

Abstract
Mucopolysaccharidosis (MPS) II, or Hunter syndrome, is a lysosomal storage disease characterized by multi-systemic involvement and a progressive clinical course. Enzyme replacement therapy with idursulfase has been approved in more than 50 countries worldwide; however, safety and efficacy data from clinical studies are currently only available for patients 1.4 years of age and older. Sibling case studies of infants with MPS I, II, and VI who initiated ERT in the first weeks or months of life have reported no new safety concerns and a more favorable clinical course for the sibling treated in infancy than for the later-treated sibling. Here we describe our experiences with a case series of eight MPS II patients for whom idursulfase treatment was initiated at under 1 year of age. The majority of the patients were diagnosed because of a family history of disease. All of the infants displayed abnormalities consistent with MPS II at diagnosis. The youngest age at treatment start was 10 days and the oldest was 6.5 months, with duration of treatment varying between 6 weeks and 5.5 years. No new safety concerns were observed, and none of the patients experienced an infusion-related reaction. All of the patients treated for more than 6 weeks showed improvements and/or stabilization of some somatic manifestations while on treatment. In some cases, caregivers made comparisons with other affected family members and reported that the early-treated patients experienced a less severe clinical course, although a lack of medical records for many family members precluded a rigorous comparison.
Introduction
Mucopolysaccharidosis II (MPS II or Hunter syndrome; OMIM 309900) is an X-linked lysosomal storage disorder caused by a deficiency in the lysosomal enzyme iduronate-2-sulfatase (I2S), leading to impaired glycosaminoglycan (GAG) catabolism (Neufeld and Muenzer 2001). The storage materials, heparan sulfate and dermatan sulfate, accumulate within lysosomes and are present in plasma, urine, and cerebral spinal fluid (Neufeld and Muenzer 2001; Tomatsu et al. 2005). Although patients with MPS II appear normal at birth, the disease is progressive and signs and symptoms usually become apparent during early childhood (Wraith et al. 2008). Early signs and symptoms of the disease may include dysmorphic facial features, airway obstruction, chronic rhinorrhea, recurrent respiratory infections, recurrent ear infections, hearing loss, hernia, hepatosplenomegaly, chronic watery diarrhea, and joint stiffness and contractures (Martin et al. 2008).
Clinical heterogeneity is a hallmark of MPS II, with patients presenting on a wide spectrum of severity. Not all patients display all associated signs and symptoms (Young et al. 1982a). About two-thirds of patients have cognitive impairment and/or behavioral abnormalities (Froissart et al. 1998, Vafiadaki et al. 1998). These patients are often described as having the severe phenotype, and they typically only survive into the second and occasionally the third decade of life (Jones et al. 2009, Young and Harper 1983; Young et al. 1982b). Patients with the attenuated phenotype do not have cognitive impairment; however, they can experience all of the somatic signs and symptoms of the disease, although often with a more gradual onset. This includes neurological manifestations such as communicating hydrocephalus and carpal tunnel syndrome (Martin et al. 2008). Such patients may live into the fifth or sixth decade of life or longer (Jones et al. 2009).
In 2006, enzyme replacement therapy (ERT) with recombinant human idursulfase (Elaprase®, Shire Human Genetic Therapies, Inc., Lexington, MA) was approved for the treatment of patients with MPS II based on phase II/III clinical trial data from patients aged 5 years and older (Muenzer et al. 2006). Some evidence reported since that time has suggested that earlier treatment before irreversible organ damage occurs may improve clinical outcomes and growth (Muenzer et al. 2012; Muenzer et al. 2009, Schulze-Frenking et al. 2011). A recent 53-week, open-label, safety trial of 0.5 mg/kg weekly idursulfase in children aged 1.4–7.5 years (n = 28) has been completed (Giugliani et al. 2013). Adverse events experienced by the younger patients were similar to those experienced by patients aged 5 years and older in the pivotal phase II/III trial of idursulfase (Muenzer et al. 2006). The most common adverse events (57 %) were infusion-related reactions (IRRs). The most common serious adverse reactions occurring in at least 10 % of patients (≥3 patients) included bronchopneumonia/pneumonia (18 %), ear infection (11 %), and pyrexia (11 %). Available genotype data from 27/28 patients demonstrated that patients with a complete gene deletion, large gene rearrangement, nonsense, frameshift, or splice site mutation were more likely to experience IRRs and serious adverse events after idursulfase administration than were patients with missense mutations (idursulfase [Elaprase] prescribing information, Shire 2013).
