

Abstract
Bacterial meningitis is, despite progress in research and the development of new treatment strategies, still a cause of severe neuronal sequelae. The brain is protected from penetrating pathogens by both the blood–brain barrier and the innate immune system. The invading pathogens are recognized by pattern recognition receptors including the G-protein coupled formyl peptide receptors (FPRs), which are expressed by immune cells of the central nervous system. The expression of FPRs is up-regulated during bacterial meningitis, but the consequence on the progression of inflammation and impact on mortality are far from clear. Therefore, we used mFPR1 and mFPR2-deficient mice to investigate the effects on inflammation, bacterial growth and mortality in a mouse model of pneumococcal meningitis. Our results revealed increased bacterial burden, increased neutrophil infiltration and higher mortality in mFPR1/2-deficient mice in comparison to wild-type mice. The mFPR1- or mFPR2-deficient mice also showed significantly increased glial cell density, whereas the immune responses including the expression of anti-inflammatory cytokines and antimicrobial peptides were decreased in bacterial meningitis. Taken together, the results suggest that FPR1 and FPR2 play an important role in the innate immune responses against Streptococcus pneumoniae within the central nervous system and the lack of the receptors leads to a dysregulation of the inflammatory response compared with wild-type mice.
Keywords: bacterial meningitis, formyl peptide receptor, glia cell, innate immunity, Streptococcus pneumoniae
Introduction
The bacterium Streptococcus pneumoniae is the most frequent pathogen responsible for bacterial meningitis in adults and, despite effective antibiotic treatment, causes the highest mortality rates, up to 60% in Africa.1,2 Furthermore, S. pneumoniae causes the highest rates of neurological sequelae.3 The pathophysiological process in pneumococcal meningitis is complex with the immune response being a major key factor for both positive and negative effects on the outcome.4 For the initiation of the inflammatory response and the detection of pathogens and their components, the cells of the innate immune system – glial cells, astrocytes and microglia – play a very important role. Astrocytes and microglia are involved in the pathogen recognition and the activation of the innate immune response. They are the key regulators of the innate immune response within the brain because of their ability to release various factors supporting the host's immune defence and helping to coordinate the activation of different immune cells.5 Astrocytes are furthermore required for structural support and the maintenance of the blood–brain barrier. Microglia are considered to be central nervous system (CNS) -resident macrophages and sensor cells that function as principal innate immune effector cells.6
Pattern-recognition receptors (PRR) on immune cells are responsible for the detection of pathogens and their components. These receptors recognize pathogen-associated-molecular patterns as an important mechanism for the recognition of pathogens such as bacteria by the innate immunity.7 One of these secreted pathogen-associated-molecular patterns is N-formyl-methionyl-leucyl-phenylalanine peptide (fMLF) from the bacterial cell wall. The fMLF is known as an inducer of chemotaxis for neutrophil granulocytes and monocytes.8,9 Known receptors for fMLF are the formyl peptide receptors (FPR). These belong to the family of G-protein coupled receptors. The murine FPR gene family has at least six members in contrast to only three in humans. The two most important members are the Fpr1 and Fpr2. Fpr1 encodes murine Fpr1, which is considered the murine orthologue of human FPR, whereas Fpr-rs2 (mFPR2) encodes a receptor that is most similar to human formyl peptide receptor like 1 (FPRL1) or FPR2.10,11 It is known that the receptor interacts with a menagerie of structurally diverse ligands including some from bacterial origins beside fMLF, such as Hp(2-20), derived from Helicobacter pylori, HIV envelope peptides, or is associated with different neurodegenerative disorders, including Alzheimer's and prion disease.12 Previous works revealed the expression of FPRs in the brain by glial cells.13,14 In addition, the receptors are important for the chemotatic movement and mobilization of neutrophils, and FPR-deficient mice showed an impaired antibacterial host defence against Listeria monocytogenes infection.15,16 Furthermore, a recent work revealed involvement of FPRL1 (FPR2) in the inflammatory response of glial cells after bacterial exposure.17 To the knowledge of the authors, there are no data in the context of bacterial meningitis. However, it is unknown whether the FPRs play a pro-inflammatory or anti-inflammatory role in the inflammatory responses. Although the results of Dufton et al.18 suggest anti-inflammatory properties of FPRL1 (FPR2), the work of Chen et al.19 showed a pro-inflammatory role of FPR2.
This study was designed to determine the role of FPRs in a mouse model of pneumococcal meningitis. We examined bacterial growth and mortality of infected mFPR1-deficient, mFPR2-deficient and wild-type (WT) mice and characterized inflammatory responses by evaluating neutrophil infiltration, glial cell density and cytokine/chemokine expression We also investigated the activation of mFPR1-, mFPR2-deficient and WT glial cells by bacterial supernatants and components.
Materials and methods
Reagents
Bacterial supernatants were produced as described in detail by Brandenburg et al.20 Briefly, for the production of bacterial culture supernatants, bacteria were grown in stationary broth cultures until they had reached an optical density of 1·0. Brain–heart infusion broth was used for the cultivation of S. pneumoniae, and thioglycollate broth supplemented with K1 and haemin was used for Neisseria meningitides. Broth cultures of the test bacteria were centrifuged at 5000 g for 15 min, and the resulting supernatants were filter-sterilized. In these experiments, bacterial supernatants of Neisseria meningitidis (NM; ATCC 13077) and S. pneumoniae (SP; ATCC 6303) were used, all at a dilution of 1 : 100. Lipopolysaccharide (LPS; L9516) from Salmonella enterica serovar Typhimurium (used at 100 ng/ml) and peptidoglycan (PGN; 77140) from Staphylococcus aureus (used at 10 μg/ml) were obtained from Sigma-Aldrich (Munich, Germany).
Animals
FPR1-deficient (knockout; KO) mice were a kind gift from Dr Philip Murphy of the National Institute for Allergy and Infectious Diseases, NIH, Bethesda, MD.16 The FPR2-KO mice were generated as described previously.19 Both mouse strains have a C57BL/6 background. The wild-type (WT) mice were back-crossed on the C57BL/6J background for at least five generations.
Cell culture
Isolated cerebral cortices and rostral mesencephali from mFPR1-deficient, mFPR2-deficient or WT mice (P2 to P3) were stripped of the meninges, minced and dissociated enzymatically with trypsin from bovine pancreas (Sigma-Aldrich, Taufkirchen, Germany) in PBS and 1 mg/ml DNase I (Roche Molecular Biochemicals, Mannheim, Germany) for 30 min at 37° and crushed mechanically with Pasteur pipettes. Astrocytes were prepared according to the protocol of McCarthy and de Vellis,21 which allows the preparation of nearly pure cultures of astrocytes (> 97%), and cultivated in Dulbecco's modified Eagle's medium (DMEM; PAA Laboratories, Pasching, Austria) supplemented with 10% fetal calf serum (FCS) and 1% penicillin and streptomycin (Sigma-Aldrich). Contaminating microglial cells were removed by pre-plating the cells on plastic dishes for 5–10 min and discarding the unattached cells. Microglia were collected from the medium of primary cell cultures of astrocytes. As cell culture medium we used microglial cell growth medium [DMEM contains 10% FCS (heat inactivating from 44 to 53°) and antibiotics (penicillin and streptomycin)].22 After about 10 days, the cells begin to move away from the cell layer and swim in the supernatant. The cells were collected by centrifugation of the supernatants and then seeded in normal medium (DMEM, 10% FCS heat-inactivated at 56°, penicillin and streptomycin). Before replating microglial cells for different assays, cell number and viability were estimated by trypan blue exclusion. This procedure increased the purity of the microglial preparation to > 98% with only a few remaining astrocytes.
Mouse model of experimental pneumococcal meningitis
Male mFPR1-deficient, mFPR2-deficient or WT mice (weight 19–23 g, aged 2–3 months) were anaesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg) and infected by injecting 104 colony-forming units (CFU) of an S. pneumoniae D39 (type 2) strain in the subarachnoid space through the right frontolateral skull.23 Uninfected control animals were injected with 10 μl of a sterile saline solution. All infected mice developed clinical signs of infection within 24 hr. The most sensitive sign of meningitis in mice was weight loss. There were no apparent behavioural abnormalities until c.18 hr after infection, but mice gradually became lethargic between 18 and 24 hr. Later than 24 hr after infection, mice became severely lethargic, unable to walk and developed an opisthotonus. Epileptic seizures were observed but were infrequent. For the first set of experiments, the clinical state and survival time were closely monitored without antibiotic treatment. In a second set of experiments with the same infection regimen, male mFPR1-deficienct, mFPR2-deficient or WT mice were infected and killed 30 hr after induction of meningitis without antibiotic treatment and were either perfused with 4% formalin for immunohistochemical analysis or with 0·9% NaCl solution for RNA isolation. Bacterial titres were evaluated 30 hr after infection in tissue homogenates of cerebellum and spleen and in blood samples by plating 10-fold dilutions on blood agar plates and incubation for 24 hr at 37° and 5% CO2 (detection limit 102 CFU/ml in tissue homogenates and 103 CFU/ml in blood samples24). For meningeal inflammation, the mice were infected for 48 hr. At this time-point, a strong infiltration of neutrophil granulocytes was observed. All animal experiments were approved by the Animal Care Committee of the University Hospital of Aachen and by the District Government in Recklinghausen, North Rhine-Westphalia, Germany.
