

Abstract
Systemic sclerosis is an autoimmune idiopathic connective tissue disease, characterized by vasculopathy, inflammation and fibrosis. There appears to be a link between inflammation and fibrosis, although the exact nature of the relationship is unknown. Serum amyloid A (SAA) is an acute-phase protein that is elevated up to 1000-fold in times of infection or inflammation. This acute-phase reactant, as well as being a marker of inflammation, may initiate signals in a cytokine-like manner, possibly through toll-like receptors (TLRs) promoting inflammation. This study addressed the role of SAA in initiating interleukin-6 (IL-6) production in dermal fibroblasts and the role of TLR2 in this system. We show that SAA induces IL-6 secretion in healthy dermal fibroblasts and that blockade of TLR2 with a neutralizing antibody to TLR2 or specific small interfering RNA attenuated the SAA-induced IL-6 secretion and that this was also mediated through the TLR adaptor protein IL-1 receptor-associated kinase 4. The effect is nuclear factor-κB-mediated because blockade of nuclear factor-κB reduced the induction. We also demonstrate that dermal fibroblasts express TLR2; this is functional and over-expressed in the fibroblasts of patients with systemic sclerosis. Taken together these data suggest that SAA is a danger signal that initiates IL-6 signalling in systemic sclerosis via enhanced TLR2 signalling.
Keywords: danger signal, interleukin-6, serum amyloid A, Toll-like receptor 2
Introduction
Systemic sclerosis (SSc) is a rare, debilitating autoimmune disease characterized by inflammation, vascular dysfunction and fibrosis of the skin and internal organs leading to premature morbidity and mortality.1,2 The disease is unresponsive to most current therapeutics. The aetiology of SSc is unknown but immunity appears to play a pivotal role as shown by the presence of an inflammatory infiltrate in the affected dermis of patients,3 activation of Toll-like receptors (TLRs)4 and an increase in the presence of inflammatory proteins such as cytokines, chemokines and acute-phase proteins in the serum and affected organs of SSc patients.5–9 Although it is known that activation of the immune system and inflammation precedes fibrosis, the exact molecular mechanisms governing the inflammation-driven fibrosis are still not elucidated.
Serum amyloid A (SAA) is an acute-phase protein that has been shown to increase 1000-fold after infection or injury.10–12 Usually synthesized and secreted by hepatocytes, which produce SAA in response to inflammatory stimuli such as tumour necrosis factor-α, SAA has been suggested to signal through multiple receptors including receptor advanced glycosylated end products, formyl peptide receptor like -1, CD36 and TLR2/4.13–16 That it signals through the TLR pathway suggests that SAA is an endogenous danger signal protein. The TLRs are germ-line-encoded pattern recognition receptors that have evolved to recognize conserved motifs in pathogens to activate signalling pathways that lead to activation of the innate immune system, which then directs adaptive immunity. However, they can also recognize ‘endogenous’ molecules that are termed danger-associated molecular pattern molecules (DAMPs). The DAMPs are important molecules released from dead or ‘stressed’ cells to initiate signalling to promote sterile inflammation17 and include a variety of molecules including mitochondrial DNA18 and various matrix molecules.17 TLR2 has recently emerged as a receptor for danger signals. SAA has multiple functions including transportation of cholesterol from sites of inflammation,15 metabolism of high-density lipoproteins,11 induction of inflammatory cytokines16 and granulocyte colony-stimulating factor,10 chemotaxis of neutrophils,10 induction of fibroblast proliferation and the proliferation of regulatory T cells.13 It is suggested that the ‘lipid-poor’ form of SAA is pro-inflammatory. Extra-hepatic synthesis of SAA can occur in synovial fibroblasts and chronic elevation of SAA levels can contribute to the pathogenesis of diseases16,19 including rheumatoid arthritis (RA),20 Crohn's disease15 and certain cancers.21 Indeed, in RA, SAA levels correlate with swollen joint counts and it has been demonstrated that SAA can induce the proliferation of RA-fibroblast-like synovial cells,20 and induce matrix metalloprotease production and activation14 and cytoskeletal disassembly in RA-fibroblast-like synovial cells.22 In SSc patients an increase in SAA levels in the serum has been reported,8 although the mechanisms by which SAA could contribute to pathogenesis of disease have not been investigated.
In SSc patients it is postulated that inflammatory mediators such as interleukin-6 (IL-6) and tumour necrosis factor-α promote the activation and proliferation of fibroblasts, resulting in excessive accumulation of extracellular matrix components, particularly collagen 1A1.23 Here, we demonstrate that stimulation of dermal fibroblasts with SAA results in the induction of IL-6 by these cells. We also show that TLR2 is expressed on dermal fibroblasts and illustrate that SAA signals via this receptor to induce IL-6 production in an IL-1 receptor-associated kinase 4 (IRAK4) and nuclear factor-κB (NF-κB) -dependent manner. Furthermore, SSc dermal fibroblasts have elevated TLR2, which enhances SAA signalling.
Methods
Cell culture
Local ethics approval was granted by the Tyneside research ethics committee code 10/H0906/22. Healthy primary dermal fibroblasts, SSc dermal fibroblasts (n = 3 separate donors) and IRAK4-deficient fibroblasts were isolated from skin biopsies (4 mm) using the skin explant method as previously described24 and cultured in 75-cm2 culture flasks in RPMI-1640 medium (Sigma-Aldrich, Poole, UK) supplemented with heat-inactivated 10% fetal calf serum (FCS), l-glutamine (2 mm), penicillin (100 U/ml) and streptomycin (100 μg/ml) (all Sigma) in an incubator at 5% CO2 at 37°. Dermal fibroblasts were also isolated from an IRAK4-deficient patient (as described in ref. 4). This patient has a mutation in the IRAK4 gene in exon 8 that leads to no IRAK4 protein expression. This patient has increased susceptibility to Gram-positive bacterial infections.
Stimulation of fibroblasts with A-SAA and inhibition of TLR2
Dermal fibroblasts from healthy controls (HC) and the IRAK4-deficient patient were trypsinized and seeded on a 24-well plate at a cell density of 1 × 105 cells/well until confluent. Confluent dermal fibroblasts were washed with sterile PBS (Sigma) and serum starved for 24 hr in serum-free RPMI-1640 medium (Sigma-Aldrich) at 5% CO2 at 37° before stimulation with acute serum amyloid A-1 (A-SAA-1) (Peprotech, Rocky Hill, CT) at concentrations of 1–10 μg/ml for 24 hr. Recombinant A-SAA amino acid sequence corresponds to the sequence of SAA 1α isotype, with the exception of the addition of Met at the N terminus, substitution of Asp for Asn at position 60, and substitution of His for Arg at position 71. According to the manufacturer the endotoxin level of A-SAA is < 0·1 ng/μl.