There are currently no clinical trial data addressing the treatment of children under 1 year of age. Benefits with early ERT in MPS I and MPS VI have been suggested by sibling case reports (Gabrielli et al. 2010; McGill et al. 2010). Tylki-Szymańska and colleagues published an unusual case study of twin boys, one of whom has MPS II (Tylki-Szymanska et al. 2012). The twins are the younger siblings of an older affected sister. MPS II in the sister was caused by the heterozygous IDS gene mutation c.1568A>G in association with almost totally skewed X-chromosome inactivation of the X chromosome carrying the wild-type IDS allele of paternal origin. The affected twin was treated with idursulfase from 3 months of age with no significant adverse events, and after 3 years of treatment, was reported to have normal somatic development. The only somatic sign of the disease is that the affected twin has a mild deformity of one vertebra. Although comparisons are difficult, the older affected sister at the age of 3 experienced the following somatic manifestations: mild coarse facial features; decreased range of motion in elbow, hip and ankle joints; slight hepatomegaly; and a small umbilical hernia.
Tajima and colleagues have very recently published a case study of two Japanese brothers with MPS II caused by an inversion mutation (Tajima et al. 2013). The older brother initiated treatment with idursulfase at 3.0 years of age, while the younger sibling initiated treatment at 4 months. At the start of treatment, the older brother showed typical somatic features of MPS II, including mitral valve regurgitation, gibbus deformity, joint stiffness, umbilical hernia, coarse facies, short stature, hepatomegaly, and cognitive impairment. After 2 years of treatment, the older brother’s somatic disease was stable or improved, while cognitive decline continued. By comparison, after 32 months of ERT, the younger brother remained free from most of the somatic features that had already appeared in his brother at the same age. His only apparent disease manifestations were otitis media with effusion, slight findings of dysostosis multiplex, and mild cognitive impairment.
Here, we present data about the treatment of very young patients with MPS II in a case series of eight patients who received ERT with idursulfase beginning before 1 year of age, including a patient who began therapy at 10 days of age. All of the patients had non-consanguineous parents. A summary of patient demographics; birth weight, height, and head circumference; signs/symptoms at diagnosis, and details of treatment with enzyme replacement therapy are presented in Table 1.
Table 1.
Summary of patient demographics and details of treatment with enzyme replacement therapy (ERT)
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | |
|---|---|---|---|---|---|---|---|---|
| Consanguinity | N | N | N | N | N | N | N | N |
| Positive MPS II family history (Relationship to Case) | Sibling, maternal uncle | Sibling | Maternal second cousin | Sibling | Maternal half-uncle, maternal great-uncles, maternal first cousin once removed | None | Maternal uncles, maternal first cousin once removed | Maternal great-uncle |
| Gestational ultrasound findings | Normal | Ventriculomegaly | ND | ND | Polyhydramnios | Normal | Normal | Large for gestational age |
| Apgar score 1′/5′ | 9/10 | 8/9 | 8/10 | 9/9 | 6/9 | 9/10 | ND | ND |
| Birth weight in g, (percentile) | 3,320 (54) | 3,856 (92) | 3,050 (27) | 3,884 (93) | 3,005 (24) | 3,920 (95) | 2,900 (16) | 4,366 (>99) |
| Birth length in cm, (percentile) | 51 (72) | 52 (86) | 49 (31) | 52 (89) | 48 (16) | 51 (73) | 47 (7) | 53 (96) |
