

Allostery is the coupling of conformational changes between two widely separated sites. It is one of the most common and powerful means to regulate protein function and was referred as “the second secret of life”.[1, 2] Modulation of the allosteric sites of protein targets provides opportunities for identifying unique molecules with therapeutic advantages over classic active site inhibitors, such as improved subtype selectivity, reduced drug resistance and the ability to selectively tune (activate or inhibit) the response of target protein.[3] With increasing emphasis on cellular functional screens, more allosteric ligands are being discovered as potential drugs. However, discovery and characterization of allosteric sites is still very challenging due to the intrinsic complexity of protein allostery and the experimental difficulties in detecting and verifying allosteric effects and binding sites. Here we present an easily applicable STD-NMR/Docking/CORCEMA-ST method for efficient identification and validation of novel allosteric sites.
Protein flexibility and conformational dynamics are the key elements of allosteric regulations. Molecular dynamics (MD) simulation has proven to be a powerful approach for studying protein dynamics as well as the related intramolecular interactions to understand the underlying regulation mechanisms of known allosteric ligands[4–7] as well as to predict novel allosteric sites.[7–10] However, currently there is no method that can efficiently validate the computationally predicted results. This has significantly dampened the enthusiasm of devoting computational effort to explore unknown allosteric sites and thus far very few novel allosteric sites have been discovered through theoretical studies. Methods that can verify novel allosteric binding sites are therefore highly desirable for understanding protein allosteric regulation and for advancing structure-based drug design, discovery and development.
The Saturation Transfer Difference (STD) NMR technique was originally developed by Mayer and Meyer[11] as a binding assay for screening small compound libraries and for the qualitative analysis of group epitopes.[12–14] The application of STD-NMR to drug discovery has been significantly enhanced following the development of the CORCEMA-ST (COmplete Relaxation and Conformational Exchange Matrix Analysis of Saturation Transfer) theory[15–17] which allows quantitative analysis of STD-NMR data. CORCEMA-ST program calculates the predicted STD-NMR intensities for any proposed molecular model of a ligand-receptor complex based on the knowledge of saturated protein protons and using parameters such as the correlation times, exchange rates, and spectrometer frequency. The STD intensities were calculated as percentage fractional intensity changes ([(I0(k)−It(k))*100]/I0(k), where k is a particular proton in the complex, and I0(k) is its thermal equilibrium intensity, and It(k) is its intensity when the protein is saturated. The experimental and calculated STD values are compared using an NOE R-factor defined as:[18]
where, Sexp,k and Scal,k refer to experimental and calculated STD values, respectively, for proton k. The STD-NMR/CORCEMA-ST method has been successfully applied with molecular modelling studies to predict protein-ligand interaction, differentiate binding modes and refine ligand-bound conformation.[15–17,19–21] Compared to the traditionally utilized X-ray and NMR methods for binding site verification in ligand-protein complexes, which can be very time consuming and limited by experimental difficulties, such as crystallization difficulties, protein size, sample preparation, spectra assignments of labelled proteins etc., STD-NMR requires only a small amount of unlabeled target protein and 1D-NMR data acquisition, and can be easily applied in a rather wide range of binding conditions with Kd values ranging from 10−3 M to 10−7 M.[15] It is therefore a very attractive approach for obtaining structural information of ligand-protein complexes. Nevertheless, each of the reported STD-NMR/CORCEMA-ST applications so far has been limited to the study of ligand binding at a specific known binding site; the feasibility of using this method to differentiate the “real” ligand binding site from multiple putative sites, which is of great interest for the discovery of novel allosteric binding sites, has not been explored. In the present study, we investigated the combined STD-NMR/Docking/CORCEMA-ST method for its capability of identifying the proper binding site of an allosteric ligand of the protein Eg5.
Eg5 is a plus-end directed member of the kinesin-5 subfamily. Inhibition of Eg5 function blocks centrosome migration and leads to cell cycle arrest and eventually to apoptotic cell death.[22, 23] Discovery of small molecule inhibitors of Eg5 has attracted significant attention in the past decade and a number of ligand-bound Eg5 co-crystal structures have been solved in the last several years.[24–28] Interestingly, all these ligands in crystal structures bind at the same allosteric site where monastrol, the first Eg5 inhibitor identified a decade ago,[29] binds. Nevertheless, both experimental and modelling studies have demonstrated the existence of other allosteric sites on the Eg5 kinesin domain and allosteric Eg5 inhibitors that bind to non-monastrol sites have also been reported.[30–32] The presence of multiple allosteric sites in the Eg5 system thus provides an ideal template for the present study. We have previously identified several novel allosteric sites on Eg5 based on molecular dynamics simulations.[10] Two of the identified allosteric sites, S1 and S2, along with the monastrol- and ADP-binding sites, were selected in this study to evaluate the STD-NMR/Docking/CORCEMA-ST method (Figure 1).
