

Abstract
Increasing evidence suggests that cellular stress may underlie mood disorders such as bipolar disorder and major depression, particularly since lithium and its targets can protect against neuronal cell death. Here we describe NIFAR (NMDA-induced filamentous actin reorganization) as a useful in vitro model for studying acute neurocellular stress and investigating the effects of mood stabilizers. Brief incubation of cultured neurons with NMDA (50μM, 5 min) induces a dramatic reorganization of F-actin within the somatodendritic domain of a majority of neurons. During NIFAR, F-actin is rapidly depleted from dendritic spines and aberrantly aggregates within the dendrite shaft. The widely used mood stabilizer lithium chloride prevented NIFAR in a time- and dose-dependent manner, consistent with its known efficacy in treating bipolar disorder. Inhibitors of the lithium target glycogen synthase kinase 3 (GSK3) and its upstream activator phosphoinositide-3-kinase also prevented NIFAR. The anti-depressant compounds imipramine and fluoxetine also attenuated NIFAR. These findings have potential relevance to neuropsychiatric diseases characterized by excessive glutamate receptor activity and synaptotoxicity. We propose that protection of the dendritic actin cytoskeleton may be a common mechanism shared by various mood stabilizers.
Keywords: glutamate, NMDA, cytoskeleton, neuronal activity, lithium, actin, mood stabilizers, bipolar disorder, cellular stress, neuroprotection
Introduction
Actin filaments are the main cytoskeletal elements that regulate dendritic spine stability [1]. Actin cytoskeletal changes play a fundamental role in diseases of the central nervous system [2–4], and actin-based mechanisms are critical in the neural circuit function needed for learning and memory [5].
Acute stimulation of NMDA receptors induces dendritic spine loss and F-actin reorganization within 5–10 min [6–8]. Such NMDA-induced F-actin reorganization (NIFAR) is characterized by a rapid depolymerization of F-actin in dendritic spines and accumulation of aberrant filaments within the dendritic shaft. Here we used NIFAR as an in vitro model of sub-lethal neuronal damage to test the hypothesis that the mood stabilizer lithium can prevent or reduce sub-lethal injury to excitatory neurons.
Lithium is a commonly-used treatment for mood disorders, particularly bipolar disorder. However, despite being in clinical use for over fifty years, its mechanisms of action as a mood stabilizer still remain unclear [9–13]. Numerous studies have demonstrated that lithium has significant neuroprotective actions against a variety of insults [9–13], including traumatic brain injury [14].
This neuroprotective property of LiCl may underlie its therapeutic efficacy in mood disorders [9–13]. However, although volume reductions in specific brain areas have been described [12], for most neuropsychiatric disorders there is little direct evidence that cell death is responsible for the cognitive and behavioral disturbances that characterize the disease. It seems more likely that any physical disruption to neural circuitry associated with such disorders might occur on a more subtle level, involving dendritic and axonal branches and their synapses. Indeed, several neuropsychiatric disorders exhibit loss or deformation of dendritic spines – the dynamic, actin-rich membrane protrusions of excitatory synapses [1,2,4].
Although lithium has multiple molecular targets, its ability to inhibit glycogen synthase kinase 3 (GSK3) activity has consistently been implicated in its neuroprotective properties [9–13,15]. Our findings described here implicate a role for GSK3, as one of the potential mechanisms of lithium’s ability to protect against early, sub-lethal changes to the actin cytoskeleton induced by NMDA. Moreover, results using specific antidepressants suggest that stabilization of the actin cytoskeleton may be a common mechanism shared by multiple classes of mood stabilizing compounds.
Methods
Ethics Statement
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego specifically approved this study under protocol #S07290. All surgical procedures were terminal, and deep anesthesia with isoflurane was used to prevent any pain or discomfort.
Cell culture
Hippocampal cultures containing a natural mixture of neurons and glia were prepared and maintained according to [16]. Cells were plated at a density of 300 cells/mm2 and maintained in Neurobasal medium (Gibco), supplemented with B27 (Invitrogen) or with Neurocult SM1 neuronal supplement (STEMCELL Technologies) and 0.5 mM L-glutamine (Sigma).