| Birth head circumference in cm, (percentile) | 34 (35) | 36 (89) | 33 (12) | 37 (98) | 33 (12) | 36 (89) | 33.6 (24) | ND |
| Age at diagnosis | Prenatal | 1 week | 6 weeks | 1 day | 4 weeks | 11 weeks | 1 week | 5.5 months |
| Genotype | p.R88H | p.R95G | p.P86L | p.R493P | c.1270insCC | p.G336E | c.1133A>G | c.1362-1365dup |
| Signs and symptoms at ERT baseline | Lumbar gibbus | Mild frontal bossing, hepatomegaly, lumbar kyphosis, pectus excavatum | Diastasis recti abdominis, hepatosplenomegaly, umbilical hernia | Mild coarse facies, hepatosplenomegaly, hearing loss, lumbar kyphosis | Coarse facies, Diastasis recti abdominis, hepatomegaly, umbilical hernia | Hydrocele, inguinal hernia, hepatomegaly, congestive heart failure | Mild frontal bossing, chronic otitis media with effusion, scapular flaring with right shoulder abduction | Mild coarse facies, small thickened ears, hepatomegaly, bilateral syndactyly of the second and third toes, gibbus; bilateral foot adduction, frequent upper respiratory infections, recurrent episodes of acute otitis media |
| X-Ray/MRI skeletal findings at ERT baseline | Lumbar gibbus at L3-L5 | Lumbar kyphosis, pectus excavatum, mild broadening of the ribs | ND | Mild focal kyphosis (apex at L2), anterior beaking of the caudal aspect of L2 | Normal | ND | ND | Thoracolumbar gibbus with the apex at L2, bilateral forefoot adductus |
| Echocardiography at ERT baseline | Normal | Mildly dilated left ventricle with low-normal systolic function and increased apical trabeculations | Normal | ND | Small atrial septal defect or stretched patent foramen ovale | Abnormal (See Table 2) | Normal | Normal |
| Age at ERT start | 1.5 weeks | 6 weeks | 8 weeks | 10 weeks | 11 weeks | 12 weeks | 6 months | 6.5 months |
| Dose in mg/kg/weekly infusion | 0.5 | 1.5 at first, reduced to 0.5 as the patient gained weightb | 0.6 mg for the first 8 infusions, and 0.5 mg for 9th and 10th infusionsb | 1.4 at first, reduced to 0.5 as the patient grewb | 0.5 | 0.5 | 0.5 | 0.66 at first, reduced to 0.5 as the patient gained weightb |
| Total duration of treatment | 6 weeks | 2 yearsa | 10 weeks | 3 yearsa | 5.5 yearsa | 20 monthsa | 4 yearsa | 3.5 yearsa |
| HSTC | Yes | No | Yes | No | No | No | No | No |
ERT enzyme replacement therapy, HSTC Hematopoietic stem cell transplantation, MPS II mucopolysaccharidosis II, ND no data
aDuration of treatment to date of last follow-up examination; patient currently continues on treatment
bOff-label use of the medication. The approved dose is 0.5 mg/kg weekly (Elaprase® prescribing information 2011)
Case 1
Patient 1, a Brazilian male, has a family history of an affected older half-brother and an affected maternal uncle, both with severe MPS II. He was diagnosed at 16 weeks 5 days gestation by an I2S enzyme activity assay that revealed undetectable I2S activity in amniotic fluid cells. A molecular genetic analysis revealed the familial p.R88H mutation, confirming the diagnosis. The prenatal course was uneventful and no abnormalities were noted on ultrasound. At 37 weeks 2 days gestation, Patient 1 was delivered via cesarean section due to labor failure to progress. Physical examination of the infant revealed a very subtle lumbar gibbus. A lumbar X-ray was notable for L3-L5 abnormality (Fig. 1a). An echocardiogram was normal. I2S enzyme activity was measured in plasma [1.2 nmol/4 h/mL (ref = 122–463 nmol/4 h/mL)] and in leukocytes [4.3 nmol/4h/mg/ptn (ref = 31–110 nmol/4 h/mg/ptn)], reconfirming the diagnosis of MPS II. Placental analysis by electronic microscopy revealed discrete lysosomal storage in endothelial cells and in pericytes, as has been previously reported (Baldo et al. 2011).