Meningeal inflammation score
Meningeal inflammation was estimated by the invasion of granulocytes into the frontal interhemispheric region, the whole hippocampal fissure (both sides), three superficial meningeal regions over the convexities, and the third ventricle (complete distribution). One high-power field (20 × objective) was scored in each region: no granulocytes: 0; < 10 granulocytes: 1; 10–50 granulocytes: 2; and > 50 granulocytes: 3. The scores of the individual regions were added (range of the score: 0–21).25
RNA isolation and real-time RT-PCR and PCR array
Total RNA was isolated using the peqGold Trifast reagent (Peqlab, Erlangen, Germany) according to the manufacturer's instructions. RNA samples were reverse-transcribed by Moloney murine leukaemia virus reverse transcriptase (Fermentas, Burlington, ON, Canada) and random hexamer primers (Invitrogen, Darmstadt, Germany). The cDNA products were used immediately for SYBR green (Applied Biosystems, Darmstadt, Germany) real-time RT-PCR. Gene expression was monitored using the StepOne Plus apparatus (Applied Biosystems) according to the manufacturer's protocol.22 Relative quantification was performed using the ΔCt method, which results in ratios between target genes and a housekeeping reference gene (TATA box binding protein for brain tissue, 18s for cells). The primers for glial fibrillary acid protein (GFAP), Integrin alpha M (Itgam), Cathelin-related antimicrobial peptide (CRAMP) and defensin b4 (Defb4) were manufactured by Qiagen (QT00101143, QT00156471, QT00156571 and QT00257656; QuantiTect Primer Assay; Qiagen, Hilden, Germany). The primers for interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), Toll-like receptor 2 (TLR2), IL-1 receptor antagonist (IL-1RA), haemoxygenase 1 (HO-1), chemokine (C-C motif) ligand 2 (CCL2) and chemokine (C-C motif) ligand 3 (CCL3) were manufactured by Eurofins MWG Operon (Ebersberg, Germany; for primer sequences of TATA box binding protein, IL-6 and TNF-α see ref. 26; for 18s see ref. 27; for CCL2 and CCL3 see ref. 28; for TLR2, IL-1RA and HO-1 see Table1). The specificity of the amplification reaction was determined by melting curve analysis.
Table 1.
Primer sequences for real-time RT-PCR gene analysis
| Primer | Sequence | Annealing temp. [°] | |
|---|---|---|---|
| IL-1RA | For Rev | 5′-GGGGGCAAGCTGTGCCTGTC-3′ 5′-TCAAAGCTGGTGGTGGGGCC-3′ | 60 |
| HO-1 | For Rev | 5′-AAGCCGAGAATGCTGAGTTCA-3′ 5′-GCCGTGTAGATATGGTACAAGGA-3′ | 61·5 |
| TLR2 | For Rev | 5′-AACAGTCCTCTTCAGCAAACGC-3′ 5′-TAGAGGTGAAAGACCTGGAGCG-3′ | 60 |
Immunohistochemistry
Formalin-fixed and paraffin-embedded 5-μm whole coronary brain sections were examined. For immunofluorescence staining, sections were deparaffinized, for Iba-1 staining they were pre-treated for 10 min with microwaving in Tris/EDTA/Tween-20 buffer and after blocking with 5% normal goat serum in PBS incubated with either polyclonal rabbit anti-GFAP (1 : 9000; EnCor, Gainesville, FL) or anti-Iba1 (1 : 10 000; Wako, Neuss, Germany) overnight at 4°. This was followed by incubation with the biotinylated secondary antibody (1 : 400; DAKO, Hamburg, Germany) and peroxidase-labelled streptavidin–biotin staining technique. For staining, aminoethyl carbazole was used. After counterstaining with haematoxylin, the slides were finally mounted with Aquatex (Boehringer, Mannheim, Germany). For neutrophil granulocyte staining, slices were stained with naphthol AS-D chloroacetate esterase (91-C Kit; Sigma-Aldrich) according to the manufacturer's protocol. For the negative controls the primary antibodies were omitted.
Quantification of immunoreactive cells
The sections were examined blind using a 10 × objective up to a 40 × objective. Only immunoreactive cells within the hippocampal formation were counted. An analysis software imaging system (microscope Keyence BZ-9000; Keyence, Neu-Isenburg, Germany) was used to measure the area of the hippocampal formation. The densities of immunolabelled cells were expressed as the number of marked cells per square millimetre of the area measured. The density of labelled cells was evaluated in four coronal sections from each mouse.
CellTiter-Blue cell viability assay
The CellTiter-Blue assay (Promega, Mannheim, Germany) was used to measure the viability of glial cells after bacterial treatment. The cells were seeded in 96-well plates and starved overnight by adding FCS-free medium. The cells were treated with different bacterial supernatants (NM and SP) or components (LPS and PGN) for 24 hr. A CellTiter-Blue assay was used according to the manufacturer's recommendations. Spectrophotometric evaluations were performed after 1, 2 and 4 hr, and treated cells were compared with untreated cells.
Statistical analysis
Survival time was expressed in hours and evaluated by generating a Kaplan–Meier plot that was statistically analysed using the log-rank test. Bacterial titres were converted into log CFU/ml and compared using non-parametric U-test. Immunohistochemical data were expressed as median and interquartile range. For statistical comparison, the non-parametric Mann–Whitney U-test was used. All real-time RT-PCR experiments were performed in duplicate. The values were expressed as mean ± SEM. For statistical comparison, of analysis of variance tests followed by Bonferroni multiple comparison test were used. A value of P < 0·05 was considered statistically significant. For statistical calculation, graphpad prism 5.0 was used (Graph Pad Software, San Diego, CA).
Results
FPR1- or FPR2-deficiency resulted in increased bacterial burden, increased neutrophil infiltration and mortality after pneumococcal meningitis
In a first set of experiments, mortality after pneumococcal meningitis was investigated. The survival time after infection was significantly shorter in FPR1- or FPR2-deficient (KO mice) compared with WT mice [58 (55/68) hr and 48 (38/67) versus 72 (59/77) hr; medians (25th/75th centiles); Mann–Whitney U-test; P = 0·0023 and P = 0·004, Fig.1a). Fifty hours after infection, 94·1% of the WT animals were still alive whereas only 76·9% of FPR1-KO and 80% of FPR2-KO mice were still alive (Fig.1b). The Kaplan–Meier curve revealed a significantly increased mortality in FPR1-KO in comparison with WT mice (P =0·0005; Logrank-Test). In addition, we determined bacterial titres in homogenates of cerebellum and spleen and in blood samples from a separate set of FPR1-KO, FPR2-KO or WT mice 30 hr after infection. The results showed that FPR1-KO animals had a significantly higher bacterial burden in all investigated tissue compartments compared with WT mice, whereas the FPR2-KO mice were only found in the blood with a significantly increased bacterial load (Table2). Furthermore, neutrophil granulocyte invasion was evaluated histochemically by chloroacetate esterase staining. 48 hr after infection, FPR1-KO or FPR2-KO mice displayed a higher degree of neutrophil granulocyte infiltration than WT mice (Fig.1c). To verify the results, meningeal inflammation was determined by a semi-quantitative score: a strong granulocytic invasion of the meninges was observed in all infected mice 48 hr after inoculation, whereas infiltration of granulocytes was absent in non-infected animals. For WT mice, the meningeal inflammation score was 14·8 ± 1·3, whereas for FPR1-KO or FPR2-KO mice the scores were 23·4 ± 1·5 or 23 ± 1 (each group n = 4). The differences between infected FPR1-KO or FPR2-KO and WT mice were significant (P = 0·0243 for FPR1-KO versus WT; P = 0·0498 for FPR2-KO versus WT, Mann–Whitney U-test).
Figure 1.