Drugs
All drugs were purchased from Sigma-Aldrich. All experiments used coverslips from the same preparation as controls, which were treated with vehicle alone.
Immunocytochemistry
Cultures were fixed using 3.7% formaldehyde in phosphate-buffered saline (PBS) plus 120 mM sucrose for 20 min at 37°C. Cultures were incubated in 20 mM glycine for 5 min, rinsed and permeabilized with 0.2% Triton X-100 for 5 min at room temperature, and then blocked for 30 min with 2% bovine serum albumin (BSA). Primary rabbit anti-MAP2 antibody 1:2000 [17] was incubated for 1 hr at room temperature. Following rinsing with PBS, cells were incubated in the presence of 2% BSA with AlexaFluor488-conjugated secondary antibody (Invitrogen), and with AlexaFluor568-phalloidin at 1:1000 (Invitrogen, Molecular Probes) for 45 min at 37°C to label F-actin. Coverslips were washed twice with PBS and mounted using Aqua-Mount (Thermo Scientific).
Quantitative Image Analysis
The fraction of neurons characterized by NIFAR was quantified directly on the microscope, by an investigator blind to experimental condition, using a manual cell counter to count all MAP2 positive neurons and all NIFAR positive cells while scanning the entire 12-mm round coverslip, excluding area within ~600 μm of the edge. Sample sizes are indicated in all figure legends.
Statistics
Statistical significance was set at the 95% confidence level (two tailed) and calculated using Prism (Graphpad Software). Values are presented as mean ± S.E.M. Statistical tests for each assay are defined in the corresponding figure legends. Pharmacological experiments were statistically compared to their corresponding vehicle control.
Results
Lithium prevents NMDA-induced F-actin reorganization
In control neurons, phalloidin staining reveals the typical distribution pattern for F-actin within glutamatergic pyramidal neurons, the prevalent neuronal cell type in these cultures. F-actin exhibits a characteristic enrichment in dendritic spines, with greatly reduced concentrations within the soma and dendrites (Fig. 1a, control; Fig. 1b, left). Upon incubation for 5 min with 50μM NMDA this typical phalloidin staining pattern is disrupted in 50–60% of neurons. This NMDA-induced F-actin reorganization (NIFAR) is characterized by extensive loss of F-actin from the majority of spines, and an accumulation of phalloidin-positive filaments within the proximal somatodendritic domain (Fig.1a, NMDA; Fig. 1b, right).
Figure 1. Lithium prevents NMDA-induced F-actin reorganization.
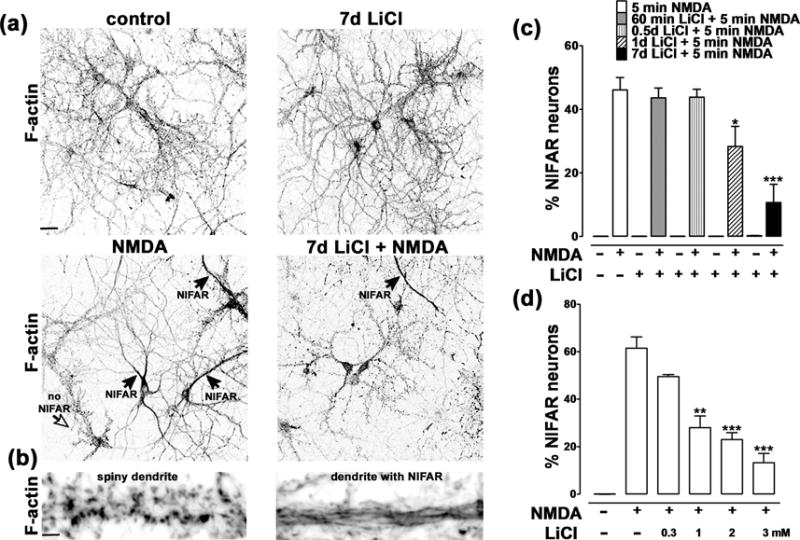
(a) Cultured hippocampal neurons incubated in the absence (top left) or presence (bottom left) of NMDA (5min; 50 μM), with (right) or without (left) LiCl and then fixed and stained for F-actin using Alexa568-phalloidin. The black arrows indicate neurons that have undergone NMDA-induced F-actin reorganization (NIFAR), while the open arrow indicates a no-NIFAR neuron. We observed that NIFAR occurs only in the presence of NMDA and that it is significantly attenuated by preincubation with LiCl. Scale bar: 50 μm. (b) Higher magnification images from dendritic regions of a control (left) and a NIFAR neuron (right). Scale bar: 3 μm.