Fig. 1.
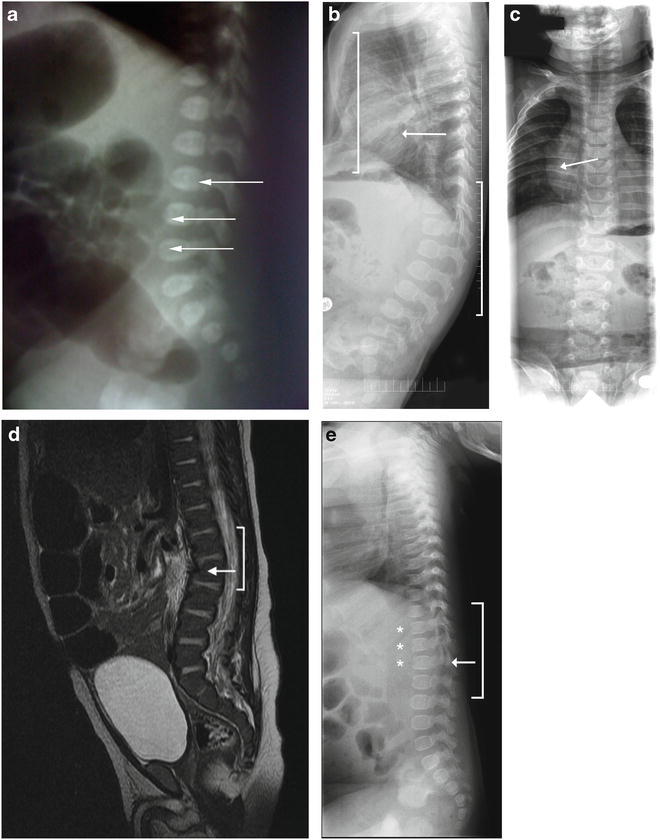
Skeletal abnormalities in infants with MPS II. (a) Skeletal X-ray image of Patient 1 at 1 day of age showing L3-L5 abnormality (arrows). b and c) Skeletal X-ray images of Patient 2 at 6 months of age showing pectus excavatum (panel b , left bracket), mild lumbar kyphosis (panel b , right bracket), and mild broadening of the ribs (panels b and c , arrows). (d) A computed tomography scan of Patient 4’s abdomen performed at 8 weeks of age showing upper lumbar kyphosis (bracket) with mild inferior beaking at L2 (arrow). (e) Skeletal X-ray images of Patient 8 at 5.5 months of age showing a thoracolumbar gibbus (bracket) with the apex at L2 (arrows) and beaked vertebrae (stars)
ERT with 0.5 mg/kg idursulfase weekly infused over 3 h via a peripheral IV line was initiated when the patient was 10 days old. Infusions were given weekly in the hospital setting and were continued for a total of six infusions, at which point ERT was stopped in preparation for hematopoietic stem cell transplantation (HSCT). At 19 days of age, the infant presented with a fever of 39°C, significant nasal obstruction, and yellow nasal discharge. He was admitted to the hospital and a 10-day course of intravenous antibiotics was prescribed. The fever and respiratory infection resolved without sequelae. ERT was not discontinued during this episode, as the child was not febrile on the day of the scheduled infusion.
The response to ERT was evaluated with a uGAG analysis. The baseline uGAG level at 5 days of age was 2,000 mg/g creatinine (upper limit of normal: 170 mg/g creatinine), which fell to 1,300 mg/g creatinine after 6 infusions of ERT. No adverse events were noted.
At 70 days of age, the patient underwent hematopoietic stem cell transplantation (HSCT) with umbilical cord blood from an unrelated donor. After transplantation, no additional ERT was given. His development has continued to be followed, and the patient’s mother reports that he is doing much better than his older half-brother at the same age. A detailed account of the patient’s post-transplant development will be published elsewhere.
