberg equilibrium (HWE) with the respective P-value (P-HWE), variance explained by the polymorphism for EBV-PAT (R2) and P-values for allelic (P-Allele) and genotypic (P-Genotype) association as well as after Bonferroni correction for multiple testing (P-Bonf) are given.
Haplotype association for SNPs within PLCz1
In the subsequent haplotype analysis, we identified seven haplotype blocks using Haploview 4.2 [23] and the four gamete rule algorithm. Pairwise linkage disequilibria (r2) varied widely, but 24 SNPs reached r2-values >0.80 (Figure S5). Four adjacent haplotype blocks (block 2-block 5) were significantly (P = 0.005) associated with EBV-PAT (Table S4). The three significantly EBV-PAT-associated SNPs were contained in these haplotype blocks. Together, these haplotype blocks explained 17.36% of the variance (R2) of EBV-PAT in Hanoverian stallions. Variances explained by single haplotype blocks were at 4–11% (Table 3).
Table 3. Haplotype blocks within PLCz1 significantly associated with estimated breeding values for the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in Hanoverian stallions.
| Haplotype block | POS | SNP | R2 (%) | P |
| Block 2- Block 5 | 45,586,245–45,599,091 | 17.36 | 0.005 | |
| Block 2 | 45,586,245–45,586,601 | 4.09 | 0.013 | |
| Block 3 | 45,586,682–45,594,075 | BIEC2-952439 | 10.88 | 0.002 |
| Block 4 | 45,594,143–45,595,060 | g.45595060G>T | 4.86 | 0.015 |
| Block 5 | 45,595,352–45,599,091 | g.45599001G>A | 7.69 | 0.011 |
The start and end of the haplotype block in base pairs (POS), the variance explained (R2) by each haplotype block, significantly associated SNPs within haplotype blocks (SNP) and P-values (P) are given.
Discussion
Herein, we report a successful application of Equine SNP50 Genotyping Beadchip and GWAS for stallion fertility, implicating equine PLCz1 as a locus for stallion fertility. The haplotype analysis corroborated the associations for single SNPs. We assume that several SNPs within PLCz1 may have effects on trait expression and particularly, SNPs within the significantly associated haplotype blocks may be important for EBV-PAT in Hanoverian stallions. In comparison to the previous candidate gene studies [6]–[10], the variance explained by single PLCz1-associated SNPs and haplotypes was much larger. This may indicate the higher power of genome-wide studies contrasted to candidate gene studies. The associated SNPs do not influence the coding sequence of PLCz1, but may exhibit regulatory functions. Intronic mutations can affect transcription factor binding sites (TF) [24] and may cause significant phenotype diversity [25]. The highest associated SNP BIEC2-952439 influences the TF for estradiol receptor-alpha and -beta (ER-alpha, ER-beta). ER-alpha and ER-beta activated by estradiol influence male sperm metabolism and are involved in the pathophysiology of varicocele-associated male infertility [26]. Furthermore, estradiol exerts its cellular effects through estrogen receptors and acts as a germ cell survival factor in the human testis [27]. We propose, therefore, that intronic PLCz1 SNPs associated with EBV-PAT may have regulatory functions via TFs and transcription levels of PLCz1.
Testis-specific mammalian PLCz1 occupies an important key role in the cascade of male fertility. During fertilization sperm-induced [Ca2+] oscillation is essential for oocyte activation and support of embryonic development [28]–[30]. In mammals, this highly conserved mechanism [31]–[35] is triggered by PLCz1 released from the sperm during fertilization [28], [36]. In infertile men, defects in expression and abnormal patterns of PLCz1 were observed [37]–[39]. Furthermore, missense mutations within human PLCz1 confer male infertility [17], [40]–[42]. This could be confirmed in an equivalent mouse model [37]–[39]. The D202R (D210R in mice) mutation of in X-catalytic domain abrogated the Ca2+ oscillatory ability of PLCz1 by failure of nuclear translocation [43], [44]. Recently, species-specific differences in sequence and expression patterns of equine PLCz1 were reported [35], [45]. Subfertile stallions showed a reduced PLCz1 expression [45], [46] as well as abnormal patterns of localization of PLCz1 in the sperm [45]. In pigs, higher expression levels of PLCz1 were related with better sperm quality [47]. A positive correlation of PLCz1 expression levels with sire conception rate was observed in Holstein bulls [48]. In Chinese Holstein bulls, semen quality traits were shown significantly associated with single SNPs in the 5′ flanking region of PLCz1 and the haplotypes constructed from these SNPs [49].
Out of the 47 PLCz1 polymorphisms, three SNPs (g.45595060G>T, g.45599001G>A and BIEC2-952439) reached significant associations with EBV-PAT in the validation sample after accounting for multiple testing. Using in-silico predictions, potential regulatory functions may be assumed for all 14 EBV-PAT-associated SNPs (Table S5). Searches for splice-modifying SNPs predicted g.45595060G>T as a candidate to disrupt the binding sites for putative exonic splicing enhancers (ESE) octamer motifs. ESEs stimulate alternative and constitutive splicing and serve as binding sites for splicing factors like the serine/arginine-rich proteins (SR proteins) [50], [51]. ESEs are exonic sequence elements. However, less frequent abundance of ESEs sequences in introns and their specific interaction with splicing factors have also been documented [51]–[53]. There are reports about deep intron mutations creating ESE motifs and splice factor binding sites which lead to pseudoexon activation [54]–[56]. Consequently, even little is known about the mechanisms by which intronic sequence variations can activate pseudoexons and may alter trait expression, we can speculate that the deep intron mutation g.45595060G>T could lead to inaccurate recognition of exon/intron boundaries and therefore modulate the splicing pattern of equine PLCz1.
Disruption of an intronic microRNA (miRNA) motif was predicted for the SNP g.45599001G>A. These small non-coding regulatory RNAs are able to interfere post-transcriptionally with the protein production of their targets by suppressing translation or destabilizing mRNAs [57]. They are involved in mammal core cellular processes [57], [58] as well as in many human diseases [59]–[61]. Numerous intronic sequences have been found to encode miRNAs, responsible for gene silencing [62]. Mammalian miRNAs decrease target mRNA levels and translational efficiency resulting in reduced protein output [63]. In mammalian reproduction pathways, miRNAs play a potential role in testicular and ovarian physiology [64]. MiRNAs are associated with functional regulation of Leydig cells and Sertoli cells in testis in steroid synthesis which suggest the potential involvement of miRNAs in translational control during spermatogenesis [64]. The pattern of miRNAs expressed in infertile men suggested their role in regulating male germ and somatic cells and that their alteration is associated with reproductive abnormalities [65]. A recent study in livestock animals including horses showed a profound effect of miRNA polymorphisms on a wide range of phenotypic traits in animal production [66]. The miRNA motif when removed by the SNP g.45599001G>A may alter protein expression level of PLCz1.