Figure 1.

Graphic presentation of the binding sites of Eg5 motor based on the crystal structure of monastrol-Eg5 complex (PDB ID: 1Q0B). Eg5 motor was shown in ribbon and colored by secondary structures (α-helix: red; β-sheets: cyan; loops: grey). Monastrol and ADP molecules were represented as solid sticks. The two putative allosteric binding sites, S1 and S2, were illustrated as meshed surfaces.
Figure 2 shows the 1D 1H STD-NMR spectrum of the monastrol-Eg5 mixture sample. All the protons of monastrol exhibited strong STD signals, suggesting they are in close contacts with protein protons. This is consistent with the crystal structure of monastrol-Eg5 complex where monastrol is fully buried within a pocket surrounded by helix α3, helix α2 and loop 5 (Figure 1). Using the monastrol-Eg5 crystal structure as a starting model, we were able to obtain a low NOE R-factor value of 0.12 by optimizing the CORCEMA-ST parameters (see supplementary information for experimental details), which indicated an overall excellent agreement between the predicted intensities for the crystal structure of the complex and experimental STD intensities (Figure 3a). According to the Eg5-monastrol crystal structure, the methyl and ethyl groups of monastrol are located at the solvent exposed side of the binding pocket and are relatively farther from the receptor residues, which well explains the observed relatively weaker STDs of HA, HB and HC. While the relatively larger differences between the calculated and measured STDs were observed for HA, HB and HC, they can be explained by the fact that the crystal structure only represents one snapshot of a conformational ensemble. It is likely that these three protons spend a part of their lifetime closer to the protein than seen in the crystal structure.
Figure 2.
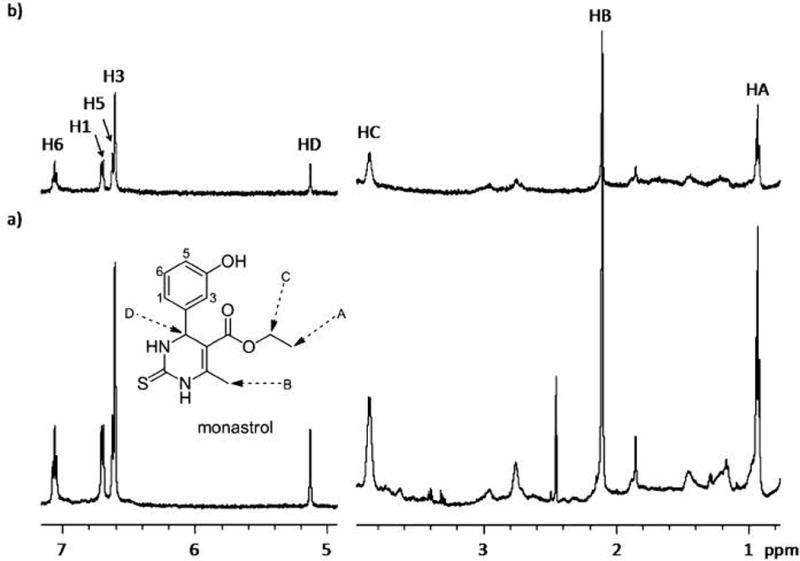
a) The reference 1D 1H NMR spectra of monastrol in the presence of Eg5, at 600 MHz and 298 K. b) The corresponding STD-NMR spectrum [I0−It] (×1) obtained by saturating at a δ value 1.67 ppm. The structure with proton numbering of monastrol is given for reference, and the corresponding signals are assigned.
Figure 3.

Comparison between experimental and CORCEMA-ST calculated STD values (by saturating at 1.67 ppm) at different binding sites. a) Monastrol binding site: the Eg5-monastrol crystal structure as well as the selected docked model-a and model-b at the monastrol-binding site were used for CORCEMA-calculations. b) S1, S2 and ATP-binding site: for each site, two best scored models with different binding mode (-a and -b) were selected for the CORCEMA-ST calculations and compared with the STD-NMR results.
While the above results demonstrated that reasonably low R-factor can be obtained based on known ligand-receptor conformation, the more intriguing question we investigated in this study is whether the STD-NMR/CORCEMA-ST approach can be used to identify the “real” binding mode when it is unknown. This further diverges into two equally important issues: 1) whether it can identify the real binding mode from among multiple binding modes at the same binding site; and 2) whether it can identify the real binding mode from multiple binding sites, which is of special interest to the discovery of novel allosteric site(s). The first issue was already elegantly addressed by Mario Pinto and coworkers[19–21] who demonstrated that a combined STD-NMR/Docking/CORCEMA-ST approach can identify the proper binding mode from among multiple binding modes predicted by docking programs for a given binding pocket. The second issue dealing with multiple binding pockets is the main focus of our study.