Case 2
Patient 2 is an American male of Caucasian race. An ultrasound performed in the second trimester of the pregnancy revealed ventriculomegaly in the fetus. An amniocentesis was then performed; the chromosome analysis was normal. A fetal magnetic resonance imaging (MRI) study of the brain revealed mild prominence of the right ventricle and a midline cystic structure of the cavum vergae. Neurosurgical consultation was obtained, and it was felt these were likely benign findings. No prenatal testing for MPS II was performed because the older brother was not diagnosed until the mother’s seventh month of gestation with Patient 2. There was no other family history of the disorder. Patient 2 was born at 39 weeks gestation via a planned cesarean section.
Six hours after the birth, the infant experienced respiratory distress and was transferred to the neonatal intensive care unit. The infant was initially given supplemental oxygen, then was intubated and given surfactant. After 24 h, the infant was extubated and eventually weaned to room air. Laboratory testing revealed elevated C-reactive protein levels, but cultures of blood, urine, and cerebral spinal fluid were negative. Nonetheless, the infant received antibiotic treatment for 7 days. The infant was discharged from the hospital at 2 weeks of age.
The diagnosis of MPS II was suspected while the infant was in the hospital due to mild frontal bossing (Fig. 2a) and hepatomegaly (liver edge palpable 4 cm below the right costal margin). A skeletal survey showed mild lumbar kyphosis, pectus excavatum, and mild broadening of the ribs (Fig. 1b, c). The child also failed the initial newborn hearing screen, but the follow-up assessment with an audiologist was normal. An echocardiogram performed at 2 days of age showed a non-obstructive band across the left ventricle. A follow-up echocardiogram at 1 month of age revealed a mildly dilated left ventricle with low-normal systolic function and increased apical trabeculation, possibly compatible with mild left ventricular non-compaction.
Fig. 2.
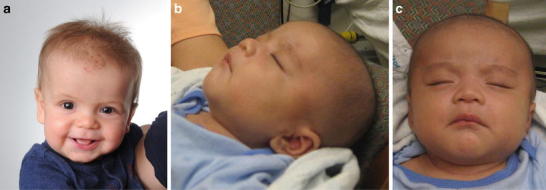
Mild coarse facial features and frontal bossing characteristic of MPS II. (a) Patient 2 at 7 months of age. b and c) Patient 4 at 6 weeks of age. Images used with permission
A uGAG analysis was performed at 6 days of age, revealing an elevated level of 151.6 mg/mmol creatinine (upper limit of normal: 53 mg/mmol creatinine). An I2S enzyme activity assay in leukocytes revealed no activity at 7 days of age, confirming the diagnosis. A molecular genetic analysis revealed a p.R95G mutation.
ERT was initiated when the child was 6 weeks of age. The starting dose was 1.5 mg/kg idursulfase given over 3 h via peripheral IV line; a full 6 mg vial was completely used in order not to waste medication, an off-label usage of the medication (Elaprase prescribing information 2011). As the infant gained weight, the dose per kilogram body weight decreased until a 0.5 mg/kg dose was achieved. He received weekly infusions in the infusion center at the hospital until 7 months of age, when he was transitioned to home therapy. At 10 months of age, a central line was placed. He remains on treatment at 2 years of age, and no adverse events have been seen.
At 7 months of age, the physical exam was essentially within normal limits except for mild frontal bossing and pectus excavatum (as noted at birth) and the palpable liver edge 2–3 cm below the right costal margin. The uGAG level, tested in a different hospital from the birth hospital, was 185 mg/g creatinine (normal < 148 mg/g creatinine). When last examined at 2 years of age, the physical examination was completely normal except for a somewhat broad forehead with mild frontal bossing. Development was appropriate for age.