The SNP g.45599001G>A creates a TF for the hepatocyte nuclear factor 3-alpha (HNF-3alpha). HNF-3alpha is involved in embryonic development, establishment of tissue-specific gene expression and regulation of gene expression in differentiated tissues [67].
Comparison of the sequences generated in the present study to the annotated equine PLCz1 sequences showed several differences. Ensembl transcript 1 (ENSECAT00000012304) and NCBI transcript (XM_001497766.3) were annotated with 13 exons whereas Ensembl transcript 2 (ENSECAT00000012372) consists of 15 exons. Non-coding exon 1 homologs were not annotated in the horse genome reference assembly EquCab2.0. The coding sequences (CDS) and protein sequences of non-PTC-containing transcripts are consistent with the Ensembl (ENSECAT00000012304 and ENSECAP00000009710) annotation, however, different to NCBI variants XM_001497766.2 and XP_001497816.2 (Figure S4). The mRNA sequences of non-PTC-containing transcripts exhibit 85% homology to human (NM_033123.3) and 75% to murine PLCz1 (NM_054066.4) mRNA, respectively. They are predicted to encode a protein of 638 aa which displays 82% similarity to the human PLCz1 protein (NP_149114.2) and 70% sequence similarity to the murine protein (NP_473407.2). CLUSTAL OMEGA alignment displayed high interspecies conservation across PLCz1, except for the X-Y linker domain.
Sequencing of CAPZA3 cDNA revealed a transcript consistent with the annotation in Ensembl (ENSECAT00000004119). The mRNA sequence exhibits 90.3% homology to human (ENST00000317658) and 82.0% to murine CAPZA3 (ENSMUST00000043797) mRNA, respectively. The predicted equine CAPZA3 protein displays 84.9% similarity to the human CAPZA3 protein (ENSP00000326238) and 85.2% similarity to the murine protein (ENSMUSP00000038562).
In human, two isoforms of PLCz1 have been described. Human transcript variant 1 (NM_033123.3) encodes the functional protein whereas transcript variant 2 (NR_073075.1) is considered to be non-protein coding due to the existence of a PTC. We determined two equine transcript variants of PLCz1 (JX545318, JX545320) that harbour a PTC at the same position as shown in human PLCz1 transcript variant 2 (NR_073075.1). Both equine PTC-containing transcript variants are predicted to result in truncated proteins and to elicit NMD. Comparison of equine ORF2 to human PLCz1 pinpointed a truncated protein (ENSP00000443320) with a similar size (412 aa) (Figure S4). A PTC is located >50 nucleotides upstream of the 3′most exon–exon junction [68]. PTC-containing transcripts frequently arise from alternative splicing events and elicit NMD [68], [69] in order to prevent production of potentially damaging truncated proteins [70]–[73] and to regulate normal gene expression by controlling the frequency of transcripts [74], [75]. An example for alternative splicing pattern activated by a PTC was recently described in the human GABRG2 gene associated with childhood absence epilepsy [76], [77]. Furthermore, alternative splicing induced intron retention in human ribosomal protein L3 (rpL3) gene was considered to activate PTC and targeted mRNA for NMD. Overexpression of rpL3 caused downregulation of canonical spliced mRNA and upregulation of alternative spliced mRNA, promoted by hnRNP H1 and other splicing factors for selection of alternative splice site [78]. Therefore, we propose that the equine PTC-containing PLCz1 transcripts may also respond to NMD.
Even if most of the eukaryotic mRNAs contain only one ORF, translation can be initiated at a downstream start codon due to leaky scanning [79], [80]. Leaky scanning skips the major ORF and enables initiation of translation of internal second ORFs [81]. A strong identity to kozak (GXXATGa) [81], [82] represented by downstream translation start of the PTC-containing transcripts may give evidence to the existence of ORF2. Nevertheless, the 534 aa protein derivated from ORF2 lacks 2/3 of N-terminal EF-hand domains (helix-loop-helix structural domains). Loss of EF-hand domains sequence is crucial for the ability of enzymes to initiate timely Ca2+ oscillation within the oocytes [35], [37], [44]. Thus, a potentially not fully functional protein will arise from ORF2. Even if all the different transcript variants could be observed in the six Hanoverian warmblood horses sequenced, it is unlikely that PLCz1 splicing patterns share a relationship with EBV-PAT. In summary, we can hypothesize that the equine PLCz1 transcript without a PTC may be dominantly expressed.
Even if it has been demonstrated that the closely neighbouring sperm-specific intronless CAPZA3 is of notably interest in human [19] and mice fertility [20], [21], no significant association with EBV-PAT was found for the CAPZA3-related SNP in Hanoverian stallions. Based on failing validation, this gene had to be eliminated as candidate locus for Hanoverian stallions.
In conclusion, we identified PLCz1 as an important gene for equine male fertility. The polymorphisms significantly associated with EBV-PAT may have regulatory effects on transcription levels of the gene. Several SNPs may influence the effects of PLCz1 and these SNPs are located in three adjacent haplotype blocks.
Materials and Methods
Ethics statement
All animal work has been conducted according to the national and international guidelines for animal welfare. All horses included in this study were owned by the National State Stud of Lower Saxony in Celle and the Landstallmeister Dr. Brockmann of the National State Stud of Lower Saxony in Celle gave his explicit consent for this study. The Lower Saxony state veterinary office at the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany, was the responsible Institutional Animal Care and Use Committee (IACUC) for this specific study. The EDTA-blood sampling for the present study had been approved by the IACUC of Lower Saxony, the state veterinary office Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (registration number 33.42502-05-07A482).
When a male horse should be castrated for reasons other than research, we were informed by the National State Stud of Lower Saxony in Celle. The castration was performed by a veterinarian specialized in equine reproductive medicine (GM). Then, we could collect testis samples from these horses after castration and therefore, a specific approval through the IACUC was not necessary according to the German Animal Welfare Law (released on 05/18/2006, last changes on 07/08/2013). All testis tissue samples used in the present study were from horses that were owned by the National State Stud of Lower Saxony in Celle.