For TLR signalling inhibition experiments, dermal fibroblasts were incubated for 1 hr with neutralizing anti-TLR2 antibody (clone T2.5; eBioscience, San Diego, CA) or a matched IgG isotype control (clone P3.6.2.8.1; eBioscience) both at a concentration of 1 μg/ml before treatment with A-SAA 10 μg/ml (Peprotech) for 24 hr. In addition, dermal fibroblasts were pre-incubated for 2 hr with an inhibitor of NF-κB kinase 2 (IKK2) inhibitor 300 nm (EMD Millipore; Billerica, MD) and subsequently exposed to A-SAA 10 μg/ml stimulation.
Small interfering Rna (siRNA) was employed to knockdown TLR2 using 100 nm siRNA TLR2 smartpool (Dharmacon, Lafayette, CO) transfected with Dharmafect 1 (Thermo Scientific, London, UK) transfection reagent or non-targeting control. Twenty-four hours following transfection, the transfection reagent containing medium was removed and SAA 10 μg/ml was added to the cultures.
ELISA
After 24 hr of SAA stimulation, cell culture supernatants were collected and IL-6 (in-house as previously described 25) concentration was determined by ELISA. The ELISAs were developed using o-phenylenediamine dihydrochloride substrate (Sigma-Aldrich) at room temperature. Samples were analysed in duplicate and fluorescence was measured on a Tecan plate reader at 490 nm.
Western blot
Cells were cultured in serum-free medium before the incubation with SAA (10 μg/ml) for 30 min, 1 hr or 2 hr, cells were harvested in lysis buffer and run on a 10% SDS–PAGE. Proteins were transferred to a nitrocellulose membrane, blocked and probed with anti-phosphoP65 NF-κB (Cell Signalling, London, UK) 1 : 1000 and re-probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Abcam, Cambridge, UK) 1 : 100 000 as a loading control for total protein.
Flow cytometry
Healthy dermal and SSc fibroblasts and U937 monocytoid cells were analysed for TLR2 expression by flow cytometry. Cells were resuspended and incubated for 30 min with mouse anti-human TLR2 antibody (clone T2.5; eBioscience) at 1·25 μg/ml in PBS on ice for 30 min before washing three times and incubation with Alexa Fluor 488 goat anti-mouse IgG antibody (Invitrogen, Paisley, UK) for 30 min. Cells were then analysed using a BDLSRII cytometer and analysed using FlowJo analysis software. Mean fluorescence intensity (MFI) was calculated by subtracting the ‘secondary only stained’ MFI from the ‘TLR2-stained’ MFI.
HEK293 BLUE reporter assays
HEK293 Blue™ (Invivogen, Paisley, UK) are HEK293 cells stably transfected with either TLR2 or TLR4 receptors and have an NF-κB reporter system that secretes secreted embryonic alkaline phosphotase (SEAP) into the media following induced activation. HEK293 cells were harvested and cultured in a 96-well plate at 100 000 cells/well in complete medium overnight. The medium was removed and replaced with FCS-free complete medium containing nothing else (Control), or FCS-free complete medium with SAA (10 μg/ml) or heat-killed Listeria monocytogenes (HKLM) (108 cells/ml), a natural agonist of TLR2. In some experiments a neutralising antibody to TLR1 mouse monoclonal antibody (1 μg/ml) (Invivogen) was added to the HEK TLR2 Blue cells to block TLR1 signalling. After 24 hr in culture the medium was removed and released SEAP was measured on a Tecan plate reader at 620 nm. In the case of TLR4-HEK293 cells the positive control was 100 ng/ml of pure lipopolysaccharide (LPS) reconstituted in ultrapure water (Invivogen).
Transfection experiments
HEK-TLR2 cells were transfected with DNA fragments of the 3X NF-κB cloned downstream of the luciferase plasmid in a pGL2-basic vector at a final concentration of 300 ng using 1·5 μl of Fugene HD (Promega, Southampton, UK) transfection reagent per well of a 24-well plate. Renilla plasmid at a final concentration of 30 ng was used to normalize transfection efficiency. Twenty-four hours after transfection, cells were stimulated with SAA (10 μg/ml) or HKLM (108 cells/ml) for an additional 6 hr. Following stimulation, assays for reporter gene activity were performed according to Promega's Dual-luciferase protocol and analysed using a Glomax multi-detection system (Promega). A dominant negative IκB-α plasmid described previously,26 was also co-transfected with NF-κB luciferase reporter and renilla to normalize transfection efficacy, this plasmid acts as a repressor of NF-κB activity. Twenty-four hours after transfection, cells were stimulated with SAA (10 μg/ml) for an additional 6 hr and reporter activity was determined.
Quantitative RT-PCR
Healthy and diseased dermal fibroblasts were cultured and then harvested, RNA was isolated using TRizol according to the manufacturer's instructions and 1 μg of RNA was DNAse treated and reverse transcribed to cDNA using reverse transcriptase (Invitrogen). In some experiments healthy dermal fibroblasts were incubated with Trichostatin A (500 ng/ml) or DMSO vehicle control 0·01% (v/v). Quantitative RT-PCR was performed using SYBR™ green (Sigma-Aldrich) in triplicate using primers detailed in Table1. Relative differences were calculated normalized to 18S by the comparative threshold cycle method.
Table 1.
Primers used in this study
| Gene | Primer forward | Primer reverse | Annealing temp ° |
|---|---|---|---|
| TLR2 | 5′-TGCTTTCCTGCTGGAGATTT-3′ | 5′-TGTAACGCAACAGCTTCAGG-3′ | 54 |
| TLR4 | 5′-TGGACAATTTGGGCTAGAGG-3′ | 5′-GATCCCAGCCATCTGTGTCT-3′ | 56 |
| 18S | 5′ CGA ATG GCT CAT TAA ATC AGT TAT GG-3′ | 5′ TAT TAG CTC TAG AAT TAC CAC AGT TAT CC-3′ | 55 |
Statistical analysis
Data are presented as mean ± SEM. Differences between groups were analysed using either an unpaired Student's t-test or two-way analysis of variance. A P < 0·05 was considered statistically significant.