Case 3
Patient 3 is a German male. The infant was delivered spontaneously at 38 weeks’ gestation after an uncomplicated pregnancy. The presence of diastasis recti abdominis was noted on physical examination at birth. No complications were reported in the postnatal period. When the infant was 6 weeks of age, the mother requested I2S testing because her maternal cousin’s son (the infant’s second cousin) had just been diagnosed with MPS II at 5 years of age. The mother also recalled that her maternal great-aunt had a son who had died at the age of 15 years from presumed Hurler syndrome (severe MPS I). As severe MPS I may appear clinically very similar to MPS II, it seems likely that this boy was also affected by MPS II and was incorrectly diagnosed. The uGAG level at 6 weeks of age was 55.80 mg/mmol creatinine (upper limit of normal: 27.20 mg/mmol creatinine), and an I2S activity assay revealed an activity of 8.2 nM/4 h/mL (normal range: 300–800 nM/4 h/mL). Molecular genetic testing revealed a p.P86L mutation, which was also shared by the infant’s second cousin.
On clinical examination at 6 weeks of age, the boy appeared healthy and active. Diastasis recti abdominis was still present and a small umbilical hernia was seen. The liver was palpable 1 cm below the costal arch, but the spleen was not palpable. Reflexes were normal. The boy showed no paresis or contractures and had normal muscle tone and joint range of motion. Echocardiogram and electrocardiogram findings were both normal. An abdominal ultrasound revealed spleen enlargement and marginal liver enlargement. A cranial ultrasound was normal. Cognitive testing using the Bayley Scales of Infant Development was within normal ranges.
ERT was begun when the infant was 2 months of age. A total of 10 doses were given by peripheral IV infusion over 3.5 h. Idursulfase was used off-label at a dosage of 0.6 mg/kg weekly for the first eight infusions (Elaprase prescribing information 2011). During this time the infant’s weight increased, so a dose of 0.5 mg/kg was used for the ninth and tenth infusions. No adverse events were seen during ERT, and no premedication was given. Antibody testing for IgE and IgG antibodies to idursulfase was negative throughout. After 10 doses of idursulfase, ERT was discontinued, and bone marrow transplantation with a matched donor was performed.
At 18 weeks of age, after 10 weeks on idursulfase treatment, the uGAG level was 27.61 mg/mmol creatinine, which was within normal range. Growth was appropriate for age. The liver remained palpable 1 cm below the costal arch. There were no evident developmental delays. An MRI of the spinal column revealed mild left convex scoliosis but no dysostosis multiplex. No hearing loss was found on audiological exam.
Case 4
Patient 4, a Hmong male born in the USA, has a family history of an older affected brother who was diagnosed at 2 years and 9 months of age. There were no prenatal complications during the mother’s pregnancy with Patient 4, other than a known MPS II carrier status for the mother, and no prenatal testing for MPS II was performed. The infant was delivered vaginally at 40 weeks’ gestation. At the time of delivery, umbilical cord blood was obtained and sent for genetic testing for MPS II. The enzyme activity assay on the umbilical cord blood measured an I2S activity level of 36 cpm/h/ml plasma (normal value: >14,376 cpm/h/ml/plasma), confirming the diagnosis of MPS II. The molecular genetic analysis revealed a p.R493P mutation, which is also found in the mother and the older affected brother.
The infant failed the routine newborn hearing test bilaterally. At 2 weeks of age, mild frontal bossing and slightly coarse facial features were present (Fig. 2b and c). A skin examination revealed several areas of dermal melanocytosis on his right ankle, left thigh, and buttocks. A computed tomography scan of the abdomen performed at 8 weeks of age demonstrated hepatosplenomegaly with a liver volume of 244 mL (normal: 125 mL) and spleen volume of 36 mL (normal: 10 mL) as well as upper lumbar kyphosis with mild inferior beaking at L2 (Fig. 1d). The kyphosis was only observed upon imaging and was not evident clinically. An MRI of the spine at 10 weeks of age revealed mild focal kyphosis with apex at L2 and anterior beaking of the caudal aspect of L2. An MRI of the brain at 10 weeks of age was normal.