Animals, phenotypes data, DNA and RNA extraction
EDTA-blood samples of 228 Hanoverian warmblood stallions from the National State Stud of Lower Saxony in Celle were collected for the present GWAS-study. Fertility data contained >20,000 mares with >200,000 artificial inseminations (AI) records of >600 stallions for calculation of the pregnancy rate per estrus and estimation of breeding values for the paternal component of the pregnancy rate per estrus cycle (EBV-PAT). The model used for prediction of EBV-PAT are described elsewhere [83]. Briefly, this model included an additive genetic component for the stallion and an additive genetic component for the maternal effect. Non-genetic effects taken into account included month and year of insemination, management of insemination and semen, age of the mare and number of foals given birth by the mare. Calculations were performed using an animal threshold model.
Genomic DNA from EDTA-blood samples was isolated using standard methods with red blood cell lysis buffer and sodium EDTA buffer. The DNA concentration of the samples was adjusted to 50 ng/µl using Nanodrop ND-1000 (Peqlab Biotechnology, Erlangen, Germany).
For sequence analysis of genomic DNA, we prepared genomic DNA from EDTA-blood samples of 19 Hanoverian warmblood stallions using the NucleoSplin 96 Blood DNA kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions. This sample contained 19 fertile Hanoverian stallions with low, moderate, high or very high EBV-PAT (Table S1). These 19 stallions were not contained in the detection and validation sample.
Testis tissue samples were collected from six male Hanoverian horses that were castrated due to the owner's decision. For sequencing cDNA, testis tissue was conserved in RNAlater solution (Qiagen, Hilden, Germany) or liquid nitrogen. The RNeasy 96 Lipid Tissue Kit (Qiagen) was used to extract total RNA from testis tissue. The RNA was transcribed into cDNA using Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Fermentas, St. Leon-Rot, Germany) according to the manufacturer's protocol.
For validation of candidate gene-associated polymorphisms, genomic DNA samples of 237 Hanoverian stallions were analysed. This sample included the 228 Hanoverian stallions employed for the GWAS.
Genome-wide association study
A total of 228 Hanoverian stallions were genotyped using the Equine SNP50 Beadchip (Illumina, San Diego, CA, USA) including 54,602 SNPs. Quality criteria were minor allele frequency (MAF) >0.05, genotyping rate per SNP and animal >0.90 and HWE (P<0.00001). After filtering for quality criteria, 46,074 SNPs remained for the final analysis. A mixed linear model (MLM) was employed to control spurious associations caused by population structure. The MLM included genotype as fixed effect and a random animal effect through an identity-by-state-kinship (IBS) matrix. A Q-matrix to detect population structure was estimated using principal components (PCAs). We tested several extended models employing up to six PCAs to show the robustness of the outcome of the GWAS. All these models yielded the same associated SNPs as the final model. Thus, adding principal components for cryptic data structure did not change the results of the final model. Thus, the IBS matrix reflected the genomic relationship matrix among all individuals genotyped and captured the relatedness among animals as well as the cryptic family structure. The analysis was run using TASSEL, version 3.0.146 [85]. The adaptive false discovery rate according to Benjamini-Hochberg and the Bonferroni correction were calculated using the MULTIPLE TEST procedure of SAS, version 9.3, to determine the threshold for experiment-wide significance. The haplotype structure of the associated genomic regions was analyzed using Haploview 4.2 [23].
Bioinformatic analyses and sequencing
In order to build an equine gene model, we employed human mRNA as a reference sequence. Using BLAST (http://www.ncbi.nlm.nih.gov/BLAST/) and SPIDEY (http://www.ncbi.nlm.nih.gov/spidey/), we determined the equine sequence and its genomic location on equine chromosome 6. To compare horse, human and murine mRNAs and protein sequences we used the multiple-alignment program CLUSTALW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The splice site prediction tools Human Splicing Finder (HSF) 2.4.1 (www.umd.be/HSF/) and NetGene2 (www.cbs.dtu.dk/services/NetGene2) were used to predict the effect of intronic variants on splicing processes and to analyze the effect of polymorphisms on putative splicing regulatory sequences. Further, we used HSF 2.4.1 sequence analysis matrices to identify branch point, enhancer and silencer motifs. SNPs influencing transcription factor binding sites were predicted using Alibaba 2.1 (www.gene-regulation.com/pub/programs/alibaba2/bin/index.html) and Patch1.0 (www.gene-regulation.com/cgi-bin/pub/programs/patch/bin/patch.cgi). To predict potentially regulatory micro RNA (miRNA) motifs in human and equine sequences with essential roles in transcriptional and post-transcriptional regulation of gene expression, we employed miRBase, a miRNA database (http://www.mirbase.org/) [84], [85]. The software PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [22] was used to predict possible impact of an amino acid substitution on the structure and function of the equine protein. Open reading frame prediction was performed using ORF finder (http://www.ncbi.nlm.nih.gov/projects/gorf/), prediction of initiation codons in cDNA sequences was performed using ATGPR (http://atgpr.dbcls.jp/).
The genes PLCz1 and CAPZA3 were selected for sequencing using genomic DNA and cDNA. We amplified all exons, exon-intron boundaries and the complete intron 8 of PLCz1 containing the EBV-PAT-associated SNP BIEC2-952439 (Table S6). The primer sequences were obtained using equine reference mRNA and genomic sequence of ECA6 (ENSECAT00000012304 and ENSECAG00000011373) of the horse genome reference assembly EquCab2.0. The primer design was carried out with Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) after masking repetitive sequences using the RepeatMasker (http://www.repeatmasker.org/). In order to detect variation in non-annotated equine exon 1 of PLCz1, we designed several 5′UTR primer combinations as shown in Table S6. PCR reactions were performed in 20-µl reaction volumes using 10 ng DNA, 100 µM dNTPS, 10 pmol of each primer and 2 units/µl Taq DNA polymerase (MP Biomedicals, Eschwege). After 4 min initial denaturation at 94°C, 35 cycles of 30 s at 94°C, 30 s annealing temperature and 45 s at 72°C and 10 min final extension at 72°C were performed on a PTC 100 thermal cyclers (MJ Research, Watertown, MA, USA). After purification the PCR products using EXO-SAP, amplicons were directly sequenced using the Big Dye Sequencing Kit (Life Technologies, Darmstadt, Germany) on an ABI Prism 3500 (Life Technologies).