Results
Serum amyloid A induces IL-6 in a dose-dependent manner
It has previously been demonstrated that SAA can induce the production of pro-inflammatory cytokines in specific cell types including synovial fibroblasts from patients with RA. Here, we examined the role of SAA in inducing IL-6 secretion in dermal fibroblasts. Healthy dermal fibroblasts were stimulated with SAA (0·5–10 μg/ml) for 24 hr before cell-free supernatants were analysed for IL-6 levels by ELISA. As shown in Fig.1(a), SAA induces IL-6 production in dermal fibroblasts in a dose-dependent manner, with 10 μg/ml being the optimum concentration for IL-6 secretion. Concentrations of SAA above this plateaued (data not shown). Boiling the recombinant protein abolished the induction of IL-6. HKLM was also tested as a positive control and this was slightly more effective at IL-6 induction than 10 μg/ml SAA.
Figure 1.
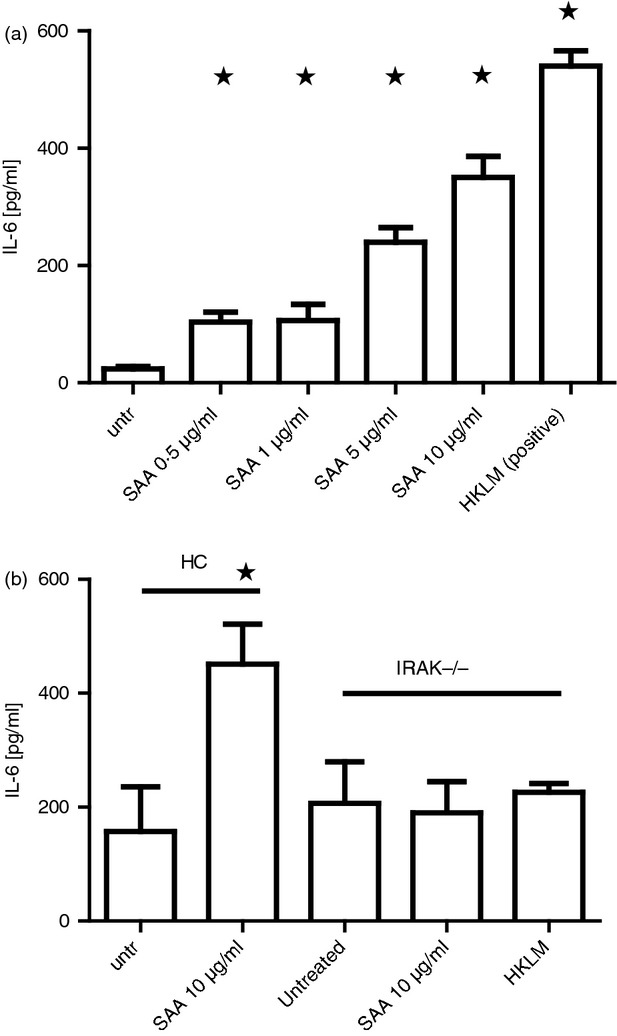
Serum amyloid A (SAA) stimulates interleukin-6 (IL-6) secretion in dermal fibroblasts (a) Healthy dermal fibroblasts were cultured with increasing concentrations of recombinant SAA and heat-killed Listeria monocytogenes (HKLM) as a positive control and after 24 hr the supernatant was removed and measured for IL-6 by ELISA. (b) Healthy or IRAK4 deficient fibroblasts were incubated with 10 μg/ml SAA for 24 hr after which the supernatant was removed and IL-6 was measured by ELISA. Data are the mean and SEM are the error bars (n = 5).*P ≤ 0·05.
Serum amyloid A induction of IL-6 is mediated by IRAK4 and TLR2
Next, we sought to determine if SAA signalling is facilitated through TLR2 to induce IL-6 production. After stimulation of TLR by a TLR ligand signalling usually occurs via the MyD88-dependent pathway.27 Signalling via this pathway requires the activation of the protein kinase IRAK4. Using dermal fibroblasts isolated from a patient deficient in the IRAK4 protein, due to a genetic defect, and healthy controls we determined the role of IRAK4 in SAA stimulation and subsequent IL-6 expression. As shown in Fig.1(b), stimulation of dermal fibroblasts with SAA (10 μg/ml) resulted in a significant increase in IL-6 production, whereas there was no change in IL-6 secretion in IRAK4-deficient fibroblasts after stimulation with SAA (10 μg/ml) (P ≤ 0·05) or stimulated with the positive control HKLM. Therefore, IRAK4 is a critical downstream mediator of SAA-induced effects.
Because it has been shown that TLR2 mediates the effects of SAA in another experimental system we used an anti-human neutralizing antibody to TLR2 to see if the effects of SAA addition were mediated by TLR2. As Fig.2(a) demonstrates, SAA incubation results in induction of IL-6; however, co incubation of an anti-TLR2 antibody at a concentration of 1 μg/ml results in reduction of SAA-induced IL-6 (P ≤ 0·05). Incubation with a matched isotype control antibody at the same concentration (1 μg/ml) did not lead to a significant reduction of IL-6 production. We further used specific validated TLR2 siRNA to knockdown TLR2 and as Fig.2(b) demonstrates siRNA of TLR2 leads to a significant reduction of SAA-induced IL-6; however, non-targeting siRNA does not lead to a reduction (P ≤ 0·05). Figure2(c) shows the efficacy of the TLR2 knockdown.
Figure 2.