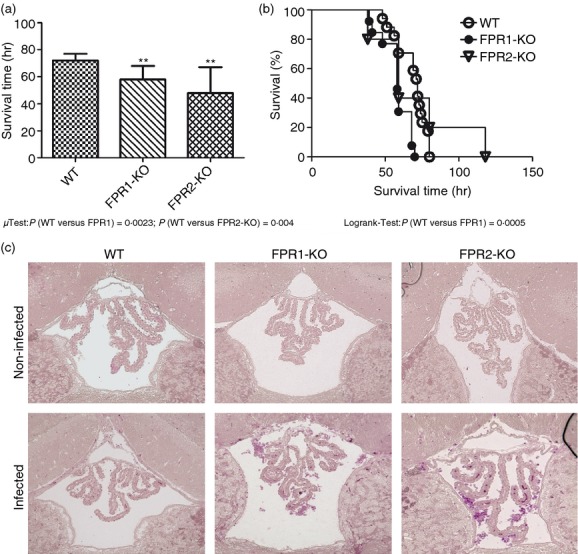
Mortality after pneumococcal meningitis. Wild-type (WT) and formyl peptide receptor 1 (FPR1) or FPR2 knock out (KO) mice were infected by injection of 104 colony-forming units (CFU) of Streptococcus pneumoniae D39 (type 2) strain in the subarachnoid space. (a) Comparison of survival times revealed that WT mice lived significantly longer (WT, n = 18; FPR1-KO, n = 15; FPR2-KO, n = 17; median with percentile). (b) Mortality was significantly increased in FPR1-KO animals (Kaplan–Meier curve). (c) Detection of neutrophil granulocytes by naphthol AS-D chloroacetate esterase (NCAE) reaction in the third ventricle revealing stronger neutrophil infiltration in FPR1-KO and FPR2-KO and WT mice 48 hr after pneumococcal infection. The figures show representative results from one of three mice per group.
Table 2.
Bacterial titres after pneumococcal meningitis
| Bacterial titre (log CFU/ml) | ||||
|---|---|---|---|---|
| Cerebellum | Blood | Spleen | ||
| WT | 5 (4·3/5·48) | 5 (3·98/5·4) | 5·9 (5·3/6·3) | |
| FPR1-KO | 8·3 (7/10)** | 7 (7/9)*** | 9·6 (6·6/10·8)*** | |
| FPR2-KO | 5·65 (4·9/8·25) | 5·78 (4·95/7·25)* | 6·65 (5·75/8·9) | |
CFU, colony forming units; FPR, formyl peptide receptor 1; WT, wild-type; KO, knockout.
Data are presented as median (25th/75th centile).
Please note significantly increased bacterial titres in infected FPR1- or 2-KO compared to infected WT mice (n = 9 each group;
P = 0·0301 for blood;
P = 0·0041 for cerebellum;
P = 0·0006 for blood and P = 0·0009 for spleen; Mann–Whitney U-test).
Increased glial cell density during pneumococcal meningitis in FPR1- or FPR2-deficient mice
Thirty hours after infection, the degree of glial cell density was determined by immunohistochemistry of the astrocyte marker GFAP and the microglia marker ionized calcium-binding adaptor molecule 1 (Iba-1) in the hippocampal formation as a clearly definable brain region especially involved in the pathogenesis of bacterial meningitis. As shown in Fig.2(a,c), the hippocampal formation of non-infected FPR1-KO or FPR2-KO as well as WT mice showed very few GFAP or Iba-1-positive cells whereas the infection with S. pneumoniae resulted – as expected – in a strong increase in the number of GFAP or Iba-1-immunoreactive cells. Quantification of astrocytes in the hippocampal formation revealed more astrocytes/mm2 in all three mouse strains compared with non-infected mice after pneumococcal meningitis (Fig.2b). However, the comparison between FPR1- or FPR2-deficient and WT mice did not show significant differences. As expected, the number of microglial cells strongly increased in all mice within the hippocampal formation 30 hr after infection. In contrast to astroglia, the density of microglia was higher in FPR1-KO or FPR2-KO mice with statistical difference for FPR2-KO in comparison with WT mice (Fig.2d). The level of the GFAP or Iba-1 quantification of the non-infected animals showed no significant differences (data not shown).
Figure 2.

Glial cell density in the hippocampus after pneumococcal meningitis. Coronal brain sections stained with anti-GFAP to identify astrocytes (a) or anti-Iba-1 to identify activated microglial cells (c) 30 hr after infection with Streptococcus pneumoniae. The figures show representative results from one of three independent experiments. A detailed section was included. GFAP (b) and Iba-1 (d) immunoreactive cells were quantified per mm2 area of the hippocampal formation of infected and healthy mice. Immunoreactive cells were normalized to non-infected controls. The asterisk indicates a significant difference between infected wild-type (WT) and formyl peptide receptor 1 knockout (FPR1-KO) or FPR2-KO (each group n = 6) mice as determined by analysis of variance followed by Bonferroni test (*P < 0·05).
Next, we analysed the mRNA expression of GFAP as well as integrin αM (Itgam or CD11b29; as a marker for microglial cell activation) in the hippocampal formation and frontal cortex of FPR1-KO, FPR2-KO or WT mice 30 hr after infection with S. pneumoniae. As shown in Fig.3(a), FPR1-KO mice showed a significant increase of GFAP mRNA expression in the hippocampal formation compared with the infected WT (281·7 ± 98-fold compared with 23·8 ± 9-fold increase in expression, P < 0·05). In the hippocampal formation of FPR2-KO mice and in the cortex of FPR1-KO or FPR2-KO mice, there was a tendency to higher levels of GFAP mRNA but this difference did not reach statistical significance (Fig.3a,c). Similarly, the expression of Itgam mRNA was higher under pathological conditions in both the hippocampal formation and the frontal cortex but there was no statistically significant difference between KO and WT mice, (Fig3b,d).
Figure 3.
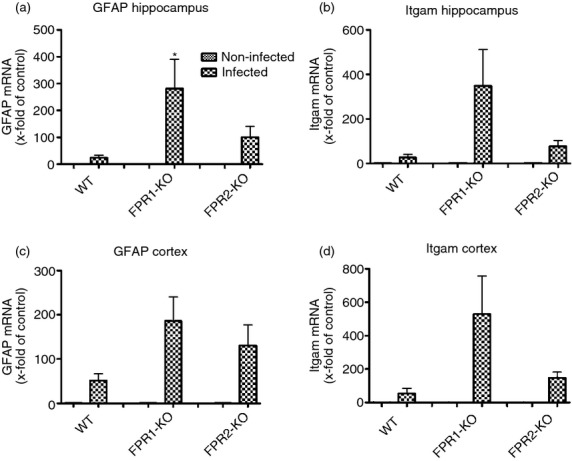
Gene expression of glial cell marker after pneumococcal meningitis. Analysis of mRNA expression of the astrocyte marker GFAP (a, c) and the activated microglia marker Itgam (b, d) in the hippocampal formation and cortex of formyl peptide receptor 1 knockout (FPR1-KO) or FPR2-KO) and wild-type (WT) mice 30 hr after induction of pneumococcal meningitis by real-time RT-PCR were assessed from six independent experiments in duplicate. The asterisks indicate a significant difference between infected WT and infected FPR1-KO mice as determined by analysis of variance followed by Bonferroni test (*P < 0·05).