Mutation analysis and genotyping of polymorphisms in the validation sample
Sequence data were analysed using Sequencher 4.8. (Gene Code, Ann Arbor, MI, USA). All 48 polymorphisms identified were tested for association in the detection sample (n = 19) and validated in 237 Hanoverian stallions. Genotyping of polymorphisms was performed using PCR-RFLP or TaqMan SNP Genotyping Assay (Table S7). For enzymatic digestion, we used 15-µl reaction volume containing 1.5 µl buffer, 1.5 U endonucleases and 5 µl PCR amplicons. A mismatch PCR-RFLP technique [86] was used in the case that no restriction site was present. For genotyping the exonic SNP g.45599207G>A, a custom TaqMan SNP Genotyping Assay (Life Technologies) was applied. The assay incorporates 5′ allele-specific labeled TaqMan MGB probes (FAM and VIC dye-labeled) and unlabeled assay primers. The assay contained in 12-µl reaction volume Maxima Probe qPCR Master Mix 2× (Fermentas, St. Leon-Rot), 0.07 ROX solution 50 µM (Fermentas, St. Leon-Rot), 0.25 µl Custom TaqMan SNP genotyping assay and 2 µl template DNA. RT-PCR was carried out using the Applied Biosystems 7300 Real-Time PCR System (Life Technologies). The indel mutation g.45581388delTTAA was genotyped using DY-682-tagged primers (Eurofins MWG Operon, Ebersberg, Germany). PCR reactions were performed as described above. The PCR products were analyzed by gel electrophoresis on the automated sequencer LI-COR 4300 (LI-COR, Bad Homburg, Germany) using a 4% polyacrylamide denaturing gels (Rotiphorese Gel40, Carl Roth, Karlsruhe, Germany). Differences in allele sizes caused by deleted or inserted bases were defined by visual examination.
Statistical analysis of the validation samples
The ALLELE procedure of SAS/Genetics was used to calculate allele and genotype frequencies, the minor allele frequency (MAF), the polymorphism information content (PIC), the observed heterozygosity (HET), allelic diversity and χ2-tests for Hardy-Weinberg equilibrium for the SNPs genotyped in the validation sample. Calculation of association was performed using PLINK, version 1.07, and the procedure GLM of SAS, version 9.3. Correction for multiple testing according to Bonferroni was done using the MULTIPLE TEST procedure of SAS, version 9.3. Imputation of genotypes was carried out using BEAGLE 3.3.2 [87], [88]. We applied a population linkage disequilibrium based localized haplotype clustering method. We selected all polymorphisms sequenced (n = 48) in 19 stallion and a subset of 16 SNPs genotyped in 237 animals as reference cohort to impute the remaining genetic variants within PLCz1.
Calculation of pairwise linkage disequilibria (LD) was performed and visualized using Haploview 4.2 [23]. To define haplotype blocks and to analyse the haplotype structure of the PLCz1-associated polymorphisms, we used the four gamete rule algorithm with a D' value >0.8. We excluded polymorphisms with a MAF<0.01. The proportion of variance explained for EBV-PAT by polymorphisms or haplotypes was estimated using the procedure GLM of SAS, version 9.3.
Supporting Information
Haplotype structure of the genomic region at 44,000,970–46,952,651 base pairs on horse chromosome (ECA) 6 and genes annotated on the horse genome reference assembly EquCab2.0. Protein-coding genes are highlighted in bold, pseudogenes are marked in grey and non-coding RNAs are marked in light purple. Haplotype blocks were defined using the four gamete rule algorithm. The haplotype block 9 in intron 8 of the equine PLCz1 contains the SNP BIEC2-952439 significantly associated with estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in Hanoverian stallions. The figure displays Hendrige's multiallelic D, which represent the degree of linkage disequilibrium between each two SNPs. Red fields display LOD≥2 (D′ = 1), shades of red show the same LOD with D′<1. White and blue fields display LOD<2 with D′<1 and D′ = 1, respectively.
(TIF)
Transcript variants of equine PLCz1 detected. (A) Usage of variant-specific 5′ UTR primer revealed non-coding exon 1 variation shown in transcripts. Blue errors indicate position of specific primers and evaluated start for exon 1a and exon 1b. Exons show overlapping position, with an exon 1b position more downstream of exon 1a. Due to study design, definitely 5′ start of exons 1 was not determined, indicated by dashed lines. (B) Comparison of mature mRNA transcript variants. They differ in size due to usage of variable exon1 (1a and 1b) and exon 4 (4a and 4b).
(TIF)
Transcript variants of equine PLCz1 detected in testis tissue. Sequence analyses revealed four different transcripts variants. Translation start and stop are given for each transcript (A: JX545319, B: JX545320, C: JX545317, D: JX545318). Variants show different size of non-coding exon 1. Exon 1a represented in the first primary transcript (A) and in derived transcript (B) contains 114 base pairs (bp), whereas exon 1b represented in second primary transcript (C) and in derived transcript (D) consists of 174 bp. The non- premature-terminating-codon (PTC) containing variants (A, C) consist of the translated exon 4a whereas truncated variants (B, D) show a PTC in exon 4b leading to two potential open reading frames (ORFs, ORF1 and ORF2). Translated exons are shown as solid black boxes. Untranslated exon regions are shown as open boxes. The scale is in bp.
(TIF)
Protein sequence alignments of equine and human PLCz1 variants. (1,2) Sequences highlighted in yellow list truncated PLCz1 protein of 45 amino acids (aa) in human (ENSP00000326397) and equine (JX545318, JX545320). (3–6) Sequence variations of predicted equine protein annotation (3: JX545317, JX545319; 4: ENSECAP00000009710; 5: ENSECAP00000009768; 6: XP_001497816.2) are shown. (7) Interspecies alignment with reviewed human peptide sequence of PLCz1 (NP_149114.2). (8,9) Sequences highlighted in green list truncated PLCz1 protein of 412 aa and 534 aa in human (ENSP00000443320) and equine (JX545318, JX545320). Truncated sequences may be generated due to activation of the premature termination codon (PTC) or are thought to undergo nonsense mediated decay (NMD). The open boxed region corresponds to the N-terminus located EF-hand domain, whereas the boxed region marks the X-Y linker domain. Human sequences are highlighted in bold.
(TIF)
Linkage disequilibria (LD) among 47 polymorphisms within PLCz1 in 237 Hanoverian stallions. (A) The LD display shows Hedrige's multiallelic D which represents the degree of LD between two blocks. Red fields display LOD≥2 (D′ = 1), shades of red show the same LOD with D′<1. White and blue fields display LOD<2 with D′<1 and D′ = 1, respectively. The pairwise LD coefficients (r2) values are shown for each SNP pair in fields. (B) Haplotype blocks were defined using the four gamete rule algorithm. Each haplotype in a block with its frequency and connections from one block to the next block is displayed. In the crossing areas, a value for the multiallelic D′ is shown, representing the level of recombination between the two blocks. Significantly associated haplotype blocks and significantly associated SNPs within haplotype blocks are framed in red.
(TIF)
Hanoverian stallions used in the detection sample and their estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) on the natural scale and standardized scale (R-EBV-PAT: 100±20) as well as the accuracy of EBV-PAT. Stallions were sorted in four groups according to their EBV-PAT.