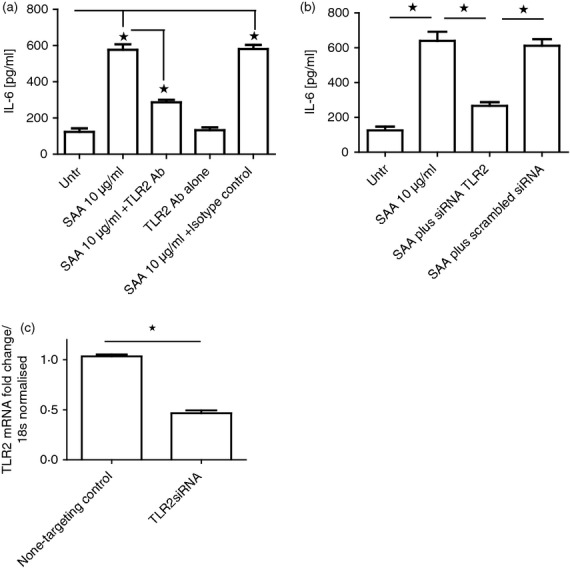
Serum amyloid A (SAA) -induced interleukin-6 (IL-6) production is mediated via Toll-like receptor 2 (TLR2). (a) Healthy dermal fibroblasts were cultured in the presence of SAA (10 μg/ml) or SAA plus a neutralizing antibody to TLR2 (1 μg/ml) or anti-TLR2 alone (1 μg/ml) or isotype-matched control and SAA. After 24 hr incubation the supernatants were removed and IL-6 was quantified by ELISA. (b) Healthy dermal fibroblasts were transfected with dharmfect1 reagent and specific siRNA for TLR2 (100 nm) was validated for 24 hr after which time the medium was replaced and incubated with or without SAA (10 μg/ml) for 24 hr and the supernatants were removed and IL-6 was quantified by ELISA. Data are the mean and SEM are the error bars (n = 3). *P ≤ 0·05. (c) TLR2 expression in dermal fibroblasts after small interfering RNA (siRNA; 100 nm) for non-targeting control or specific TLR2 siRNA 24 hr after transfection. Data are normalized to 18s and non-targeting control siRNA was set as the calibrator. Data are the mean and SEM are the error bars (n = 4). *P ≤ 0·0001.
Interleukin-6 induction by SAA is NF-κB dependent
Stimulation of TLRs via the MyD88-dependent pathway results in the activation of the central transcription factor NF-κB, downstream of IRAK4. It has previously been demonstrated that stimulation of TLR2 induces NF-κB activation and subsequent IL-6 expression and secretion in dermal fibroblasts. To determine if NF-κB activation is required in IL-6 secretion following SAA stimulation in dermal fibroblasts, we blocked NF-κB activation by pre-treatment of healthy dermal fibroblasts for 2 hr with an IKK-2 inhibitor before stimulation with SAA (10 μg/ml). As shown in Fig.3(a) we confirmed that SAA-stimulated IL-6 induction by SAA is dependent on NF-κB activation as blockade of NF-κB significantly attenuated IL-6 production (P ≤ 0·05). 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay demonstrated no increase in cell death with the incubation of the IKK-β inhibitor (data not shown). We also employed the use of an NF-κB ‘super repressor’; this is a dominant negative inhibitor of NF-κB and co-transfection of this with the NF-κB luciferase plasmid resulted in a significant inhibition of luciferase activity following SAA stimulation in TLR2 over-expressing HEK293 cells (P = 0·03) (Fig.3b). Figure3(c) demonstrates that SAA induces phosphorylation of the P65 subunit of NF-κB with maximal phosphorylation at 1 hr after stimulation in dermal fibroblasts.
Figure 3.
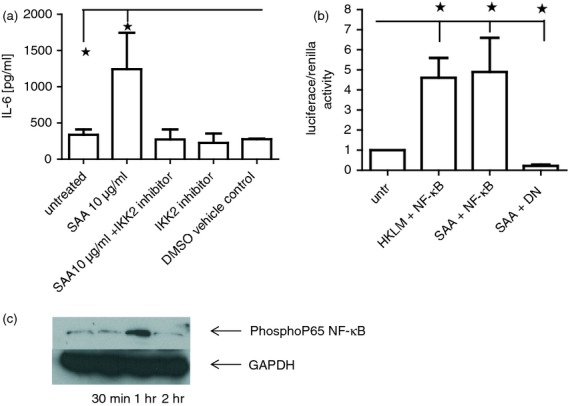
Serum amyloid A (SAA) induction of interleukin-6 (IL-6) is NF-κB dependent. (a) Healthy dermal fibroblasts were cultured in the presence of SAA (10 μg/ml) or pre-treated with NF-κB inhibitor (300 nm) for 2 hr or NF-κB inhibitor alone or DMSO vehicle control for 24 hr, after which the supernatants were removed and IL-6 was quantified by ELISA. (b) Healthy dermal fibroblasts were co-transfected with both NF-κB luciferase reporter construct (300 ng) and also a ‘dominant negative’ IκBα plasmid and renilla. After 24 hr the cells were then incubated with SAA and cells were analysed for luciferase reporter activity. Data were normalized to renilla and shown as a ratio. Data are the mean with SEM as the error bars (n = 4). (c). Representative immunoblot of phosphorylated P65 NF-κB after SAA stimulation up to 2 hr. GAPDH is used as a loading control (n = 4). *P ≤ 0·05.
TLR2 is expressed on dermal fibroblasts
As SAA has been speculated to bind and stimulate via TLR2,15 the expression of TLR2 by healthy dermal fibroblasts was analysed using flow cytometry. As shown in Fig.4(a), TLR2 was expressed on approximately 46% of healthy unstimulated dermal fibroblasts. Figure4(b) shows elevated TLR2 expression on SSc dermal fibroblasts from one patient compared with control. Healthy CD14+ monocytes were included as a positive control and are shown in Fig.4(c). Figure4(d) shows the mean MFI for TLR2 expression between healthy and SSc patients. SSc dermal fibroblasts have significantly elevated expression of TLR2.
Figure 4.