(c) Time course of effect of LiCl preincubation on prevention of NIFAR. Hippocampal neurons were incubated with 3mM LiCl for the indicated times prior to addition of 50 μM NMDA for 5 min. Data are expressed as the percentage of neurons exhibiting NIFAR and graphed as mean ± SEM (*p<0.05, *** p<0.001 one-way ANOVA, followed by Bonferroni post hoc test vs NMDA +; number of coverslips: 6 per condition; number of neurons counted per coverslip = 400–800). NIFAR was absent in untreated cultures or cultures treated with lithium alone.
(d) Dose-dependence of the effect of LiCl preincubation on prevention of NIFAR. Hippocampal neurons were pre-incubated for 7 days with the indicated concentrations of LiCl prior to addition of 50 μM NMDA for 5 min. Data are expressed as percentage of neurons exhibiting NIFAR, and graphed as mean ± SEM. (*p<0.05, *** p<0.001 one-way ANOVA, followed by Bonferroni post hoc test vs NMDA +; number of coverslips: 6 for each condition; number of neurons counted per coverslip = 400–800). NIFAR was absent in untreated cultures.
We found that a seven day preincubation with 3 mM LiCl substantially reduced NIFAR (Fig. 1a and c);a one day preincubation had a lesser but still statistically significant effect (Fig.1c). Shorter preincubations with LiCl (12 hrs and 1 hr) did not protect against NIFAR. Using a 7 day preincubation, we observed a dose-dependent effect of LiCl in preventing NIFAR, with both 2 mM and 1 mM, but not 0.3 mM, LiCl showing significant protection (Fig.1d). Higher concentrations of LiCl (5–20 mM) induced extensive neuronal cell death, without detectable astrocyte cell death.
GSK3 inhibition prevents NIFAR
Since many of lithium’s actions are known to be mediated via its direct and/or indirect inhibition of GSK3 [9–11,15], we tested whether inhibition of GSK3 shows a similar neuroprotective effect against NIFAR. Consistent with this hypothesis, preincubation for 1 hr or 7 days with the specific GSK3 inhibitor SB216763 (10 μM) significantly protected against NIFAR (Fig.2a). Another GSK3 inhibitor, inhibitor VIII (16 μM), also substantially protected against NIFAR, although slightly less effectively (Table 1). Inhibition of cyclin dependent kinase 5 (cdk5) has been reported to mediate some of lithium’s effects [18]. However, the cdk inhibitor roscovitine (10 μM;1hr to 7 days) did not protect against NIFAR (Table 1).
Figure 2. Inhibition of GSK3 prevents NIFAR.
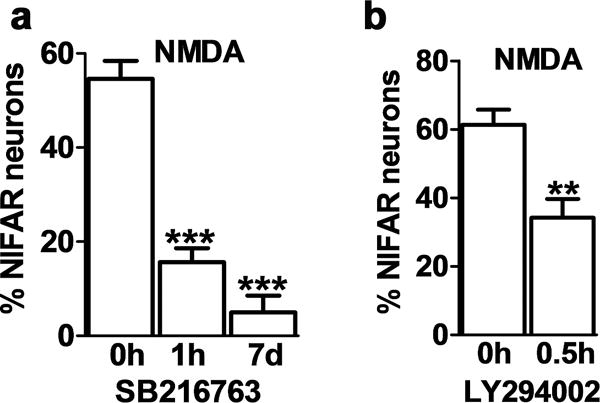
Data are expressed as the percentage of neurons exhibiting NIFAR and graphed as mean ± SEM. (number of coverslips: 6 per condition; number of neurons counted per coverslip = 400–800). (a) Hippocampal neurons were pre-incubated with SB216763 for the indicated times prior to addition of 50 μM NMDA for 5 min. *** p<0.001 one-way ANOVA, followed by Bonferroni post hoc test vs NMDA +. (b) Hippocampal neurons were pre-incubated with LY 294002 for the indicated time prior to addition of 50 μM NMDA for 5 min. **p<0.01 unpaired t-test.