Comparison of gene expression of innate immunity markers after bacterial meningitis in the hippocampal formation and frontal cortex of FPR1- or FPR2-deficient and WT mice by real-time RT-PCR
In a next set of experiments, we analysed the mRNA expression of pro-inflammatory and anti-inflammatory as well as antimicrobial genes involved in the innate immune response in the hippocampal formation and frontal cortex of FPR1-deficient, FPR2-deficient or WT mice 30 hr after infection. At first, we examined the gene expression of the pro-inflammatory cytokines IL-6 and TNF-α as representatives of inflammatory mediators in pneumococcal meningitis. The infection resulted in a strong increase of the mRNA expression of TNF-α in all three strains with a significant increase in the cortex of FPR2-KO mice (24 ± 8-fold induction compared with 7 ± 2-fold of expression of infected WT mice, P < 0·05; Fig.4a,f). The expression of IL-6 was increased in all three strains, too and most pronounced in the hippocampal formation of FPR1-deficient mice (125 ± 55-fold induction compared with 38·1 ± 11-fold of expression of infected WT mice; Fig.4b,g). Furthermore, we analysed the mRNA expression of antimicrobial peptides as important mediators of the innate immunity.30 The first antimicrobial peptide to be investigated was β-defensin 4. In contrast to WT mice where the β-defensin 4 expression was distinctly increased in both hippocampal formation and frontal cortex (55·8 ± 25- and 52·7 ± 18-fold increase of expression in WT mice; Fig.4c,h), FPR1- or FPR2-deficient mice displayed attenuated increase in expression. The second antimicrobial peptide to be investigated was CRAMP: which is the active peptide from the only known cathelicidin gene present in mice.31 Whereas all three strains displayed an increase of CRAMP in the hippocampal formation – reaching the highest levels in WT mice, mRNA expression was significantly attenuated in the frontal cortex of FPR2-deficient mice (28 ± 6-fold induction compared with 80 ± 19-fold of expression of infected WT mice, P < 0·05; Fig.4d,i). Besides pro-inflammatory and antimicrobial peptide genes, we analysed the mRNA expression of TLR2. TLRs are an important receptor family of the PRRs and TLR2 specifically recognizes cell wall components of Gram-positive bacteria, such as peptidoglycan.32 As shown in 4(e,j), the analysis revealed a significant increase of TLR2 in the hippocampal formation of infected FPR1-deficient mice compared with infected WT mice (92 ± 22-fold induction compared with 4·7 ± 1·5-fold expression of infected WT mice, P < 0·01; Fig.4e,j). In contrast, the results of the frontal cortex showed only a slight increase of TLR2 in FPR1-deficient mice but a strong increase in FPR2-deficient mice (70 ± 13-fold induction for FPR2-KO or 16 ± 6-fold induction for FPR1-KO compared with 8 ± 3-fold of expression of infected WT mice, P < 0·001; Fig.4e,j). Interestingly, the differences in the hippocampus as well as cortex between infected FPR1-KO and FPR2-KO mice are significant (P < 0·01 for Fig.4e and P < 0·001 for Fig.4j).
Figure 4.
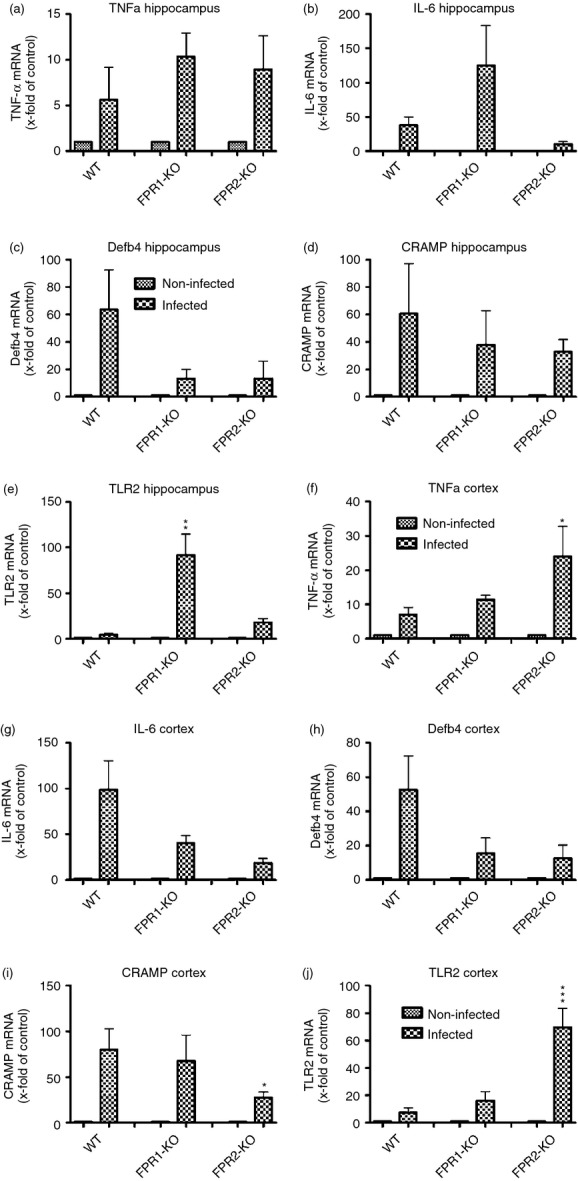
Expression of different innate immunity markers after pneumococcal meningitis in hippocampus and cortex. Thirty hours after subarachnoid infection with Streptococcus pneumoniae, mRNA expression levels of tumour necrosis factor-α (TNF-α) (a, f), interleukin-6 (IL-6) (b, g), β-defensin 4 (c, h), cathelicidin-related antimicrobial peptide (CRAMP) (d, i) and Toll-like receptor 2 (TLR2) (e, j) were determined in the hippocampal formation and cortex of formyl peptide receptor 1 knockout (FPR1-KO) or FPR2-KO and wild-type (WT) mice by real-time RT-PCR. Data were assessed from six independent experiments in duplicate. An asterisk indicates a significant difference between infected WT and infected FPR1-KO or FPR2-KO mice as determined by anova followed by Bonferroni test (*P < 0·05; **P < 0·01; ***P < 0·001).
Comparison of anti-inflammatory agents and chemokine gene expression after bacterial meningitis in the hippocampal formation and frontal cortex of FPR1- or FPR2-deficient and WT mice by real-time RT-PCR
To further characterize the innate immune response in FPR1- or FPR2-deficient mice during pneumococcal meningitis, mRNA expression of cytokines/chemokines with anti-inflammatory properties was conducted. At first, mRNA expression of the anti-inflammatory cytokine IL-1 receptor antagonist (IL-1RA) was determined. IL-1RA inhibits the pro-inflammatory response of IL-1β, which is released primarily in response to exogenous agents.33 While IL-1RA expression was similarly increased in infected WT and FPR1-KO mice, no increase was observed in FPR2-deficient mice. The difference between infected WT and infected FPR2-KO and also between infected FPR1-KO and FPR2-KO in the frontal cortex reached statistical significance (P < 0·05; Fig.5a,e). Furthermore, the expression of the anti-inflammatory enzyme HO-134 was determined. As expected, in WT mice a significant increase of HO-1 expression was noted whereas no increase could be observed in both the hippocampal formation and frontal cortex of both KO strains. For the FPR2-deficient animals, the difference between infected WT and infected FPR2-KO mice was significant in both compartments (P < 0·05, Fig.5b,f). The formyl peptide receptors are chemotactic G-protein coupled receptors for the bacterial cell wall component fMLF.14 To analyse whether the deficiency of FPRs resulted in compensatory reactions of other chemotactic agents, we revealed the mRNA expression of CCL2, which recruits monocytes, memory T cells and dendritic cells to the sites of inflammation,35 and CCL3, which is involved in the acute inflammatory state in the recruitment and activation of polymorphonuclear leucocytes.36 CCL2 expression was significantly increased in both the hippocampal formation as well as the frontal cortex of FPR1-deficient mice only (12 664 ± 4734-fold and 616 ± 132-fold induction of expression; P < 0·05 and P < 0·01; Fig.5c,g) whereas in FPR2-deficient mice, no significant changes were determined (1563 ± 695-fold induction compared with 100 ± 45-fold of expression of infected WT mice). Also the difference in the hippocampus and cortex between infected FPR1-KO and FPR2-KO were significant (P < 0·05 for Fig.5c and P < 0·01 for Fig.5g). The analysis of CCL3 revealed a similar increase of expression in the cortex of all three strains (Fig.5h) whereas in the hippocampal formation of FPR1-deficient mice, the expression was significantly increased compared with the infected WT but also compared with the infected FPR2-KO (1434 ± 328-fold induction compared with 13·4 ± 5-fold of expression of infected WT mice; P < 0·01; Fig.5d).
Figure 5.