(DOCX)
Polymorphisms identified in equine PLCz1 and CAPZA3 . Identification of polymorphisms, their accession numbers, their intragenic locations and their possible effects on the coding sequence including the nomenclature for the protein changing mutations are given.
(DOCX)
Association analysis for 48 polymorphisms with the estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in the detection sample using 19 Hanoverian stallions. The variance explained (R2) by each polymorphism, the P-values (P) for association with EBV-PAT and minor allele frequencies (MAF) are given.
(DOCX)
Haplotypes within PLCz1 significantly associated with the estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in 237 Hanoverian stallions. Significant haplotypes, their frequencies, corresponding haplotype blocks, least-square means (LSM) with their standard errors (SE) and P-values (P) are given.
(DOCX)
Predicted influence of EBV-PAT-associated polymorphisms within PLCz1 on transcription factor (TF) binding sites, microRNA (miRNA) and splice-site modifying motifs.
(DOCX)
Primer sequences and their positions, product sizes and annealing temperatures (AT) for sequencing the equine genomic DNA and cDNA of PLCz1 and CAPZA3 .
(DOCX)
Genotyping techniques used for validation of polymorphisms in 237 Hanoverian stallions. All single nucleotide polymorphisms (SNPs) genotyped using RFLPs (restriction fragment length polymorphisms) or IRD (infrared dye) labeled gel electrophoresis are given.
(DOCX)
Acknowledgments
We acknowledge the support of the National State Stud of Lower Saxony, Celle, and the German Hanoverian studbook society for providing data and samples of Hanoverian warmblood stallions. We thank Ute Philipp and Jörn Wrede for expert technical support.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files. Sequence data of this article has been deposited at the NCBI/GenBank Data Libraries for the Equus caballus genomic sequence of PLCz1 (JX545317-JX545320). Newly discovered polymorphisms were submitted to NCBI dbSNP database. Their accession numbers are in Table S2.
Funding Statement
This study was supported by the Mehl-Mülhens Stiftung, Köln (DI-MM/1-1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Layman LC (2002) Human gene mutations causing infertility. J Med Genet 39: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Layman LC (2003) Genetic causes of human infertility. Endocrinol Metab Clin North Am 32: 549–572. [DOI] [PubMed] [Google Scholar]
- 3. Truong BN, Moses EK, Armes JE, Venter DJ, Baker HW (2003) Searching for candidate genes for male infertility. Asian J Androl 5: 137–147. [PubMed] [Google Scholar]
- 4. Ferlin A, Bettella A, Tessari A, Salata E, Dallapiccola B, et al. (2004) Analysis of the DAZ gene family in cryptorchidism and idiopathic male infertility. Fertil Steril 81: 1013–1018. [DOI] [PubMed] [Google Scholar]
- 5. Peterlin B, Zorn B, Volk M, Kunej T (2006) Association between the apolipoprotein B signal peptide gene insertion/deletion polymorphism and male infertility. Molecular Human Reproduction 12: 777–779. [DOI] [PubMed] [Google Scholar]
- 6. Giesecke K, Hamann H, Sieme H, Distl O (2010) Evaluation of prolactin receptor (PRLR) as candidate gene for male fertility in Hanoverian warmblood horses. Reprod Domest Anim 45: e124–130. [DOI] [PubMed] [Google Scholar]
- 7. Giesecke K, Hamann H, Sieme H, Distl O (2010) INHBA-associated markers as candidates for stallion fertility. Reprod Domest Anim 45: 342–347. [DOI] [PubMed] [Google Scholar]
- 8. Giesecke K, Hamann H, Stock KF, Klewitz J, Martinsson G, et al. (2011) Evaluation of ACE, SP17, and FSHB as candidates for stallion fertility in Hanoverian warmblood horses. Anim Reprod Sci 126: 200–206. [DOI] [PubMed] [Google Scholar]
- 9. Giesecke K, Hamann H, Stock KF, Wöhlke A, Sieme H, et al. (2009) Evaluation of SPATA1-associated markers for stallion fertility. Anim Genet 40: 359–365. [DOI] [PubMed] [Google Scholar]
- 10. Hamann H, Jude R, Sieme H, Mertens U, Töpfer-Petersen E, et al. (2007) A polymorphism within the equine CRISP3 gene is associated with stallion fertility in Hanoverian warmblood horses. Anim Genet 38: 259–264. [DOI] [PubMed] [Google Scholar]
- 11. Penagaricano F, Weigel KA, Khatib H (2012) Genome-wide association study identifies candidate markers for bull fertility in Holstein dairy cattle. Anim Genet 43 Suppl 1: 65–71. [DOI] [PubMed] [Google Scholar]
- 12. Sieme H, Distl O (2012) Genomics and Fertility in Stallions. JEVS 32: 467–470. [Google Scholar]
- 13. Kosova G, Scott NM, Niederberger C, Prins GS, Ober C (2012) Genome-wide association study identifies candidate genes for male fertility traits in humans. Am J Hum Genet 90: 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raudsepp T, McCue ME, Das PJ, Dobson L, Vishnoi M, et al. (2012) Genome-wide association study implicates testis-sperm specific FKBP6 as a susceptibility locus for impaired acrosome reaction in stallions. PLoS Genet 8: e1003139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, et al. (1996) Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res 24: 3439–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, et al. (2009) Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Research 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heytens E, Parrington J, Coward K, Young C, Lambrecht S, et al. (2009) Reduced amounts and abnormal forms of phospholipase C zeta (PLCzeta) in spermatozoa from infertile men. Hum Reprod 24: 2417–2428. [DOI] [PubMed] [Google Scholar]
- 18. Swann K, Lai FA (2013) PLCzeta and the initiation of Ca(2+) oscillations in fertilizing mammalian eggs. Cell Calcium 53: 55–62. [DOI] [PubMed] [Google Scholar]
- 19. Miyagawa Y, Tanaka H, Iguchi N, Kitamura K, Nakamura Y, et al. (2002) Molecular cloning and characterization of the human orthologue of male germ cell-specific actin capping protein alpha3 (cpalpha3). Mol Hum Reprod 8: 531–539. [DOI] [PubMed] [Google Scholar]
- 20. L'Hote D, Serres C, Laissue P, Oulmouden A, Rogel-Gaillard C, et al. (2007) Centimorgan-range one-step mapping of fertility traits using interspecific recombinant congenic mice. Genetics 176: 1907–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geyer CB, Inselman AL, Sunman JA, Bornstein S, Handel MA, et al. (2009) A missense mutation in the Capza3 gene and disruption of F-actin organization in spermatids of repro32 infertile male mice. Dev Biol 330: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21: 263–265. [DOI] [PubMed] [Google Scholar]
- 24. Johnson AD, Wang D, Sadee W (2005) Polymorphisms affecting gene regulation and mRNA processing: broad implications for pharmacogenetics. Pharmacol Ther 106: 19–38. [DOI] [PubMed] [Google Scholar]