Table 1.
Effects of drug treatments in preventing NIFAR
| Treatment prior NMDA | Function | NIFAR reduction (%)* |
|---|---|---|
| LiCl (3 mM), 1d | mood stabilizer | 39 |
| LiCl (3 mM), 7d | 78 | |
| “ “ + Myo-inositol (5 mM), 7d | 2nd messenger precursor (inhibited by LiCl) | 75 |
| LiCl (2 mM), 7d | 62 | |
| LiCl (1 mM), 7d | 54 | |
| SB216763 (10 μM), 1h | GSK3 inhibitor | 71 |
| SB216763 (10 μM), 7d | 91 | |
| LY 294002 (20 μM), 0.5h | PI3 kinase inhibitor | 44 |
| Fluoxetine (10 μM), 0.5h | antidepressant (SSRI) | 52 |
| Fluoxetine (10 μM), 1h | 35 | |
| Fluoxetine (10 μM), 4h | 33 | |
| Imipramine (20 μM), 0.5h | tricyclic antidepressant | 61 |
| Imipramine (20 μM), 1h | 51 | |
| Imipramine (20 μM), 4h | −11 | |
| Inhibitor VIII (16 μM), 1h | GSK3 inhibitor | 31 |
| Inhibitor VIII (16 μM), 3d | 40 | |
| Drugs with no detectable effect on NIFAR | ||
| Roscovitine (10 μM), 1h | Cdk5 inhibitor | 2 |
| Roscovitine (10 μM), 7d | 1 | |
| Valproate (1 mM), 1h–7d | anticonvulsant | 1–3 |
| Carbamazepine (100 μM), 1h–7d | anticonvulsant | 1–3 |
Hippocampal neurons grown 3 weeks in culture were treated with a variety of drugs prior NMDA, fixed and stained for F-actin. Neurons with NMDA-induced F-actin reorganization (NIFAR) were scored as described in the Materials and methods.
NIFAR reduction (%) = [% NIFAR (NMDA − treatment)/%NIFAR (NMDA)]*100. Values indicate average percent reduction over multiple replicants. SSRI=selective serotonin reuptake inhibitor.
We next asked whether phosphoinositide-3-kinase (PI3 kinase), which acts via Akt kinases as an upstream activator of GSK3 [11,15], was involved in NIFAR. A 30 min preincubation with the specific PI3 kinase inhibitor LY294002 prevented NIFAR (Fig.2b). In addition to its inhibition of GSK3, lithium is known to inhibit myo-inositol monophosphatase, which can lead to myo-inositol depletion [10,11]. However, addition of myo-inositol (5 mM, 7 days) to the incubation medium, together with LiCl, did not reverse the protective effect of lithium (Table 1).
Fluoxetine and imipramine reduce NIFAR
We asked whether, in addition to lithium, other mood stabilizers or antidepressants would have a similar protective effect against NIFAR or whether this was a specific characteristic of lithium. The anticonvulsants valproate and carbamazepine are used to treat bipolar disorder [11]. In our model neither valproate (1mM;1 hr to 7 days) nor carbamazepine (100 μM; 1 hr to 7 days), protected against NIFAR (Table 1). However, two classes of antidepressants showed partial protection against NIFAR. The selective serotonin reuptake inhibitor fluoxetine (10 μM; 0.5–4 hr) partially prevented NIFAR (Fig. 3a). The tricyclic antidepressant imipramine (20 μM) strongly attenuated NIFAR; however it was only effective with 0.5 and 1 hr preincubations, as a 4 hr preincubation was ineffective (Fig. 3b).