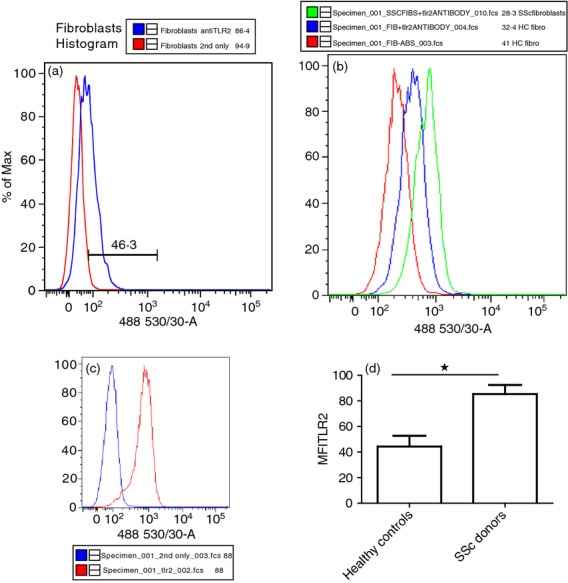
Healthy dermal fibroblasts express Toll-like receptor 2 (TLR2). (a) Representative histogram for TLR2 after staining with an anti-TLR2 antibody or primary antibody omitted and analysed using a BDLSRII cytometer (n = 5). (b). Representative histogram from one systemic sclerosis (SSc) patient and control stained with TLR2 antibody or primary antibody omitted. (c). Positive control for TLR2 staining representative histogram using U937 monocyte cell line, blue line is the secondary antibody alone. (d). Mean fluorescence intensity (MFI) of TLR2 expression from healthy and control dermal fibroblasts. MFI was calculated by subtracting the secondary only staining MFI from the TLR2 antibody staining. Data are the mean and SEM are the error bars (n = 3). *P ≤ 0·05 control versus SSc
SAA activates TLR2 but not TLR4
To confirm that SAA was binding to and activating TLR2, experiments where performed using a reporter cell line that produces a secreted alkaline phosphatase when NF-κB is activated in response to ligand stimulation in TLR2-transfected HEK293 cells. Figure5(a) shows the secreted SEAP absorbance at 620 nm which is linked to the activation of TLR2. This figure demonstrates that control untreated cells do not secrete appreciable SEAP but following SAA incubation they do secrete SEAP as effectively as the natural TLR2 ligand heat-killed bacteria, which served as a positive control. Because these cells also express TLR1 basally we also pre-incubated with a neutralising antibody to TLR1 (1 μg/ml) to inhibit TLR1 signalling. This demonstrated no inhibition of SAA-induced SEAP activity in the HEK293 TLR2 cells. We also used TLR4-transfected HEK293 cells with the same reporter system to examine if SAA or an accompanying contaminant such as LPS was activating TLR4. Incubation of SAA to these TLR4-transfected HEK293 cells did not induce any SEAP activity compared with control or that seen with LPS (the natural ligand of TLR4) (Fig.5b). Blockade of NF-κB with a specific inhibitor led to a reduction in SEAP activity in the TLR2 and SAA-stimulated reporter cells (P ≤ 0·05). Figure5(c) demonstrates that after 4 hr of stimulation with SAA NF-κB was activated using a reporter system transfected into the HEK TLR2-expressing cells.
Figure 5.
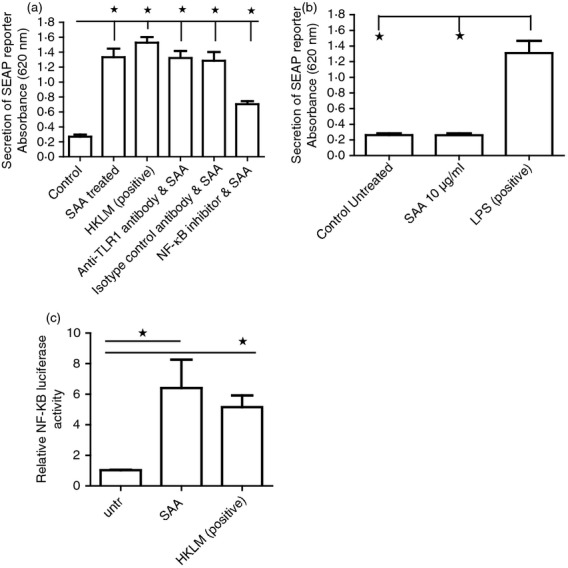
Serum amyloid A (SAA) activates Toll-like receptor 2 (TLR2) as effective as the natural ligand but does not activate TLR4. (a) HEK293 TLR2 reporter cells stably expressing TLR2 were cultured and then treated with medium alone, SAA (10 μg/ml) or heat-killed Listeria monocytogenes (HKLM), or SAA and neutralizing antibody to TLR1 (1 μg/ml) or SAA and matched isotype control antibody (1 μg/ml) or pre-treated with a nuclear factor-κB (NF-κB) inhibitor (300 nm) and SAA for 24 hr after which the supernatant was measured for SEAP absorbance (620 nm). (b) HEK293 TLR4 reporter cells were cultured and incubated with SAA or lipopolysaccharide (LPS) as a positive control for 24 hr after which time the supernatant was measured for SEAP absorbance (620 nm). Data are the mean and SEM are the error bars (n = 4). (c). HEK293 TLR2 cells were transfected with 500 ng of NF-κB plasmid carrying luciferase and also renillla after which they were exposed to SAA (10 μg/ml) or HKLM for 4 hr and then relative luciferase activity was determined and normalised to renilla (n = 3). *P ≤ 0·05 untreated versus SAA treated. P ≤ 0·05 untreated versus HKLM treated.
TLR2 gene expression in SSc dermal fibroblasts is elevated
We then decided to examine the mRNA expression of TLR2 from both healthy and SSc dermal fibroblasts derived from three donors using quantitative RT-PCR with specific TLR2 primers. Quantitative RT-PCR revealed that SSc dermal fibroblasts expressed a mean 18-fold (2·1) higher expression of TLR2 compared with healthy dermal fibroblasts. Furthermore, incubation with Trichostatin A, a broad-spectrum histone deacetylase inhibitor, in healthy dermal fibroblasts elevated TLR2 expression 5·6-fold compared with DMSO-treated control cultures (0·01% v/v) see Fig.6(a) (upper panel). However, TLR4 expression did not change with TSA treatment (Fig.6a lower panel). This indicates that acetylation is important in the regulation of TLR2 mRNA expression. However, incubation of SSc dermal fibroblasts with TSA did not result in up-regulation of TLR2, as opposed to healthy dermal fibroblasts (Fig.6b). We also did not observe up-regulation of TLR2 by chronic exposure to hydrogen peroxide to mimic the oxidative stress seen in SSc due to both hypoxia and lymphocyte infiltration.28
Figure 6.