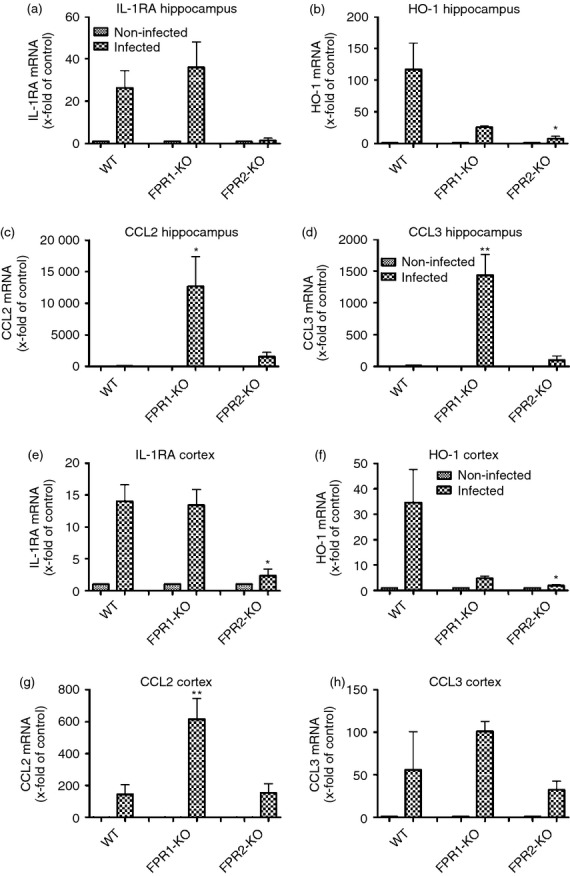
Expression of anti-inflammatory factors and chemokines after pneumococcal meningitis in different brain regions. Thirty hours after subarachnoid infection with Streptococcus pneumoniae, mRNA expression levels of interleukin-1 receptor antagonist (IL-1RA; a, e), haemoxygenase 1 (HO-1; b, f), CCL2 (c, g) and CCL3 (d, h) were determined in the hippocampal formation and cortex of formyl peptide receptor 1 knockout (FPR1-KO) or FPR2-KO and wild-type (WT) mice by real-time RT-PCR. Data were assessed from six independent experiments in duplicate. An asterisk indicates a significant difference between infected WT and infected FPR1-KO or FPR2-KO mice as determined by anova followed by Bonferroni test (*P < 0·05; **P < 0·01).
In vitro observations
Attenuated cell viability after bacterial stimulation of FPR1-deficient microglia cells
To further characterize which cell type is responsible for the different inflammatory mediations and to expand the microbial expertise, in vitro experiments with glial cells were carried out. Primary astrocytes or microglial cells from FPR1- or FPR2-deficient and WT mice were exposed to bacterial supernatants of S. pneumoniae (SP) and N. meningitidis (NM) as well as the cell wall components LPS or PGN for 24 hr. At first, the cell viability was determined using a CellTiter-Blue assay. As shown in Fig.6(a), the results show no differences for astrocytes after the treatment with bacterial supernatants (SP or NM) or components (LPS or PGN). For microglia, the FPR1 deficiency resulted in an attenuated viability after the bacterial treatment with a significant difference for SP and PGN compared with treated WT microglia (P < 0·01; Fig.6b).
Figure 6.
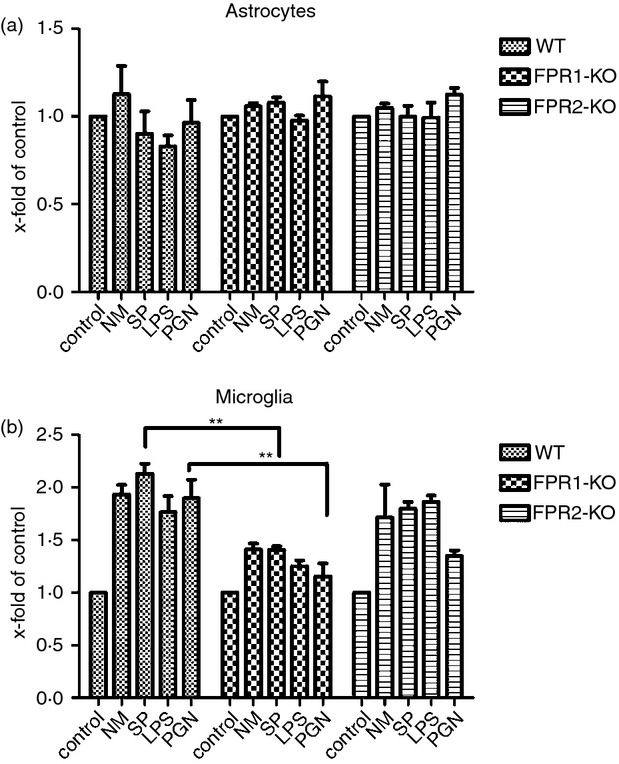
Formyl peptide receptor 1 (FPR1) deficiency resulted in attenuated cell viability after treatment of microglial cells with bacterial supernatants. Astrocytes (a) and microglial cells (b) from FPR1-knockout (KO) or FPR2-KO or wild-type (WT) mice were incubated with bacterial supernatants of Gram-positive bacterium Streptococcus pneumoniae (SP) or Gram-negative bacterium Neisseria meningitidis (NM) and bacterial cell wall components lipopolysaccharide (LPS) or peptidoglycan (PGN) for 24 hr. Cell viability was determined using CellTiter-Blue. Data were assessed from three independent experiments. An asterisk indicates a significant difference between treated WT and treated FPR1-KO or FPR2-KO glial cells as determined by analysis of variance followed by Bonferroni test (**P < 0·01).
Increased pro-inflammatory and decreased antimicrobial gene expression after bacterial stimulation of FPR-deficient glial cells
Second, the mRNA expression of pro-inflammatory cytokines TNF-α and IL-6 were examined. Both FPR1- and FPR2-deficient astrocytes displayed an increase of TNF-α expression in comparison to WT mice but most pronounced was the statistically significant increase of FPR1-deficient microglia after exposure to SP and LPS treatment (P < 0·05; Fig.7a,e), whereas no relevant change was observed in FPR2-deficient microglia. In general, IL-6 was increased in astrocytes after exposure to bacterial stimulants with only FPR1-deficient astrocytes reaching statistical significance after treatment with LPS in comparison with WT astrocytes (P < 0·001; Fig.7b). A similar observation was made in microglia with significant increases of IL-6 expression in FPR1-microlia after exposure to LPS (P < 0·001; Fig.7f). In contrast to the observations of pro-inflammatory mediators being up-regulated in FPR1- or FPR2-deficient astrocytes and microglia, the expression of antimicrobial peptides showed rather an attenuated increase: whereas no significant differences of β-defensin 4 mRNA expression were detected in astrocytes of the KO strains (Fig.7c), the expression of β-defensin 4 was significantly attenuated in microglia of FPR1- or FPR2-deficient mice stimulated by SP treatment (P < 0·05; Fig.7g). The same regulation was true for the antimicrobial peptide CRAMP, only that the regulation was significant in astrocytes instead of microglia of KO mice after LPS treatment (P < 0·05; Fig.7d,h).
Figure 7.
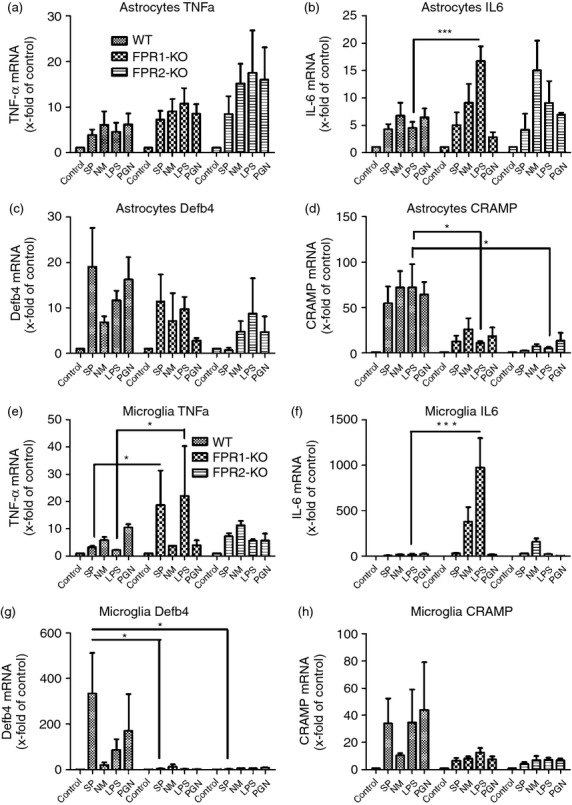
Bacterial supernatants induced pro-inflammatory cytokines and antimicrobial peptides expression in primary glial cells. Astrocytes and microglial cells from formyl peptide receptor 1 knockout (FPR1-KO) or FPR2-KO or wild-type (WT) mice were incubated with bacterial supernatants of Gram-positive bacterium Streptococcus pneumoniae (SP) or Gram-negative bacterium Neisseria meningitidis (NM) and bacterial cell wall components lipopolysaccharide (LPS) or peptidoglycan (PGN) for 24 hr. The mRNA expression levels of tumour necrosis factor-α (TNF-α) (a, e), interleukin-6 (IL-6) (b, f), β-defensin 4 (c, g) and cathelicidin-related antimicrobial peptide (CRAMP) (d, h) were determined by real-time RT-PCR. Data were assessed from six independent experiments in duplicate. An asterisk indicates a significant difference between treated WT and treated FPR1-KO or FPR2-KO glial cells as determined by analysis of variance followed by Bonferroni test (*P < 0·05; ***P < 0·001).