Figure 3. Antidepressants fluoxetine and imipramine prevent NIFAR.
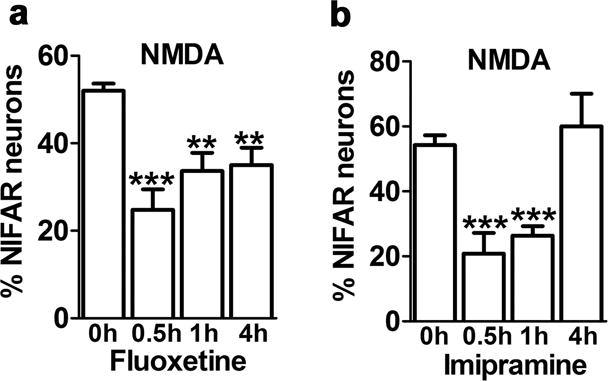
Hippocampal neurons were pre-incubated with drugs for the indicated times prior to addition of 50 μM NMDA for 5 min. Data are expressed as the percentage of neurons exhibiting NIFAR and graphed as mean ± SEM. (**p<0.01, ***p<0.001 one-way ANOVA followed by Bonferroni post hoc test, comparing all groups versus NMDA alone; number of coverslips: 6 per condition in for fluoxetine, 12 per condition for imipramine; number of neurons counted per coverslip = 400–800). (a) Hippocampal neurons were pre-incubated with 10μM fluoxetine for the indicated times prior to addition of 50 μM NMDA for 5 min. (b) Hippocampal neurons were pre-incubated with 20μM imipramine for the indicated times prior to addition of 50 μM NMDA for 5 min.
Discussion
Numerous studies have demonstrated a neuroprotective and anti-apoptotic role of lithium in neuronal cell cultures. However the precise mechanisms by which this protection occurs remain to be elucidated [9–13,15]. Although chronic lithium administration was shown to block excitotoxic cell death induced by glutamate agonists [19], our study is the first to characterize the effect of lithium on early acute changes of the actin cytoskeleton induced by increased levels of glutamate agonist. Such excess glutamate represents a cellular stress condition that potentially underlies circuit disruption in disorders ranging from neurodegeneration, stroke and epilepsy, to mood disorders and schizophrenia [20,21].
Here we describe NMDA-induced F-actin reorganization (NIFAR) as a rapid and extensive reorganization of the dendritic actin cytoskeleton in response to hyperactivation of NMDA receptors. NIFAR represents an early stage in the neuronal cellular stress response. It is thus a quantifiable read-out of glutamate-induced injury to dendrites and spines. The mechanisms underlying NIFAR are currently under investigation; however, they may involve oxidative changes, as described in other forms of cellular stress [22].
NIFAR consists of the rapid disassembly of F-actin within dendritic spines, and simultaneous accumulation of F-actin in the dendritic shaft [7]. A similar phenomenon was described for NMDA-induced relocalization of the actin binding proteins IP3 kinase [8] and drebrin [23]. Neurons that undergo NIFAR also exhibit dendritic spine shrinkage and loss [7], a potential mediator of neural circuit disruption in mood disorders. Importantly, NIFAR occurs not only in dissociated cultured neurons, but also in vivo in a rodent model of traumatic brain injury (Calabrese et al., unpublished), a clinical condition associated with prolonged elevations of extracellular glutamate and excess NMDA receptor activation [24]. Further studies are needed to examine whether a NIFAR-like phenomenon is associated with more subtle levels of cellular stress and impaired plasticity that may occur in mood disorders.