We first docked the monastrol molecule into its crystallographic binding site at the Eg5 motor using Glide with an induced-fit docking (IFD) protocol. Multiple docked results were generated by IFD and one of the docked results (model-a) well reproduced the monastrol-Eg5 crystal structure with less than 0.45 Å RMSD (Figure 4a). However, this best fitted model (model-a) was only the second best scored model among the multiple docked results, the best scored model (model-b) actually docked in a very different pose (Figure 4b). This result is not totally unexpected, because IFD allows the receptor residues near the docked ligand to be flexible to mimic ligand-induced conformational changes and thus increases the chance of identifying “correct” ligand binding mode. As a consequence, this IFD protocol, while allowing for protein flexibility, has also the potential to generate some “incorrectly” docked poses. We also tried using Glide docking without the IFD protocol, which indeed ranked the “correctly” docked pose as the best result. Nevertheless, many studies have demonstrated that, in the current state-of-art, docking score alone is not a reliable criterion for identifying the correct binding mode. For the purpose of using STD-NMR/Docking/CORCEMA-ST analysis to identify novel binding site, it is essential to have the “correctly” docked model generated and IFD is a more practical and suitable approach for that. We thus performed CORCEMA-ST calculation using the two IFD generated models and compared the results with the experimental STDs. Similar to using the crystal structure, the correctly docked model-a had a low R-factor of 0.12. In contrast, the calculated STD values using the model-b were very different from the experimental results: since the monastrol’s benzene group in model-b flipped toward the solvent exposure side of the binding pocket and became less buried, the calculated STDs of aromatic protons were significantly decreased compared to the calculated results from the crystal model; instead, the calculated STDs of the methyl and ethyl groups, which became buried inside of the protein pocket, showed relatively high values. The trend in the overall STD variations among the protons of model-b is also different from the experimental STDs (Figure 3a), which led to a significantly higher NOE R-factor value (0.35). These results demonstrate that STD-NMR/CORCEMA-ST analysis is able to identify the correct binding mode of monastrol-Eg5 from the docking results at the same site.
Figure 4.
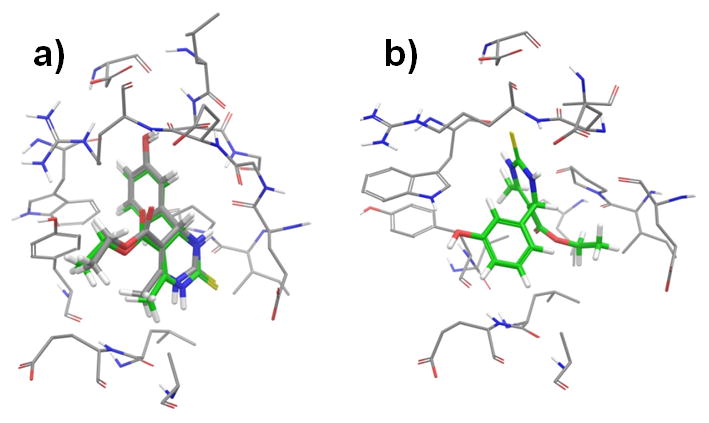
Comparison of the IFD generated docked models at the monastrol-binding site. a) Docked model-a (green-colored carbon) well reproduced the binding mode of the crystal structure (grey-colored carbon). b) Docked model-b bound very differently from model-a. The monastrol molecules were shown in solid sticks. Eg5 residues within 4 Å of the ligands were shown in thin tubes.
To further evaluate the capability of STD-NMR/CORCEMA-ST approach to differentiate the correct ligand-binding mode from docking results at multiple sites, we docked monastrol molecule into the ADP-binding site as well as two other putative allosteric sites (S1, S2) identified in our previous study[10] using the same IFD protocol. From the docked results of each site, two best-scored models with different binding modes were selected for CORCEMA-ST calculations and their calculated STDs were compared with the experimental results. Both the docking scores and the calculated NOE R-factors of selected models are listed in Table 1. Both models from the ATP site and the model-a from S2 site scored better (i.e., more negative scores) than the models from the monastrol site, which clearly indicated that docking scores were not able to differentiate the correct binding site. On the other hand, while the calculated STDs of these docked models varied from each other due to their different binding environment (Figure S1), all of them were very different from the experimental STDs, and the CORCEMA-ST calculated NOE R-factors of these models were significantly higher than the NOE R-factor obtained using the correctly docked model-a of the monastrol site. These results indicated that the STD-NMR/CORCEMA-ST analysis could be used to identify the proper ligand binding mode from the docking results at multiple binding pockets (monastrol, ADP, S1 and S2 sites in this example).