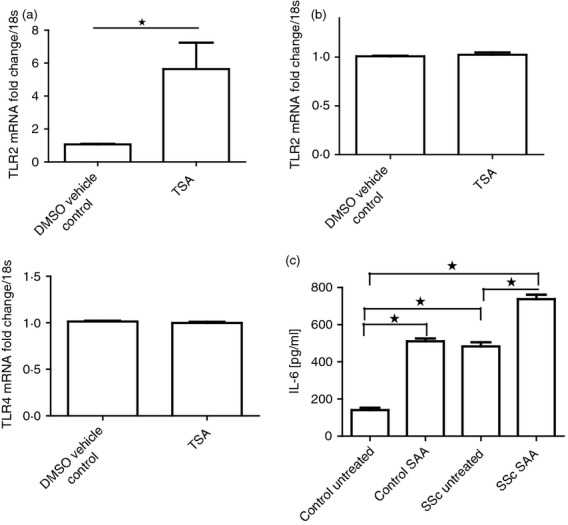
Healthy dermal fibroblasts up-regulate Toll-like receptor 2 (TLR2) in response to acetylation and systemic sclerosis (SSc) fibroblasts have higher levels of interleukin-6 (IL-6). Healthy dermal fibroblasts were cultured with vehicle control DMSO (0·01%) or TSA (500 ng/ml) for 16 hr after which time the RNA was harvested and quantiative RT-PCR was performed with TLR2-specific primers and 18S-specific primers and the data were normalized to 18S and compared with DMSO vehicle control (a, upper panel). Lower panel is the data for TLR4. (b) SSc dermal fibroblasts were cultured with vehicle control DMSO (0·01%) or TSA (500 ng/ml) for 16 hr and quantitative RT-PCR was performed with TLR2-specific primers and 18S-specific primers and the data were normalized to 18S and compared with DMSO control. (c) Healthy and SSc dermal fibroblasts were cultured with or without the addition of serum amyloid A (SAA) (10 μg/ml) for 24 hr after which time the supernatants were removed and IL-6 was quantified by ELISA. Data are the mean and SEM with error bars (n = 3). *P ≤ 0·05. Control SAA versus SSc untreated is not significant. The rest are significant as indicated.
Enhanced responsiveness of SSc fibroblasts to SAA
Dermal fibroblasts from Ssc patients where isolated and incubated with SAA (10 μg/ml) along with fibroblasts from healthy controls and levels of IL-6 were measured. This revealed that SSc fibroblasts have both a higher constitutive level of IL-6, comparable to control cultures treated with SAA (Fig.6c). (P ≤ 0·05) and can further be induced by SAA to produce IL-6.
Discussion
Serum amyloid A is an acute-phase protein which has been found to be increased in SSc,8 RA22 and certain solid cancers21 and which is the precursor of insoluble amyloid found in amyloidosis. Levels of SAA increase up to a 1000-fold after infection or injury and as such it is an acute-phase protein and is used as a biomarker in many chronic diseases. SAA has been shown to signal via multiple receptors including TLR2, TLR4 and formyl peptide receptor-like 1 to induce a variety of functions including pro-inflammatory cytokine secretion, the metabolism of high-density lipoproteins and transport of cholesterol.29 In RA an increase in SAA has been shown to induce cytoskeletal rearrangement22 and the production of matrix metalloproteases,14 and in gout SAA is thought to be involved in activation of the inflammasome.19 However, the functional effect of the increase of SAA levels in SSc patients is, to date, unknown. This study examined the role of SAA in signalling in dermal fibroblasts. SAA was demonstrated to induce IL-6, a pro-inflammatory cytokine that is known to be involved in disease progression in SSc patients, and this is mediated via the IRAK4 signalling pathway, suggesting a role of SAA in SSc pathogenesis.
First, we demonstrated that SAA induces IL-6 production in healthy dermal fibroblasts. Interleukin-6 is a critical cytokine in mediating multiple immune responses, but is also critical in multiple diseases including RA and SSc. Levels of IL-6 are increased in SSc serum30 and PBMCs from SSc patients have enhanced production of IL-6 compared with healthy controls.31 In SSc IL-6 levels correlate with the Rodnan skin score and predict worse outcome. Furthermore, we and others have shown that IL-6 mediates an increased expression of collagen I in dermal fibroblasts and that this is facilitated by the intracellular transcription factor signal transducer and activator of transcription 3.23 Blockade of signal transducer and activator of transcription 3 with the use of a small molecule inhibitor results in attenuation of the IL-6-induced collagen I expression.23 Furthermore, in an animal model of cardiac fibrosis, blockade of IL-6 signalling attenuated collagen synthesis and cardiac fibrosis.32
The innate immune system, specifically TLRs, has been implicated to play a role in the initiation and progression of disease in SSc patients. We therefore sought to determine a role for SAA signalling, via the MyD88-dependent pathway and NF-кB activation, in the activation of dermal fibroblasts. MyD88 is an adaptor protein required for signalling associated with TLRs. Previously, TLR1–9 mRNA expression has been demonstrated in dermal fibroblasts ex vivo and may contribute to fibrogenesis.33 Here, we confirm, for the first time, the novel observation that TLR2 is expressed on the plasma membrane of healthy dermal fibroblasts. We also confirmed that the functional effects of SAA on dermal fibroblasts are determined by TLR signalling as fibroblasts isolated from a patient deficient in IRAK4, a critical downstream mediator of TLR signalling, did not respond to SAA stimulation with the production of IL-6. We also used an anti-human neutralizing antibody to TLR2 to determine if SAA-mediated IL-6 secretion was facilitated by TLR2. Our data confirm this to be the case. Stimulation of TLR can induce activation of the transcription factor NF-κB, in our study we determined that SAA's affects were dependent on NF-κB activation because inhibition of this transcription factor resulted in a decrease in IL-6 production. NF-κB is a family of transcription factors that controls the transcription of various pro-inflammatory genes and is activated by TLR ligation and lies downstream of IRAK4. Hence, the TLR2-mediated effects are dependent on NF-κB. A polymorphism has been described in the NF-κB locus as conferring risk in SSc. We also demonstrated using a reporter cell line that stably expresses TLR2 and is linked to an NF-κB reporter that secretes SEAP into the medium that SAA is an endogenous ligand for TLR2. However, using the same reporter system for TLR4 we could not see an increase in activation (but LPS does). Hence, SAA does not signal through TLR4 to activate NF-κB. Furthermore, we also used a neutralizing antibody to TLR1, which is expressed by the HEK293 TLR2 cells, this did not reduce the SAA-induced reporter activity in these cells. TLR2 can dimerize with TLR1 to elicit signalling but we demonstrate that this is not necessary to elicit IL-6 production in our system.