Lithium is known to affect a multitude of biochemical and cell signaling pathways, and it is likely that its therapeutic efficacy may involve a spectrum of its many targets, including those that engage the cytoskeleton and neuroprotection [25]. Our results indicate that lithium may protect neurons against NIFAR via inhibition of GSK3 activity, although additional studies are needed to confirm this hypothesis. Here we show that GSK3 inhibitors mimic the protective effect of lithium in preventing NIFAR. GSK3 is a ubiquitous Ser/Thr protein kinase with pro-apoptotic properties that phosphorylates a variety of substrates, including cytoskeletal substrates such as the Alzheimer’s disease related microtubule associated protein tau [26], and specific actin regulatory molecules [27].
Lithium also is known to regulate specific neuromodulators, including serotonin, [25] and it is therefore possible that the protective effects of lithium against NIFAR are mediated via one or more of these systems. Indeed, the protective effect of fluoxetine and imipramine that we observed is consistent with a role for the serotonergic system in NIFAR. Lithium reportedly inhibits the presynaptic 5-HT1B autoreceptor, resulting in increased serotonin release into the synaptic cleft [28]. Direct inhibition of 5-HT reuptake by either fluoxetine or imipramine would have a similar effect on synaptic 5-HT levels. Moreover, Jope and colleagues have shown that fluoxetine and imipramine inhibit GSK3 activity [29], suggesting that GSK3 inhibition may represent a common pathway for many of the effects of lithium. Therefore, it will be of interest to further explore a potential connection between NIFAR and serotonin.
Remarkably, several compounds used in this study were rapidly effective in preventing NIFAR. The GSK3 inhibitor SB216763, the PI3 kinase inhibitor LY 294002 and the anti-depressant drugs fluoxetine and imipramine all effectively prevented NIFAR with 0.5–1 hr preincubation, in contrast to the several days of preincubation that were required for LiCl. This difference in time-course might indicate that lithium’s protective action involves additional changes in gene expression or other factors that accumulate over time. For example, lithium is reported to elevate expression of the neurotrophic factor BDNF, which has been implicated in lithium-mediated neuroprotection [9–14]. Other studies similarly reported that multiple days of lithium exposure are needed to reveal its full neuroprotective efficacy in vitro [11], and therapeutic doses of lithium in bipolar patients typically require multiple days of treatment before behavioral benefits become stabilized [11,12].
Our data appear to exclude a role for either cdk5 or myo-inositol pathways as mediating the protective effect of LiCl against NIFAR. The clinically effective mood stabilizers carbamazepine and valproate have distinct molecular targets from those of LiCl [11], and in our studies these drugs appear not to mediate protection against NIFAR at the times and dosage tested.
Conclusions
We observed that lithium, fluoxetine, and imipramine protected cultured neurons from rapid NMDA-induced aberrant changes in the dendritic actin cytoskeleton, including actin loss from dendritic spines. These findings suggest that the therapeutic potential of certain mood stabilizers may act, in part, via actin-based mechanisms that regulate postsynaptic structural stability and/or neural plasticity. GSK3 inhibitors mimicked the protective effect of lithium. Further studies will be needed to establish whether lithium and other mood stabilizers act via this pathway to stabilize neuronal structure as part of their therapeutic actions.
Acknowledgments
We thank Dr. Johannes Mosbacher for advice and encouragement in these studies. We thank Julia Braga for assisting with blinded morphological analyses.
Financial support: This work was supported in part by Novartis AG as a Specifically Funded Project at The Scripps Research Institute (SFP 1563) and by NIH grant MH087823.