Comparison of anti-inflammatory agents and chemokine gene expression after bacterial stimulation of FPR1- or FPR2-deficient and WT glial cells by real-time RT-PCR
At first, the mRNA expression of the anti-inflammatory cytokine IL-1RA was determined. The WT astrocytes reacted to bacterial stimulation by an increase of IL-1RA expression, whereas FPR1- or FPR2-deficient astrocytes displayed an attenuated increase of IL-1RA expression in answer to bacterial supernatants or components. For the stimulation with NM, LPS and PGN the differences were significant (Fig.8a). In microglia cells, the results showed a slightly attenuated increase of IL-1RA mRNA expression in FPR1- and FPR2-microglial cells with a statistically significant difference for PGN treatment between WT and FPR1- or FPR2-deficiency (P < 0·05; Fig.8e). The results of the anti-inflammatory enzyme HO-1 revealed in WT astrocytes an increased expression (Fig.8b) that was significantly reduced in comparison to FPR2-deficient astrocytes after stimulation with NM and PGN (P < 0·01 for NM and P < 0·05 for PGN). FPR1- or FPR2-deficiency inhibited the expression of HO-1 mRNA in microglia significantly in comparison to WT cells (P < 0·05 for SP and PGN, Fig.8f). In addition, the CCL2 and CCL3 mRNA expression in astrocytes as well as microglial cells were determined. For CCL2, the mRNA expression in WT astrocytes was strongly increased by the exposure to all stimulants with the exception of SP (Fig.8c). The FPR1- or FPR2-deficient astrocytes showed rather an attenuated increase of expression with a significant difference for NM stimulation between WT and FPR2-deficient astrocytes (P < 0·001). For microglial cells, the FPR1-deficient cells revealed an increase of CCL2 mRNA expression with reaching statistical significance for NM stimulation compared with NM-stimulated WT cells (P < 0·05; Fig.8g). Analysis of CCL3 mRNA expression revealed a strong increase for NM, LPS and PGN stimulation in WT astrocytes (Fig.8d) while FPR1- and FPR2-deficiency resulted in a rather attenuated increase in CCL3 mRNA expression after LPS and PGN stimulation, whereas NM treatment significantly increased the expression in FPR1-deficient astrocytes (P < 0·05). As shown in Fig.8(h), no differences in CCL3 were detected between WT and FPR1-deficient microglial cells, whereas FPR2-defiency resulted in an attenuated increase CCL3 mRNA expression.
Figure 8.
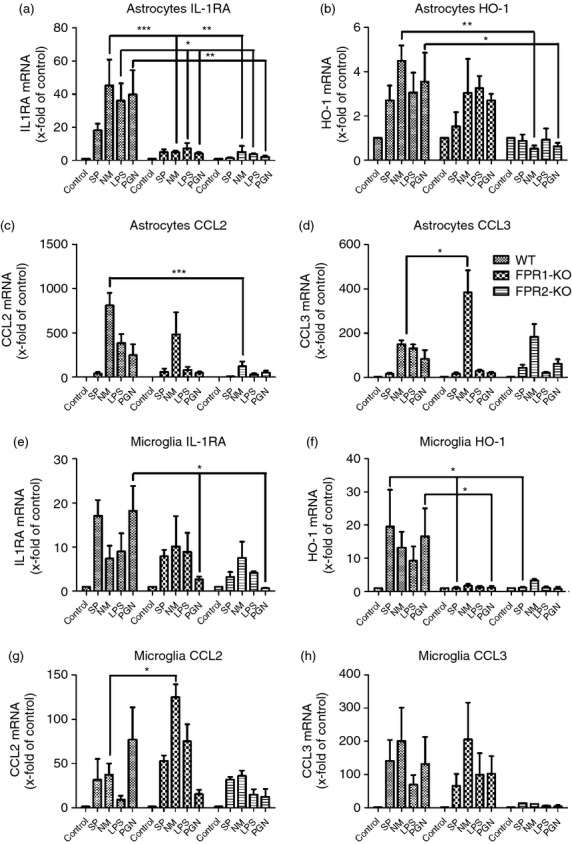
Bacterial supernatants induced anti-inflammatory factors and chemokines expression in primary glial cells. Astrocytes and microglial cells from formyl peptide receptor 1 knockout (FPR1-KO) or FPR2-KO or wild-type (WT) mice were incubated with bacterial supernatants of Gram-positive bacterium Streptococcus pneumoniae (SP) or Gram-negative bacterium Neisseria meningitidis (NM) and bacterial cell wall components lipopolysaccharide (LPS) or peptidoglycan (PGN) for 24 hr. Messenger RNA expression levels of tumour necrosis factor-α (TNF-α) (a, e), interleukin-6 (IL-6) (b, f), β-defensin 4 (c, g) and cathelicidin-related antimicrobial peptide (CRAMP) (d, h) were determined by real-time RT-PCR. Data were assessed from six independent experiments in duplicate. An asterisk indicates a significant difference between treated WT and treated FPR1-KO or FPR2-KO glial cells as determined by analysis of variance followed by Bonferroni test (*P < 0·05; **P < 0·01; ***P < 0·001).
Discussion
In the present study, the consequence of a lack of FPR1 and FPR2 was investigated in a well-established and characterized mouse model of pneumococcal meningitis.23 FPR-deficiency resulted in a higher mortality rate that was associated with increased bacterial burden and increased neutrophil granulocyte infiltration of the CNS as well as in distinct changes of the inflammatory immune reaction. Our results suggest that FPRs are an important component of the innate immune response for the defence of the CNS after invasion with S. pneumoniae and that the lack of this protein results in an unfavourable outcome.
In addition to TLRs and nucleotide-binding oligomerization domain receptors, FPRs belong also to the PRRs based on their capacity to recognize a plethora of bacteria and host-derived agonists.37 Interestingly, the chemotactic G-protein-coupled FPRs interact with a menagerie of structurally diverse pro-inflammatory and anti-inflammatory ligands associated with different diseases, including amyloidosis, Alzheimer's disease, prion disease and HIV.22,38,39 We compared the effect of mFPR1 or mFPR2 deficiency after pneumococcal meningitis. Sequence comparison of both receptors revealed a similarity of 63%.10 Whereas the mFPR1 is defined as a high-affinity receptor for fMLF, mFPR2 is defined as a low-affinity fMLF receptor, based on its activation only by high concentrations of fMLF (μm range).39 The agonist spectrum is similar, but there are affinity and ligand differences. Our own previous investigations revealed a vivid expression of mFPR2 in the mouse brain whereas the mFPR1 is endogenously only weakly detectable.22 However, the results of our mortality study revealed a significant decrease in survival after pneumococcal meningitis for both mFPR1- and mFPR2-deficient mice but there were no difference between mFPR1 and mFPR2. These results are supported by another study using infection models: Gao et al.16 reported an increased susceptibility to Listeria monocytogenes for mFPR1-deficient mice as measured by increased mortality and bacterial load in spleen and liver compared with wild-type littermates. In comparison to other PRR, TLR2 deficiency – as a receptor for bacterial cell components of Gram-positive bacteria – also resulted in incre-ased mortality after pneumococcal meningitis.40 Similarly, the bacterial burden was significantly higher in TLR2-deficient mice compared with WT. This is in accordance with our current findings of a higher bacterial load and increased neutrophil infiltration of the meninges in infected FPR1- and FPR2-deficient mice. Interestingly, the formyl peptide receptors are important for the chemotatic movement and mobilization of neutrophils.15 The cited authors detected also an increased bacterial load but a lower neutrophil migration after a Listeria infection in the liver of FPR-deficient mice. On the other hand, Gauthier et al. analysed the effect of FPR1 absence in neutrophil infiltration into the lungs of mice in a model of pneumococcal pneumonia. There is no difference between WT and KO mice and the genetic variation in the host greatly influences neutrophil recruitment.41 Maybe the difference can be explained by the immunoprivileged situation in the CNS. The low incidence of immune cells in the cerebrospinal fluid in a healthy state and the disruption of the blood–brain barrier in the course of meningitis lead to a strong immigration of neutrophils. It is also possible that the FPR deficiency resulted in a compensatory reaction by other chemotatic receptors or PRR and so led to an excessive increase in neutrophil immigration. At least the expression of TLR2 is strongly increased in infected FPR-deficient mice compared with WT mice. Interestingly, our results show a significant difference between infected FPR1-KO and FPR2-KO mice. Previous works highlighted a positive influence of TLR2 in FPR2 function in context with amyloid β 1-42-mediated microglial cell activation and internalization.42,43 Furthermore, our results showed an up-regulation of the chemokines CCL2 and CCL3, especially in FPR1-deficient mice, with a significant difference compared with infected FPR2-KO. Previous works revealed interplay between FPR and chemokine signalling. Whereas activation of FPRL1 by serum amyloid A induced CCL2 production in human endothelial cells, CCL2 acts synergistically with FPR ligand fMLF.44,45 Chen et al.46 demonstrated that interaction of the chemokine receptor CCR2, the receptor for CCL2, and FPR2 with their endogenous ligands sequentially mediated the trafficking of dendritic cells within the inflamed lung. Our in vitro results with glial cell stimulation showed that the lack of FPR1 resulted in a comparable to stronger induction of chemokine expression compared with WT glia cells, whereas FPR2 deficiency led to attenuated over-expression. This supported the earlier findings. Whereas FPR2 seems to be more complementary or synergistic with the chemokines, FPR1 influenced more dampening on the chemokine expression. Therefore, the lack of FPR1 resulted in a stronger increase of CCL2 and CCL3 in infected mice. It could be that the two receptors differ in their affinity or their ligand spectra as well as in terms of their function. However, the consequence of the interaction for the activation of the immune system in the course of meningitis is not clear and should be investigated by further studies, e.g. by investigations into the course of bacterial meningitis in chemokine-deficient mice.