That the SAA can bind and stimulate TLR2 to induce pro-inflammatory cytokine production via NF-κB suggests that SAA is a ‘danger signal’ and functions as a DAMP. HMGB-1 is a non-histone nuclear protein that in damaged cells is released and binds TLR2 and acts as a danger signal34 and has been demonstrated to promote pro-inflammatory cytokine secretion in synovial fibroblasts.35 SAA has been shown to activate the ‘inflammasome’ via TLR4.36 Hyaluron, an extracellular matrix molecule, has also been suggested to be a danger signal, regulating tissue repair processes37 and the list of DAMPs is growing and now includes mitochondrial DNA.18 We also demonstrated elevated levels of TLR2 transcript by quantitative RT-PCR in SSc patient's dermal fibroblasts and protein expression. This indicates that they have increased levels of the putative receptor and therefore maybe more responsive to the ligand SAA and that incubation with a broad-spectrum histone deacetylase inhibitor TSA at sub-toxic concentrations elevated the mRNA levels of TLR2, but not TLR4 mRNA levels, indicating a specific effect. However, incubation of SSc dermal fibroblasts with TSA did not result in up-regulation of TLR2 expression, we interpret this finding as showing that the level of TLR2 in these diseased fibroblasts is maximal and cannot be increased. This indicates that epigenetic mechanisms exist to regulate TLR2. Acetylation is a critical mechanism for the regulation of many transcription factors. Acetylation of histones is important for relaxing the chromatin structure promoting gene expression. Li et al.38 recently demonstrated that acetylation regulates TLR2 expression in THP-1 cells, a monocytic cell line. We do not identify if the acetylation of the TLR2 promoter is the mechanism underlying the increase here but this could be the mechanism. A polymorphism in TLR2 conferring risk in SSc has been described recently and was associated with elevated IL-6 secretion.39 We demonstrated that SSc fibroblasts have an elevated constitutive expression of IL-6 and respond to SAA. This suggests that the elevated TLR2 levels are enhancing signalling mediated by SAA in vivo. SAA is similar to C-reactive protein, which is also known to be elevated in SSc and often correlates with skin thickness.40 It has been demonstrated previously that SAA can signal through TLR2 in neutrophils; however, this is the first report of SAA signalling through TLR2 in dermal fibroblasts and so represents a highly novel finding.
Synthesis outside of hepatic synthesis and secretion of SAA has been reported before in synovial explant tissue from patients with RA and also by intestinal epithelial cells.41 It is suggested in SSc that the initial insult is in the vasculature and that this may lead to cell death of the endothelium.42 This cell death via necrosis may lead to the release of the endogenous ‘danger signal’ SAA, which will act on neighbouring cells to initiate a pro-inflammatory cascade, ultimately leading to the release of cytokines. These locally released cytokines may lead to the increased inflammatory environment so enhancing and perpetuating tissue fibrosis. Targeting TLR2 with therapeutic antibodies may be a novel treatment. TLR2 knockout mice are protected from pulmonary fibrosis induced by bleomycin.43 It is of interest that OPN301, a human anti-TLR2 antibody, has been used to inhibit spontaneous pro-inflammatory cytokine release from synovial explants.44
Acknowledgments
We thank Dr Andrew Knight, Immunobiology group, for the HEK Blue reporter cells. Funding by South Tees Rheumatology Research Fund.
Disclosures
JMvL has received research support and speaker's fees from Genentech, the manufacturer of tocilizumab, an anti-sIL-6R antibody.
References
- 1.O' Reilly S, Hügle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology. 2012;51:1540–9. doi: 10.1093/rheumatology/kes090. [DOI] [PubMed] [Google Scholar]
- 2.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Investig. 2007;117:557–67. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie Y, Zhang X, Wakasugi S, Makino T, Inoue Y, Ihn H. Immunohistochemical characterization of the cellular infiltrate in localized scleroderma. Int J Dermatol. 2008;47:438–42. doi: 10.1111/j.1365-4632.2008.03615.x. [DOI] [PubMed] [Google Scholar]
- 4.Ciechomska M, Huigens CA, Hügle T, et al. Toll-like receptor-mediated, enhanced production of profibrotic TIMP-1 in monocytes from patients with systemic sclerosis: role of serum factors. Ann Rheum Dis. 2012;6:6. doi: 10.1136/annrheumdis-2012-201958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Reilly S, Ciechomska M, Cant R, Hügle T, van Laar JM. Interleukin-6, its role in fibrosing conditions. Cytokine Growth Factor Rev. 2012;23:99–107. doi: 10.1016/j.cytogfr.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Chizzolini C, Brembilla NC, Montanari E, Truchetet ME. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmun Rev. 2011;10:276–81. doi: 10.1016/j.autrev.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Sato S, Hasegawa M, Takehara K. Serum levels of interleukin-6 and interleukin-10 correlate with total skin thickness score in patients with systemic sclerosis. J Dermatol Sci. 2001;27:140–6. doi: 10.1016/s0923-1811(01)00128-1. [DOI] [PubMed] [Google Scholar]
- 8.Brandwein SR, Medsger TA, Jr, Skinner M, Sipe JD, Rodnan GP, Cohen AS. Serum amyloid A protein concentration in progressive systemic sclerosis (scleroderma) Ann Rheum Dis. 1984;43:586–9. doi: 10.1136/ard.43.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciechomska M, Cant R, Finnigan J, van Laar JM, O'Reilly S. Role of toll-like receptors in systemic sclerosis. Expert Rev Mol Med. 2013;15:9. doi: 10.1017/erm.2013.10. [DOI] [PubMed] [Google Scholar]
- 10.He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood. 2009;113:429–37. doi: 10.1182/blood-2008-03-139923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen LE, Whitehead AS. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem J. 1998;334(Pt 3):489–503. doi: 10.1042/bj3340489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen KD, Macaubas C, Nadeau KC, Truong P, Yoon T, Lee T, Park JL, Mellins ED. Serum amyloid A overrides Treg anergy via monocyte-dependent and Treg-intrinsic, SOCS3-associated pathways. Blood. 2011;117:3793–8. doi: 10.1182/blood-2010-11-318832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connolly M, Mullan RH, McCormick J. Acute-phase serum amyloid A regulates tumor necrosis factor α and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012;64:1035–45. doi: 10.1002/art.33455. [DOI] [PubMed] [Google Scholar]