Table 1.
Results of STD-NMR/Docking/CORCEMA-ST calculations of models at different binding sites
| Binding site | Model | Docking Score[a] | NOE R-factor[b] |
|---|---|---|---|
| Xray | 0.12 | ||
| Monastrol site | Model-a | −7.45 | 0.12 |
| Model-b | −7.52 | 0.34 | |
| ADP site | Model-a | −7.83 | 0.38 |
| Model-b | −7.63 | 0.41 | |
| S1 site | Model-a | −5.93 | 0.32 |
| Model-b | −5.65 | 0.27 | |
| S2 Site | Model-a | −8.32 | 0.31 |
| Model-b | −6.92 | 0.29 |
Calculated using the Glide XP (extra-precision) scoring function (unit: kcal/mol).
Each NOER-factor was obtained after optimization of the parameters within 20% of reference values.
To the best of our knowledge, the present study is the first demonstration of the ability of the combined STD-NMR/Docking/CORCEMA-ST method to identify the proper binding site of an allosteric ligand from multiple binding sites. The promising results obtained in this study suggest that this method could be developed into an efficient tool that can be routinely applied for binding site validation. Meanwhile, structure-based virtual screening (SBVS) has become a widely-applied method for efficiently identifying bioactive compounds.[33,34] Along these lines, a combined method of MD simulation, SBVS and the present STD-NMR/Docking/CORCEMA-ST analysis may find wide applications in the identification and validation of novel allosteric site(s) as an aid in drug discovery research.
Experimental Section
A detailed description of molecular modelling, NMR spectroscopy and CORCEMA-ST calculations is provided in the Supporting Information.
Supplementary Material
Acknowledgments
This work was done with the support from the Southern Research Institute. The NMR measurements were performed at the High-Field NMR Facility of the UAB Cancer Center supported by the NCI CCSG grant CA-13148.
References
- 1.Mound J. Chance and Necessity: Essay on the Natural Philosophy of Modern Biology. Penguin Books; 1977. [Google Scholar]
- 2.Fenton AW. Trends Biochem Sci. 2008;33(9):420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunasekaran K, Ma BY, Nussinov R. Proteins-Structure Function and Bioinformatics. 2004;57(3):433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 4.Dixit A, Verkhivker GM. PLoS Comput Biol. 2011;7(10):e1002179. doi: 10.1371/journal.pcbi.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhuravlev PI, Papoian GA. Quarterly Reviews of Biophysics. 2010;43(3):295–332. doi: 10.1017/S0033583510000119. [DOI] [PubMed] [Google Scholar]
- 6.Ivetac A, McCammon JA. Chem Biol Drug Des. 2010;76(3):201–217. doi: 10.1111/j.1747-0285.2010.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong Y, Karplus M. Proteins. 2009;74(1):145–154. doi: 10.1002/prot.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley MJ, Chivers PT, Baker NA. J Mol Biol. 2008;378(5):1155–1173. doi: 10.1016/j.jmb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma BY, Tsai CJ, Haliloglu T, Nussinov R. Structure. 2011;19(7):907–917. doi: 10.1016/j.str.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W. J Phys Chem B. 2011;115(5):784–795. doi: 10.1021/jp107255t. [DOI] [PubMed] [Google Scholar]
- 11.Mayer M, Meyer B. Angewandte Chemie-International Edition. 1999;38(12):1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 12.Maaheimo H, Kosma P, Brade L, Brade H, Peters T. Biochemistry. 2000;39(42):12778–12788. doi: 10.1021/bi000780o. [DOI] [PubMed] [Google Scholar]
- 13.Angulo J, Nieto PM. Eur Biophys J. 2011;40(12):1357–1369. doi: 10.1007/s00249-011-0749-5. [DOI] [PubMed] [Google Scholar]
- 14.Fiege B, Rademacher C, Cartmell J, Kitov PI, Parra F, Peters T. Angew Chem Int Ed Engl. 2012;51(4):928–932. doi: 10.1002/anie.201105719. [DOI] [PubMed] [Google Scholar]