References
- 1.Penzes P, Rafalovich I. Regulation of the actin cytoskeleton in dendritic spines. Adv Exp Med Biol. 2012;970:81–95. doi: 10.1007/978-3-7091-0932-8_4. 3576144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanpied TA, Ehlers MD. Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry. 2004;55:1121–1127. doi: 10.1016/j.biopsych.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- 4.Kulkarni VA, Firestein BL. The dendritic tree and brain disorders. Mol Cell Neurosci. 2012;50:10–20. doi: 10.1016/j.mcn.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Lamprecht R. The actin cytoskeleton in memory formation. Prog Neurobiol. 2014;117C:1–19. doi: 10.1016/j.pneurobio.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Graber S, Maiti S, Halpain S. Cathepsin B-like proteolysis and MARCKS degradation in sub-lethal NMDA-induced collapse of dendritic spines. Neuropharmacology. 2004;47:706–713. doi: 10.1016/j.neuropharm.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Halpain S, Hipolito A, Saffer L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci. 1998;18:9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schell MJ, Irvine RF. Calcium-triggered exit of F-actin and IP(3) 3-kinase A from dendritic spines is rapid and reversible. Eur J Neurosci. 2006;24:2491–2503. doi: 10.1111/j.1460-9568.2006.05125.x. [DOI] [PubMed] [Google Scholar]
- 9.Bachmann RF, Schloesser RJ, Gould TD, Manji HK. Mood stabilizers target cellular plasticity and resilience cascades: implications forr the development of novel therapeutics. Mol Neurobiol. 2005;32:173–202. doi: 10.1385/MN:32:2:173. [DOI] [PubMed] [Google Scholar]
- 10.Can A, Schulze TG, Gould TD. Molecular actions and clinical pharmacogenetics of lithium therapy. Pharmacol Biochem Behav. 2014 doi: 10.1016/j.pbb.2014.1002.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65:105–142. doi: 10.1124/pr.111.005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malhi GS, Tanious M, Das P, Coulston CM, Berk M. Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs. 2013;27:135–153. doi: 10.1007/s40263-013-0039-0. [DOI] [PubMed] [Google Scholar]
- 13.Dodd S, Maes M, Anderson G, Dean OM, Moylan S, Berk M. Putative neuroprotective agents in neuropsychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:135–145. doi: 10.1016/j.pnpbp.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Leeds PR, Yu F, Wang Z, Chiu CT, Zhang Y, Leng Y, et al. A new avenue for lithium: intervention in traumatic brain injury. ACS Chem Neurosci. 2014;5:422–433. doi: 10.1021/cn500040g. 4063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology. 2010;35:2143–2154. doi: 10.1038/npp.2010.105. 3055312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calabrese B, Saffin JM, Halpain S. Activity-dependent dendritic spine shrinkage and growth involve downregulation of cofilin via distinct mechanisms. PloS one. 2014;9:e94787. doi: 10.1371/journal.pone.0094787. PMCID3989342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halpain S, Greengard P. Activation of NMDA receptors induces rapid dephosphorylation of the cytoskeletal protein MAP2. Neuron. 1990;5:237–246. doi: 10.1016/0896-6273(90)90161-8. [DOI] [PubMed] [Google Scholar]
- 18.Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, et al. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron. 2003;38:33–46. doi: 10.1016/s0896-6273(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 19.Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci U S A. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. 19446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 21.Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci. 2010;47:4–16. [PubMed] [Google Scholar]
- 22.Rajendran P, Nandakumar N, Rengarajan T, Palaniswami R, Gnanadhas EN, Lakshminarasaiah U, et al. Antioxidants and human diseases. Clin Chim Acta. 2014 doi: 10.1016/j.cca.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 23.Sekino Y, Tanaka S, Hanamura K, Yamazaki H, Sasagawa Y, Xue Y, et al. Activation of N-methyl-D-aspartate recceptor induces a shift of drebrin distribution: disappearance from dendritic spines and appearance in dendritic shafts. Mol Cell Neurosci. 2006;31:493–504. doi: 10.1016/j.mcn.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and tramautic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 26.Llorens-Maritin M, Jurado J, Hernandez F, Avila J. GSK-3b, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46–57. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun T, Rodriquez M, Kim L. Glycogen synthase kinase 3 in the world of cell migration. Dev Growth Differ. 2009;51:735–742. doi: 10.1111/j.1440-169X.2009.01141.x. [DOI] [PubMed] [Google Scholar]
- 28.Massot O, Rousselle JC, Fillion MP, Januel D, Plantefol M, Fillion G. 5-HT1B receptors: a novel target for lithium. Possible involvement in mood disorders. Neuropsychopharmacology. 1999;21:530–541. doi: 10.1016/S0893-133X(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. 1986663. [DOI] [PMC free article] [PubMed] [Google Scholar]