- 15.Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–6. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatanaka E, Dermargos A, Armelin HA, Curi R, Campa A. Serum amyloid A induces reactive oxygen species (ROS) production and proliferation of fibroblast. Clin Exp Immunol. 2011;163:362–7. doi: 10.1111/j.1365-2249.2010.04300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Migita K, Koga T, Satomura K, et al. Serum amyloid A triggers the mosodium urate -mediated mature interleukin-1β production from human synovial fibroblasts. Arthritis Res Ther. 2012;14:119. doi: 10.1186/ar3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J Immunol. 2006;177:5585–94. doi: 10.4049/jimmunol.177.8.5585. [DOI] [PubMed] [Google Scholar]
- 21.Lakota K, Thallinger GG, Cucnik S, et al. Could antibodies against serum amyloid A function as physiological regulators in humans? Autoimmunity. 2011;44:149–58. doi: 10.3109/08916934.2010.487504. [DOI] [PubMed] [Google Scholar]
- 22.Connolly M, Veale DJ, Fearon U. Acute serum amyloid A regulates cytoskeletal rearrangement, cell matrix interactions and promotes cell migration in rheumatoid arthritis. Ann Rheum Dis. 2011;70:1296–303. doi: 10.1136/ard.2010.142240. [DOI] [PubMed] [Google Scholar]
- 23.Hügle T, O'Reilly S, Simpson R, Kraaij MD, Bigley V, Collin M, Krippner-Heidenreich A, van Laar JM. Tumor necrosis factor-costimulated T lymphocytes from patients with systemic sclerosis trigger collagen production in fibroblasts. Arthritis Rheum. 2013;65:481–91. doi: 10.1002/art.37738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Shi-Wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requires transforming growth factor-β/activin-linked kinase 5, heparan sulfate-containing proteoglycans, and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699–711. doi: 10.1016/s0002-9440(10)61252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward C, Eger K, Diboll J, et al. Bronchial epithelial cells cultured from clinically stable lung allograft patients promote the development of macrophages from monocytes rather than dendritic cells. Thorax. 2009;64:430–5. doi: 10.1136/thx.2008.104067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson MR, Wallace K, Gieling RG, Manas DM, Jaffray E, Hay RT, Mann DA, Oakley F. NF-κB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J Hepatol. 2008;48:589–97. doi: 10.1016/j.jhep.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 27.O'Neill LAJ. How Toll-like receptors signal: what we know and what we don't know. Curr Opin Immunol. 2006;18:3–9. doi: 10.1016/j.coi.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Distler JH, Jüngel A, Pileckyte M, et al. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum. 2007;56:4203–15. doi: 10.1002/art.23074. [DOI] [PubMed] [Google Scholar]
- 29.de Seny D, Cobraiville G, Charlier E, et al. Acute-phase serum amyloid A in osteoarthritis: regulatory mechanism and proinflammatory properties. PLoS One. 2013;8:e66769. doi: 10.1371/journal.pone.0066769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushita T, Hasegawa M, Hamaguchi Y, Takehara K, Sato S. Longitudinal analysis of serum cytokine concentrations in systemic sclerosis: association of interleukin 12 elevation with spontaneous regression of skin sclerosis. J Rheumatol. 2006;33:275–84. [PubMed] [Google Scholar]
- 31.Hasegawa M, Sato S, Ihn H, Takehara K. Enhanced production of interleukin-6 (IL-6), oncostatin M and soluble IL-6 receptor by cultured peripheral blood mononuclear cells from patients with systemic sclerosis. Rheumatology. 1999;38:612–7. doi: 10.1093/rheumatology/38.7.612. [DOI] [PubMed] [Google Scholar]
- 32.Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J Biol Chem. 2012;287:2666–77. doi: 10.1074/jbc.M111.246173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Hori K, Ding J, Huang Y, Kwan P, Ladak A, Tredget EE. Toll-like receptors expressed by dermal fibroblasts contribute to hypertrophic scarring. J Cell Physiol. 2011;226:1265–73. doi: 10.1002/jcp.22454. [DOI] [PubMed] [Google Scholar]
- 34.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 35.Wähämaa H, Schierbeck H, Hreggvidsdottir HS, Palmblad K, Aveberger AC, Andersson U, Harris HE. High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther. 2011;13:R136. doi: 10.1186/ar3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niemi K, Teirilä L, Lappalainen J, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186:6119–28. doi: 10.4049/jimmunol.1002843. [DOI] [PubMed] [Google Scholar]
- 37.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 38.Li M, Li X, Wang E, Luo E. Upregulation of Toll-like receptor 2 gene expression by acetylation of AP-2 alpha in THP-1 cells, a human monocytic cell line. Int J Biochem Cell Biol. 2013;45:1594–9. doi: 10.1016/j.biocel.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Broen JC, Bossini-Castillo L, van Bon L, et al. A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. 2012;64:264–71. doi: 10.1002/art.33325. [DOI] [PubMed] [Google Scholar]
- 40.Muangchan C, Harding S, Khimdas S, Bonner A, Baron M, Pope J Canadian Scleroderma Research Group. Association of C-reactive protein with high disease activity in systemic sclerosis: results from the Canadian Scleroderma Research Group. Arthritis Care Res. 2012;64:1405–14. doi: 10.1002/acr.21716. [DOI] [PubMed] [Google Scholar]
- 41.Vreugdenhil AC, Dentener MA, Snoek AM, Greve JW, Buurman WA. Lipopolysaccharide binding protein and serum amyloid A secretion by human intestinal epithelial cells during the acute phase response. J Immunol. 1999;163:2792–8. [PubMed] [Google Scholar]
- 42.Kahaleh B. Vascular disease in scleroderma: mechanisms of vascular injury. Rheum Dis Clin North Am. 2008;34:57–71. doi: 10.1016/j.rdc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 43.Yang HZ, Cui B, Liu HZ, Chen ZR, Yan HM, Hua F, Hu ZW. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by reversion of suppressive immune microenvironment. J Immunol. 2009;182:692–702. doi: 10.4049/jimmunol.182.1.692. [DOI] [PubMed] [Google Scholar]
- 44.Ultaigh SN, Saber TP, McCormick J, et al. Blockade of Toll-like receptor 2 prevents spontaneous cytokine release from rheumatoid arthritis ex vivo synovial explant cultures. Arthritis Res Ther. 2011;13:R33. doi: 10.1186/ar3261. [DOI] [PMC free article] [PubMed] [Google Scholar]