- 25. Wang X, Tomso DJ, Liu X, Bell DA (2005) Single nucleotide polymorphism in transcriptional regulatory regions and expression of environmentally responsive genes. Toxicol Appl Pharmacol 207: 84–90. [DOI] [PubMed] [Google Scholar]
- 26. Guido C, Perrotta I, Panza S, Middea E, Avena P, et al. (2011) Human sperm physiology: estrogen receptor alpha (ERalpha) and estrogen receptor beta (ERbeta) influence sperm metabolism and may be involved in the pathophysiology of varicocele-associated male infertility. J Cell Physiol 226: 3403–3412. [DOI] [PubMed] [Google Scholar]
- 27. Pentikainen V, Erkkila K, Suomalainen L, Parvinen M, Dunkel L (2000) Estradiol acts as a germ cell survival factor in the human testis in vitro. Journal of Clinical Endocrinology & Metabolism 85: 2057–2067. [DOI] [PubMed] [Google Scholar]
- 28. Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, et al. (2002) PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 129: 3533–3544. [DOI] [PubMed] [Google Scholar]
- 29. Saunders CM, Swann K, Lai FA (2007) PLCzeta, a sperm-specific PLC and its potential role in fertilization. Biochem Soc Symp 74: 23–36. [DOI] [PubMed] [Google Scholar]
- 30. Swann K, Saunders CM, Rogers NT, Lai FA (2006) PLC xi (zeta): A sperm protein that triggers Ca(2+) oscillations and egg activation in mammals. Semin Cell Dev Biol 17: 264–273. [DOI] [PubMed] [Google Scholar]
- 31. Cox LJ, Larman MG, Saunders CM, Hashimoto K, Swann K, et al. (2002) Sperm phospholipase Czeta from humans and cynomolgus monkeys triggers Ca2+ oscillations, activation and development of mouse oocytes. Reproduction 124: 611–623. [DOI] [PubMed] [Google Scholar]
- 32. Coward K, Ponting CP, Chang HY, Hibbitt O, Savolainen P, et al. (2005) Phospholipase Czeta, the trigger of egg activation in mammals, is present in a non-mammalian species. Reproduction 130: 157–163. [DOI] [PubMed] [Google Scholar]
- 33. Yoneda A, Kashima M, Yoshida S, Terada K, Nakagawa S, et al. (2006) Molecular cloning, testicular postnatal expression, and oocyte-activating potential of porcine phospholipase Czeta. Reproduction 132: 393–401. [DOI] [PubMed] [Google Scholar]
- 34. Ito M, Shikano T, Oda S, Horiguchi T, Tanimoto S, et al. (2008) Difference in Ca2+ oscillation-inducing activity and nuclear translocation ability of PLCZ1, an egg-activating sperm factor candidate, between mouse, rat, human, and medaka fish. Biol Reprod 78: 1081–1090. [DOI] [PubMed] [Google Scholar]
- 35. Bedford-Guaus SJ, McPartlin LA, Xie J, Westmiller SL, Buffone MG, et al. (2011) Molecular cloning and characterization of phospholipase C zeta in equine sperm and testis reveals species-specific differences in expression of catalytically active protein. Biol Reprod 85: 78–88. [DOI] [PubMed] [Google Scholar]
- 36. Kurokawa M, Sato K, Fissore RA (2004) Mammalian fertilization: from sperm factor to phospholipase Czeta. Biol Cell 96: 37–45. [DOI] [PubMed] [Google Scholar]
- 37. Nomikos M, Elgmati K, Theodoridou M, Calver BL, Cumbes B, et al. (2011) Male infertility-linked point mutation disrupts the Ca2+ oscillation-inducing and PIP2 hydrolysis activity of sperm PLC zeta. Biochem J 434: 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nomikos M, Swann K, Lai FA (2012) Starting a new life: Sperm PLC-zeta mobilizes the Ca2+ signal that induces egg activation and embryo development. BioEssays 34: 126–134. [DOI] [PubMed] [Google Scholar]
- 39. Nomikos M, Yu Y, Elgmati K, Theodoridou M, Campbell K, et al. (2013) Phospholipase Czeta rescues failed oocyte activation in a prototype of male factor infertility. Fertil Steril 99: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kashir J, Jones C, Lee HC, Rietdorf K, Nikiforaki D, et al. (2011) Loss of activity mutations in phospholipase C zeta (PLCzeta) abolishes calcium oscillatory ability of human recombinant protein in mouse oocytes. Hum Reprod 26: 3372–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kashir J, Konstantinidis M, Jones C, Heindryckx B, De Sutter P, et al. (2012) Characterization of two heterozygous mutations of the oocyte activation factor phospholipase C zeta (PLCzeta) from an infertile man by use of minisequencing of individual sperm and expression in somatic cells. Fertil Steril 98: 423–431. [DOI] [PubMed] [Google Scholar]
- 42. Kashir J, Konstantinidis M, Jones C, Lemmon B, Lee HC, et al. (2012) A maternally inherited autosomal point mutation in human phospholipase C zeta (PLCzeta) leads to male infertility. Hum Reprod 27: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sato K, Wakai T, Seita Y, Takizawa A, Fissore RA, et al. (2013) Molecular characteristics of horse phospholipase C zeta (PLCzeta). Anim Sci J 84: 359–368. [DOI] [PubMed] [Google Scholar]
- 44. Kuroda K, Ito M, Shikano T, Awaji T, Yoda A, et al. (2006) The role of X/Y linker region and N-terminal EF-hand domain in nuclear translocation and Ca2+ oscillation-inducing activities of phospholipase Czeta, a mammalian egg-activating factor. J Biol Chem 281: 27794–27805. [DOI] [PubMed] [Google Scholar]
- 45. Bedford-Guaus SJ, McPartlin LA, Varner DD (2012) Characterization of Equine Phospholipase C Zeta: A Review and Preliminary Results on Expression Defects in Subfertile Stallions. JEVS 32: 445–450. [Google Scholar]
- 46. Gradil C, Yoon S-Y, Brown J, He C, Visconti P, et al. (2006) PLCz: A marker of fertility for stallions? Anim Reprod Sci 94: 23–25. [Google Scholar]
- 47. Kaewmala K, Uddin MJ, Cinar MU, Grosse-Brinkhaus C, Jonas E, et al. (2012) Investigation into association and expression of PLCz and COX-2 as candidate genes for boar sperm quality and fertility. Reprod Domest Anim 47: 213–223. [DOI] [PubMed] [Google Scholar]
- 48. Kasimanickam V, Kasimanickam R, Arangasamy A, Saberivand A, Stevenson JS, et al. (2012) Association between mRNA abundance of functional sperm function proteins and fertility of Holstein bulls. Theriogenology 78: 2007–2019 e2002. [DOI] [PubMed] [Google Scholar]
- 49. Pan Q, Ju Z, Huang J, Zhang Y, Qi C, et al. (2013) PLCz Functional Haplotypes Modulating Promoter Transcriptional Activity Are Associated with Semen Quality Traits in Chinese Holstein Bulls. PLoS ONE 8: e58795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu J, Mayeda A, Krainer AR (2001) Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell 8: 1351–1361. [DOI] [PubMed] [Google Scholar]