- 15.Jayalakshmi V, Krishna NR. J Magn Reson. 2002;155(1):106–118. doi: 10.1006/jmre.2001.2499. [DOI] [PubMed] [Google Scholar]
- 16.Jayalakshmi V, Krishna NR. J Magn Reson. 2004;168(1):36–45. doi: 10.1016/j.jmr.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Krishna NR, Jayalakshmi V. Prog Nucl Magn Reson Spectrosc. 2006;49(1):1–25. [Google Scholar]
- 18.Krishna NR, Agresti DG, Glickson JD, Walter R. Biophys J. 1978;24(3):791–814. doi: 10.1016/S0006-3495(78)85421-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan Y, Wen X, Sanders DAR, Pinto BM. Biochemistry. 2005;44(43):14080–14089. doi: 10.1021/bi0513406. [DOI] [PubMed] [Google Scholar]
- 20.Wen X, Yuan Y, Kuntz DA, Rose DR, Pinto BM. Biochemistry. 2005;44(18):6729–6737. doi: 10.1021/bi0500426. [DOI] [PubMed] [Google Scholar]
- 21.Yuan Y, Bleile DW, Wen X, Sanders DAR, Itoh K, Liu HW, Pinto BM. J Am Chem Soc. 2008;130(10):3157–3168. doi: 10.1021/ja7104152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tao WK, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Cancer Cell. 2005;8(1):49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Marcus AI, Peters U, Thomas SL, Garrett S, Zelnak A, Kapoor TM, Giannakakou P. J Biol Chem. 2005;280(12):11569–11577. doi: 10.1074/jbc.M413471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maliga Z, Xing J, Cheung H, Juszczak LJ, Friedman JM, Rosenfeld SS. Journal of Biological Chemistry. 2006;281(12):7977–7982. doi: 10.1074/jbc.M511955200. [DOI] [PubMed] [Google Scholar]
- 25.Yan YW, Sardana V, Xu B, Homnick C, Halczenko W, Buser CA, Schaber M, Hartman GD, Huber HE, Kuo LC. J Mol Biol. 2004;335(2):547–554. doi: 10.1016/j.jmb.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 26.Roecker AJ, Coleman PJ, Mercer SP, Schreier JD, Buser CA, Walsh ES, Hamilton K, Lobell RB, Tao WK, Diehl RE, South VJ, Davide JP, Kohl NE, Yan YW, Kuo LC, Li CZ, Fernandez-Metzler C, Mahan EA, Prueksaritanont T, Hartman GD. Bioorg Med Chem Lett. 2007;17(20):5677–5682. doi: 10.1016/j.bmcl.2007.07.074. [DOI] [PubMed] [Google Scholar]
- 27.Cox CD, Coleman PJ, Breslin MJ, Whitman DB, Garbaccio RM, Fraley ME, Buser CA, Walsh ES, Hamilton K, Schaber MD, Lobell RB, Tao W, Davide JP, Diehl RE, Abrams MT, South VJ, Huber HE, Torrent M, Prueksaritanont T, Li C, Slaughter DE, Mahan E, Fernandez-Metzler C, Yan Y, Kuo LC, Kohl NE, Hartman GD. J Med Chem. 2008;51(14):4239–4252. doi: 10.1021/jm800386y. [DOI] [PubMed] [Google Scholar]
- 28.Schiemann K, Finsinger D, Zenke F, Amendt C, Knochel T, Bruge D, Buchstaller HP, Emde U, Stahle W, Anzali S. Bioorg Med Chem Lett. 2010;20(5):1491–1495. doi: 10.1016/j.bmcl.2010.01.110. [DOI] [PubMed] [Google Scholar]
- 29.Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Science. 1999;286(5441):971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 30.Sheth PR, Shipps GW, Seghezzi W, Smith CK, Chuang CC, Sanden D, Basso AD, Vilenchik L, Gray K, Annis DA, Nickbarg E, Ma Y, Lahue B, Herbst R, Le HV. Biochemistry. 2010;49(38):8350–8358. doi: 10.1021/bi1005283. [DOI] [PubMed] [Google Scholar]
- 31.Matsuno K, Sawada J, Sugimoto M, Ogo N, Asai A. Bioorg Med Chem Lett. 2009;19(4):1058–1061. doi: 10.1016/j.bmcl.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Learman SS, Kim CD, Stevens NS, Kim S, Wojcik EJ, Walker RA. Biochemistry. 2009;48(8):1754–1762. doi: 10.1021/bi801291q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villoutreix BO, Eudes R, Miteva MA. Comb Chem High Throughput Screening. 2009;12(10):1000–1016. doi: 10.2174/138620709789824682. [DOI] [PubMed] [Google Scholar]
- 34.Andricopulo AD, Salum LB, Abraham DJ. Curr Top Med Chem. 2009;9(9):771–790. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.