The role of the FPRs for the inflammatory response is still a matter of debate. Whereas a study showed a decrease in the inflammatory response in mFPR2-deficient mice,19 another study showed an increase from which an anti-inflammatory role of the FPRs was concluded.18 The FPRs show complex functional properties, partly due to their high promiscuity, but also because their activation can stimulate several signal transduction pathways, depending on the ligand, its concentration and the cell type involved. For FPR2 in particular, the different function of the protein-binding as well as lipid-binding domain was shown. While ligands binding to protein-binding domains up-regulate pro-inflammatory transcription factors such as nuclear factor-κB (NF-κB) and activator protein 1 (AP-1), an interaction with the lipid-binding domain inhibits NF-κB and AP-1.37 As a consequence, the activation of the FPRs resulted in a rather pro-inflammatory or anti-inflammatory response. In the context of our results, the lack of the FPRs leads to an increased pro-inflammatory and attenuated anti-inflammatory as well as antimicrobial response. Our investigations revealed a stronger glial cell marker expression in FPR1-deficient mice and an increase of activated microglial cells in FPR1- or FPR2-deficient mice after pneumococcal meningitis. Several studies demonstrated the importance of glial cells in the development and regulation of inflammatory reactions in response to infection of the CNS as well as in neurodegenerative diseases.47,48 Astrocytes belong to the well-characterized innate immune neuroglia, and in addition to many other roles, their main function is the synthesis and regulation of inflammatory cytokines such as IL-1β, IL-6 and different chemokines as well as the synthesis of anti-inflammatory cytokines.49 Microglial cells, as macrophages of the brain, are activated after the invasion of pathogens into the brain and release a broad spectrum of cytokines and chemokines to activate other immune cells.29 However, our results for analysis of pro-inflammatory and anti-inflammatory markers revealed differences between astrocytes and microglial cells. Whereas the bacterial stimulation of WT or FPR-deficient astrocytes showed only minor differences for pro-inflammatory agents such as TNF-α or IL-6, the pro-inflammatory reaction in FPR1-deficient microglia was increased. A possible explanation could be the higher FPR1 expression in microglial cells compared with astrocytes.13 The difference between FPR1-KO and FPR2-KO could be explained by region-specific expression differences. Moreover, the differences could be due to a different cell density (ratio of number of cells to the total volume of the brain region). Therefore, the lack of FPR1 could result in stronger inflammation in the hippocampus of infected FPR1-deficient mice. Furthermore, the Itgam mRNA expression as a marker for activated microglial cells was higher in the hippocampus of infected FPR1-deficient mice. Hence, the FPR1 could be playing an important anti-inflammatory role in the hippocampus. However, the results provide no clear unified pattern. Interestingly, the analysis for cell viability showed that the FPR1 deficiency resulted in an attenuated viability after the bacterial treatment. It should be noted that possibly pathogen-associated-molecular patterns of Gram-positive bacteria or danger-associated-molecular patterns are responsible for the reaction. Along with the change in pro-inflammatory cytokine expression, the lack of FPRs resulted in a strong decrease of anti-inflammatory response including IL-1RA and HO-1 in vivo and in vitro. In this context, it seems to be that the difference for IL-1RA expression is mediated rather by FPR2-deficiency, whereas the decrease of the anti-inflammatory enzyme HO-1 expression is FPR1- or FPR2-dependent. Altogether, this leads to an increased inflammatory reaction and so to an unfavourable outcome. This included also the reduction of antimicrobial activity in FPR-deficient mice. Our results show a lower expression of the antimicrobial peptides CRAMP and β-defensin 4 in FPR1- or FPR2-deficient mice compared with WT after bacterial stimulation. Again, glial cells appeared to be involved in different ways. Whereas the deficiency of FPR in astrocytes apparently mediated the decrease of CRAMP expression, the lack of FPR1 as well as FPR2 led to a loss of CRAMP and β-defensin 4 expression. This confirmed our recent work for an involvement of FPRL1 in CRAMP expression in glial cells.17 In addition, several studies point to the importance of antimicrobial peptides for innate immunity. In general, defensins and cathelicidins like CRAMP have two major functions in host defence: direct inhibition of pathogens and modulation of other innate and adaptive immune responses.30 Our previous work with the CRAMP-deficient mice confirmed the importance of the antimicrobial peptide in pneumococcal meningitis. The mice suffered from increased inflammation and mortality response after infection.26
In conclusion, our results reveal the importance of the formyl peptide receptors for the host defense in a mouse model of pneumococcal meningitis. Our results provide interesting insights into the function of the innate immune system during the course of bacterial meningitis. The lack of FPRs leads to increased mortality and altered inflammation. In particular, the anti-inflammatory and anti-microbial responses appear to be severely affected. Therefore, the results suggest that formyl peptide receptors play an anti-inflammatory and pro-bactericide role in the immune response against pathogens in CNS bacterial infections.
Acknowledgments
SO and SP designed and performed experiments. EK and SJ helped to accomplish experiments. OS and JMW provided the mice and helped to draft the manuscript. TP co-conceived of the study and helped to draft the manuscript. SCJ helped to accomplish experiments and revised the manuscript. L-OB designed as well as performed experiments, and drafted the manuscript. We thank Susanne Echterhagen, Lian Shen, Michaela Nicolau and Sabine Hamm for excellent technical assistance. This study was supported by the Else Kröner-Fresenius-Stiftung (L-OB) and the START-Programme of the RWTH Aachen University (SCT and L-OB). JMW was supported by intramural research support programme of National Cancer Institute, NIH, USA.
Glossary
- CRAMP
cathelicidin-related antimicrobial peptide
- Defb4
β-defensin 4
- FPR
formyl peptide receptor
- GFAP
Glial fibrillary acidic protein
- NM
Neisseria meningitides
- Iba-1
ionized calcium binding adaptor molecule 1
- Itgam
Integrin alpha M
- KO
knock out
- SP
Streptococcus pneumoniae
- TLR
Toll-like receptor
- WT
wildtype
Disclosures
The authors declare that they have no competing interests.