- 51. Wang J, Smith PJ, Krainer AR, Zhang MQ (2005) Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res 33: 5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ke S, Shang S, Kalachikov SM, Morozova I, Yu L, et al. (2011) Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res 21: 1360–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang Z, Xiao X, Van Nostrand E, Burge CB (2006) General and specific functions of exonic splicing silencers in splicing control. Mol Cell 23: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Homolova K, Zavadakova P, Doktor TK, Schroeder LD, Kozich V, et al. (2010) The deep intronic c.903+469T>C mutation in the MTRR gene creates an SF2/ASF binding exonic splicing enhancer, which leads to pseudoexon activation and causes the cblE type of homocystinuria. Hum Mutat 31: 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Walenkamp MJ, Klammt J, Feigerlova E, Losekoot M, van Duyvenvoorde HA, et al. (2013) Genetic Analysis of GHR Should Contain Sequencing of All Coding Exons and Specific Intron Sequences, and Screening for Exon Deletions. Horm Res Paediatr 80: 406–412. [DOI] [PubMed] [Google Scholar]
- 56. Blazquez L, Aiastui A, Goicoechea M, Martins de Araujo M, Avril A, et al. (2013) In vitro correction of a pseudoexon-generating deep intronic mutation in LGMD2A by antisense oligonucleotides and modified small nuclear RNAs. Hum Mutat 34: 1387–1395. [DOI] [PubMed] [Google Scholar]
- 57. Lee CT, Risom T, Strauss WM (2006) MicroRNAs in mammalian development. Birth Defects Res C Embryo Today 78: 129–139. [DOI] [PubMed] [Google Scholar]
- 58. Hwang HW, Mendell JT (2007) MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer 96 Suppl: R40–44. [PubMed] [Google Scholar]
- 59. Vickers KC, Rye KA, Tabet F (2014) MicroRNAs in the onset and development of cardiovascular disease. Clin Sci (Lond) 126: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen X, Zhou JY, Zhou JY (2014) MicroRNA-34a: Role in cancer and cardiovascular disease. Curr Drug Targets 15: 361–373. [DOI] [PubMed] [Google Scholar]
- 61. Hartl M, Grunwald Kadow IC (2013) New roles for “old” microRNAs in nervous system function and disease. Front Mol Neurosci 6: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin SL, Miller JD, Ying SY (2006) Intronic microRNA (miRNA). J Biomed Biotechnol 2006: 26818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hossain MM, Sohel MM, Schellander K, Tesfaye D (2012) Characterization and importance of microRNAs in mammalian gonadal functions. Cell Tissue Res 349: 679–690. [DOI] [PubMed] [Google Scholar]
- 65. Abu-Halima M, Backes C, Leidinger P, Keller A, Lubbad AM, et al. (2014) MicroRNA expression profiles in human testicular tissues of infertile men with different histopathologic patterns. Fertil Steril 101: 78–86 e72. [DOI] [PubMed] [Google Scholar]
- 66. Jevsinek Skok D, Godnic I, Zorc M, Horvat S, Dovc P, et al. (2013) Genome-wide in silico screening for microRNA genetic variability in livestock species. Anim Genet 44: 669–677. [DOI] [PubMed] [Google Scholar]
- 67. Wang JC, Waltner-Law M, Yamada K, Osawa H, Stifani S, et al. (2000) Transducin-like enhancer of split proteins, the human homologs of Drosophila groucho, interact with hepatic nuclear factor 3beta. J Biol Chem 275: 18418–18423. [DOI] [PubMed] [Google Scholar]
- 68. Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23: 198–199. [DOI] [PubMed] [Google Scholar]
- 69. Lewis BP, Green RE, Brenner SE (2003) Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA 100: 189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frischmeyer PA, Dietz HC (1999) Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet 8: 1893–1900. [DOI] [PubMed] [Google Scholar]
- 71. Hentze MW, Kulozik AE (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96: 307–310. [DOI] [PubMed] [Google Scholar]
- 72. Hilleren P, Parker R (1999) Mechanisms of mRNA surveillance in eukaryotes. Annu Rev Genet 33: 229–260. [DOI] [PubMed] [Google Scholar]
- 73. Arraiano CM, Maquat LE (2003) Post-transcriptional control of gene expression: effectors of mRNA decay. Mol Microbiol 49: 267–276. [DOI] [PubMed] [Google Scholar]
- 74. Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, et al. (2010) Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67: 677–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lareau LF, Brooks AN, Soergel DA, Meng Q, Brenner SE (2007) The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv Exp Med Biol 623: 190–211. [DOI] [PubMed] [Google Scholar]
- 76. Tian M, Macdonald RL (2012) The intronic GABRG2 mutation, IVS6+2T->G, associated with childhood absence epilepsy altered subunit mRNA intron splicing, activated nonsense-mediated decay, and produced a stable truncated gamma2 subunit. J Neurosci 32: 5937–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kananura C, Haug K, Sander T, Runge U, Gu W, et al. (2002) A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol 59: 1137–1141. [DOI] [PubMed] [Google Scholar]
- 78. Russo A, Siciliano G, Catillo M, Giangrande C, Amoresano A, et al. (2010) hnRNP H1 and intronic G runs in the splicing control of the human rpL3 gene. Biochim Biophys Acta 1799: 419–428. [DOI] [PubMed] [Google Scholar]
- 79. Kochetov AV (2008) Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays 30: 683–691. [DOI] [PubMed] [Google Scholar]
- 80. Gould PS, Dyer NP, Croft W, Ott S, Easton AJ (2014) Cellular mRNAs access second ORFs using a novel amino acid sequence-dependent coupled translation termination-reinitiation mechanism. RNA 20: 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kozak M (2002) Pushing the limits of the scanning mechanism for initiation of translation. Gene 299: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kozak M (2005) Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361: 13–37. [DOI] [PubMed] [Google Scholar]
- 83. Hamann H, Sieme H, Distl O (2005) Genetic analysis of the fertility in Hanoverian Warmblood horses. Anim Reprod Sci 89: 201–203. [PubMed] [Google Scholar]
- 84. Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Quadros L, Ghosh K, Shetty S (2008) Establishment of a new mismatch PCR-RFLP technique for detection of G10430A common mutation present in moderate to mild haemophilia B patients belonging to Gujarati community from the western part of India. Haemophilia 14: 628–629. [DOI] [PubMed] [Google Scholar]
- 87. Browning BL, Browning SR (2009) A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 84: 210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Haplotype structure of the genomic region at 44,000,970–46,952,651 base pairs on horse chromosome (ECA) 6 and genes annotated on the horse genome reference assembly EquCab2.0. Protein-coding genes are highlighted in bold, pseudogenes are marked in grey and non-coding RNAs are marked in light purple. Haplotype blocks were defined using the four gamete rule algorithm. The haplotype block 9 in intron 8 of the equine PLCz1 contains the SNP BIEC2-952439 significantly associated with estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in Hanoverian stallions. The figure displays Hendrige's multiallelic D, which represent the degree of linkage disequilibrium between each two SNPs. Red fields display LOD≥2 (D′ = 1), shades of red show the same LOD with D′<1. White and blue fields display LOD<2 with D′<1 and D′ = 1, respectively.
(TIF)
Transcript variants of equine PLCz1 detected. (A) Usage of variant-specific 5′ UTR primer revealed non-coding exon 1 variation shown in transcripts. Blue errors indicate position of specific primers and evaluated start for exon 1a and exon 1b. Exons show overlapping position, with an exon 1b position more downstream of exon 1a. Due to study design, definitely 5′ start of exons 1 was not determined, indicated by dashed lines. (B) Comparison of mature mRNA transcript variants. They differ in size due to usage of variable exon1 (1a and 1b) and exon 4 (4a and 4b).
(TIF)
Transcript variants of equine PLCz1 detected in testis tissue. Sequence analyses revealed four different transcripts variants. Translation start and stop are given for each transcript (A: JX545319, B: JX545320, C: JX545317, D: JX545318). Variants show different size of non-coding exon 1. Exon 1a represented in the first primary transcript (A) and in derived transcript (B) contains 114 base pairs (bp), whereas exon 1b represented in second primary transcript (C) and in derived transcript (D) consists of 174 bp. The non- premature-terminating-codon (PTC) containing variants (A, C) consist of the translated exon 4a whereas truncated variants (B, D) show a PTC in exon 4b leading to two potential open reading frames (ORFs, ORF1 and ORF2). Translated exons are shown as solid black boxes. Untranslated exon regions are shown as open boxes. The scale is in bp.
(TIF)
Protein sequence alignments of equine and human PLCz1 variants. (1,2) Sequences highlighted in yellow list truncated PLCz1 protein of 45 amino acids (aa) in human (ENSP00000326397) and equine (JX545318, JX545320). (3–6) Sequence variations of predicted equine protein annotation (3: JX545317, JX545319; 4: ENSECAP00000009710; 5: ENSECAP00000009768; 6: XP_001497816.2) are shown. (7) Interspecies alignment with reviewed human peptide sequence of PLCz1 (NP_149114.2). (8,9) Sequences highlighted in green list truncated PLCz1 protein of 412 aa and 534 aa in human (ENSP00000443320) and equine (JX545318, JX545320). Truncated sequences may be generated due to activation of the premature termination codon (PTC) or are thought to undergo nonsense mediated decay (NMD). The open boxed region corresponds to the N-terminus located EF-hand domain, whereas the boxed region marks the X-Y linker domain. Human sequences are highlighted in bold.
(TIF)
Linkage disequilibria (LD) among 47 polymorphisms within PLCz1 in 237 Hanoverian stallions. (A) The LD display shows Hedrige's multiallelic D which represents the degree of LD between two blocks. Red fields display LOD≥2 (D′ = 1), shades of red show the same LOD with D′<1. White and blue fields display LOD<2 with D′<1 and D′ = 1, respectively. The pairwise LD coefficients (r2) values are shown for each SNP pair in fields. (B) Haplotype blocks were defined using the four gamete rule algorithm. Each haplotype in a block with its frequency and connections from one block to the next block is displayed. In the crossing areas, a value for the multiallelic D′ is shown, representing the level of recombination between the two blocks. Significantly associated haplotype blocks and significantly associated SNPs within haplotype blocks are framed in red.
(TIF)
Hanoverian stallions used in the detection sample and their estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) on the natural scale and standardized scale (R-EBV-PAT: 100±20) as well as the accuracy of EBV-PAT. Stallions were sorted in four groups according to their EBV-PAT.
(DOCX)
Polymorphisms identified in equine PLCz1 and CAPZA3 . Identification of polymorphisms, their accession numbers, their intragenic locations and their possible effects on the coding sequence including the nomenclature for the protein changing mutations are given.
(DOCX)
Association analysis for 48 polymorphisms with the estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in the detection sample using 19 Hanoverian stallions. The variance explained (R2) by each polymorphism, the P-values (P) for association with EBV-PAT and minor allele frequencies (MAF) are given.
(DOCX)
Haplotypes within PLCz1 significantly associated with the estimated breeding values of the paternal component of the pregnancy rate per estrus cycle (EBV-PAT) in 237 Hanoverian stallions. Significant haplotypes, their frequencies, corresponding haplotype blocks, least-square means (LSM) with their standard errors (SE) and P-values (P) are given.
(DOCX)
Predicted influence of EBV-PAT-associated polymorphisms within PLCz1 on transcription factor (TF) binding sites, microRNA (miRNA) and splice-site modifying motifs.
(DOCX)
Primer sequences and their positions, product sizes and annealing temperatures (AT) for sequencing the equine genomic DNA and cDNA of PLCz1 and CAPZA3 .
(DOCX)
Genotyping techniques used for validation of polymorphisms in 237 Hanoverian stallions. All single nucleotide polymorphisms (SNPs) genotyped using RFLPs (restriction fragment length polymorphisms) or IRD (infrared dye) labeled gel electrophoresis are given.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files. Sequence data of this article has been deposited at the NCBI/GenBank Data Libraries for the Equus caballus genomic sequence of PLCz1 (JX545317-JX545320). Newly discovered polymorphisms were submitted to NCBI dbSNP database. Their accession numbers are in Table S2.