References
- 1.Traore Y, Tameklo TA, Njanpop-Lafourcade BM, et al. Incidence, seasonality, age distribution, and mortality of pneumococcal meningitis in Burkina Faso and Togo. Clin Infect Dis. 2009;48(Suppl. 2):S181–9. doi: 10.1086/596498. [DOI] [PubMed] [Google Scholar]
- 2.Gessner BD, Mueller JE, Yaro S. African meningitis belt pneumococcal disease epidemiology indicates a need for an effective serotype 1 containing vaccine, including for older children and adults. BMC Infect Dis. 2010;10:22. doi: 10.1186/1471-2334-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuchat A, Robinson K, Wenger JD, Harrison LH, Farley M, Reingold AL, Lefkowitz L, Perkins BA. Bacterial meningitis in the United States in 1995. Active Surveillance Team. N Engl J Med. 1997;337:970–6. doi: 10.1056/NEJM199710023371404. [DOI] [PubMed] [Google Scholar]
- 4.Koedel U, Klein M, Pfister HW. New understandings on the pathophysiology of bacterial meningitis. Curr Opin Infect Dis. 2010;23:217–23. doi: 10.1097/QCO.0b013e328337f49e. [DOI] [PubMed] [Google Scholar]
- 5.Mariani MM, Kielian T. Microglia in infectious diseases of the central nervous system. J Neuroimmune Pharmacol. 2009;4:448–61. doi: 10.1007/s11481-009-9170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konat GW, Kielian T, Marriott I. The role of Toll-like receptors in CNS response to microbial challenge. J Neurochem. 2006;99:1–12. doi: 10.1111/j.1471-4159.2006.04076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 8.Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med. 1982;155:264–75. doi: 10.1084/jem.155.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao JL, Chen H, Filie JD, Kozak CA, Murphy PM. Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics. 1998;51:270–6. doi: 10.1006/geno.1998.5376. [DOI] [PubMed] [Google Scholar]
- 11.He HQ, Liao D, Wang ZG, Wang ZL, Zhou HC, Wang MW, Ye RD. Functional characterization of three mouse formyl peptide receptors. Mol Pharmacol. 2013;83:389–98. doi: 10.1124/mol.112.081315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–19. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Brandenburg LO, Konrad M, Wruck CJ, Koch T, Lucius R, Pufe T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid β1-42-induced signal transduction in glial cells. J Neurochem. 2010;113:749–60. doi: 10.1111/j.1471-4159.2010.06637.x. [DOI] [PubMed] [Google Scholar]
- 14.Cattaneo F, Guerra G, Ammendola R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem Res. 2010;35:2018–26. doi: 10.1007/s11064-010-0301-5. [DOI] [PubMed] [Google Scholar]
- 15.Liu M, Chen K, Yoshimura T, et al. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep. 2012;2:786. doi: 10.1038/srep00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999;189:657–62. doi: 10.1084/jem.189.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braun BJ, Slowik A, Leib SL, et al. The formyl peptide receptor like-1 and scavenger receptor MARCO are involved in glial cell activation in bacterial meningitis. J Neuroinflammation. 2011;8:11. doi: 10.1186/1742-2094-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dufton N, Hannon R, Brancaleone V, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–9. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen K, Le Y, Liu Y, et al. A critical role for the G protein-coupled receptor mFPR2 in airway inflammation and immune responses. J Immunol. 2010;184:3331–5. doi: 10.4049/jimmunol.0903022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandenburg LO, Varoga D, Nicolaeva N, et al. Expression and regulation of antimicrobial peptide rCRAMP after bacterial infection in primary rat meningeal cells. J Neuroimmunol. 2009;217:55–64. doi: 10.1016/j.jneuroim.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slowik A, Merres J, Elfgen A, Jansen S, Mohr F, Wruck CJ, Pufe T, Brandenburg LO. Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE) – and amyloid β1-42-induced signal transduction in glial cells. Mol Neurodegener. 2012;7:55. doi: 10.1186/1750-1326-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerber J, Raivich G, Wellmer A, Noeske C, Kunst T, Werner A, Bruck W, Nau R. A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathol. 2001;101:499–508. doi: 10.1007/s004010000326. [DOI] [PubMed] [Google Scholar]
- 24.Liebetanz D, Gerber J, Schiffner C, Schutze S, Klinker F, Jarry H, Nau R, Tauber SC. Pre-infection physical exercise decreases mortality and stimulates neurogenesis in bacterial meningitis. J Neuroinflamm. 2012;9:168. doi: 10.1186/1742-2094-9-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tauber SC, Stadelmann C, Spreer A, Bruck W, Nau R, Gerber J. Increased expression of BDNF and proliferation of dentate granule cells after bacterial meningitis. J Neuropathol Exp Neurol. 2005;64:806–15. doi: 10.1097/01.jnen.0000178853.21799.88. [DOI] [PubMed] [Google Scholar]
- 26.Merres J, Hoss J, Albrecht LJ, et al. Role of the cathelicidin-related antimicrobial peptide in inflammation and mortality in a mouse model of bacterial meningitis. J Innate Immun. 2014;6:205–18. doi: 10.1159/000353645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandenburg LO, Jansen S, Albrecht LJ, Merres J, Gerber J, Pufe T, Tauber SC. CpG oligodeoxynucleotides induce the expression of the antimicrobial peptide cathelicidin in glial cells. J Neuroimmunol. 2013;255:18–31. doi: 10.1016/j.jneuroim.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J Mol Neurosci. 2012;48:66–76. doi: 10.1007/s12031-012-9773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 30.Brandenburg L-O, Merres J, Albrecht L-J, Varoga D, Pufe T. Antimicrobial peptides: multifunctional drugs for different applications. Polymers. 2012;4:539–60. [Google Scholar]
- 31.Gallo RL, Kim KJ, Bernfield M, Kozak CA, Zanetti M, Merluzzi L, Gennaro R. Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J Biol Chem. 1997;272:13088–93. doi: 10.1074/jbc.272.20.13088. [DOI] [PubMed] [Google Scholar]
- 32.Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–87. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrier S, Darakhshan F, Hajduch E. IL-1 receptor antagonist in metabolic diseases: Dr Jekyll or Mr Hyde? FEBS Lett. 2006;580:6289–94. doi: 10.1016/j.febslet.2006.10.061. [DOI] [PubMed] [Google Scholar]
- 34.Cuadrado A, Rojo AI. Heme oxygenase-1 as a therapeutic target in neurodegenerative diseases and brain infections. Curr Pharm Des. 2008;14:429–42. doi: 10.2174/138161208783597407. [DOI] [PubMed] [Google Scholar]
- 35.Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411:1570–9. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Mirabelli-Badenier M, Braunersreuther V, Viviani GL, Dallegri F, Quercioli A, Veneselli E, Mach F, Montecucco F. CC and CXC chemokines are pivotal mediators of cerebral injury in ischaemic stroke. Thromb Haemost. 2011;105:409–20. doi: 10.1160/TH10-10-0662. [DOI] [PubMed] [Google Scholar]
- 37.Cattaneo F, Parisi M, Ammendola R. Distinct signaling cascades elicited by different formyl Peptide receptor 2 (FPR2) agonists. Int J Mol Sci. 2013;14:7193–230. doi: 10.3390/ijms14047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brandenburg LO, Koch T, Sievers J, Lucius R. Internalization of PrP106-126 by the formyl-peptide-receptor-like-1 in glial cells. J Neurochem. 2007;101:718–28. doi: 10.1111/j.1471-4159.2006.04351.x. [DOI] [PubMed] [Google Scholar]
- 39.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–8. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 40.Echchannaoui H, Leib SL, Neumann U, Landmann RM. Adjuvant TACE inhibitor treatment improves the outcome of TLR2–/– mice with experimental pneumococcal meningitis. BMC Infect Dis. 2007;7:25. doi: 10.1186/1471-2334-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gauthier JF, Fortin A, Bergeron Y, Dumas MC, Champagne ME, Bergeron MG. Differential contribution of bacterial N-formyl-methionyl-leucyl- phenylalanine and host-derived CXC chemokines to neutrophil infiltration into pulmonary alveoli during murine pneumococcal pneumonia. Infect Immun. 2007;75:5361–7. doi: 10.1128/IAI.02008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen K, Iribarren P, Hu J, et al. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid β peptide. J Biol Chem. 2006;281:3651–9. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 43.Chen K, Zhang L, Huang J, Gong W, Dunlop NM, Wang JM. Cooperation between NOD2 and Toll-like receptor 2 ligands in the up-regulation of mouse mFPR2, a G-protein-coupled Aβ42 peptide receptor, in microglial cells. J Leukoc Biol. 2008;83:1467–75. doi: 10.1189/jlb.0907607. [DOI] [PubMed] [Google Scholar]
- 44.Lee HY, Kim SD, Shim JW, Yun J, Kim K, Bae YS. Activation of formyl peptide receptor like-1 by serum amyloid A induces CCL2 production in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2009;380:313–7. doi: 10.1016/j.bbrc.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 45.Gouwy M, Struyf S, Verbeke H, Put W, Proost P, Opdenakker G, van Damme J. CC chemokine ligand-2 synergizes with the nonchemokine G protein-coupled receptor ligand fMLP in monocyte chemotaxis, and it cooperates with the TLR ligand LPS via induction of CXCL8. J Leukoc Biol. 2009;86:671–80. doi: 10.1189/jlb.1008638. [DOI] [PubMed] [Google Scholar]
- 46.Chen K, Liu M, Liu Y, et al. Signal relay by CC chemokine receptor 2 (CCR2) and formylpeptide receptor 2 (Fpr2) in the recruitment of monocyte-derived dendritic cells in allergic airway inflammation. J Biol Chem. 2013;288:16262–73. doi: 10.1074/jbc.M113.450635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–71. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finsen B, Owens T. Innate immune responses in central nervous system inflammation. FEBS Lett. 2011;585:3806–12. doi: 10.1016/j.febslet.2011.05.030. [DOI] [PubMed] [Google Scholar]