ERT was initiated at 10 weeks of age with a dose of 0.5 mg/kg rounded to the nearest whole vial amount (a practice that is off label) and infused over 4 h via central IV line. The infant was prophylactically pre-medicated with acetaminophen, diphenhydramine, and hydrocortisone. The child has remained on ERT, which as of the time of publication is greater than 3 years of treatment. During this time, no IRRs have been seen, and the child is currently only pre-medicated with acetaminophen. The infusion time has been reduced to 2 h. Urinary GAG levels have improved, falling from a baseline level of 1856.7 μg/mg creatinine to 272.0 μg/mg creatinine (normal: < 127 μg/mg creatinine) at last measurement after 10 months of treatment. No antibodies to idursulfase have been detected at any point during treatment.
At 7 months of age, the otolaryngologist recognized bilateral hearing loss and placed tympanostomy tubes. Some mild hearing loss persisted after placement, and hearing aids were prescribed. At 14 months of age, electromyography and nerve conduction velocity testing for carpal tunnel syndrome were both normal. An echocardiogram at 16 months of age was normal. The last physical examination was performed at 18 months of age. Mild coarse facial features, including some mild frontal bossing, were present as was some minor joint stiffness in the elbows and wrists. The rest of the examination was unremarkable. The child’s parents had no concerns for him at the time of the examination and said that he is developing much better than his older affected brother at the same age. He is a well-established walker. The orthopedist continues to monitor the spinal malformation but no further changes have been noted.
A formal developmental assessment was undertaken for the patient at 18 months of age. According to the Bayley Scales of Infant and Toddler Development, his gross motor and expressive language skills were age appropriate. His self-help, fine motor, receptive language, and verbal comprehension skills showed a 6- to 9-month delay.
Case 5
Patient 5 is an American male with an extensive positive family history of MPS II. The patient’s mother had a maternal half-brother, two maternal uncles, and a maternal male first cousin who were all affected and died in the second to third decade of life. The pregnancy was unremarkable, and no prenatal testing was performed. Two routine prenatal ultrasounds were within normal parameters, but at 36 weeks gestation, a third ultrasound revealed polyhydramnios. The patient was delivered vaginally after 38 weeks and 2 days gestation. He was noted to be hypotonic on first exam and was given blow by oxygen. At 3 h of life, he was noted to have difficulty in breathing and so was transferred to the neonatal intensive care unit (NICU) where he was treated for pneumonia and given surfactant. The infant was released after 13 days.
Enzyme testing for MPS II was ordered while the patient was in the NICU given the family history of MPS II. At 4 weeks of age, his I2S enzyme activity was found to be 0 cpm/h/mL/plasma (normal value: >64,147 cpm/h/mL/plasma). Molecular genetic testing revealed the c.1270insCC mutation in the IDS gene. On examination at 11 weeks of age, the patient was noted to have a normal growth pattern with normal head circumference. He presented with mild coarse facies (round face with periorbital fullness), diastasis recti, and fingertip umbilical hernia. The liver edge was palpable at 2 cm below the right costal margin. The rest of the examination was unremarkable. Echocardiogram findings reported either a stretched patent foramen ovale or a small secundum atrial septal defect measuring 4.5 mm, while an electrocardiogram suggested possible left ventricular hypertrophy. The skeletal survey was normal.
ERT was initiated when the infant was 11 weeks of age at a dose of 0.5 mg/kg weekly via peripheral IV initially; the patient later received infusions using a central IV line. The patient received infusions in the hospital for 15 months and then was transitioned to home therapy. He has now been receiving ERT for approximately 6 years and has not become positive for anti-idursulfase antibodies. His uGAG levels have fallen from 88.9 mg/mmol creatinine at baseline (normal: 10.9–22.3 mg/mmol creatinine) to 15.4 mg/mmol creatinine at last measurement in 2011. He has had his central line replaced twice, once due to a device malfunction and once due to an infection. One episode of hypertension was reported by a home health nurse during ERT administration in the home setting; however, this could not be replicated in the emergency room. No other adverse events to ERT have been observed.
The last follow-up examination occurred when the boy was 5.5 years of age. The patient had normal growth [height: 116.1 cm (69th percentile); weight: 21.6 kg (69th percentile)] and very minor joint range-of-motion restrictions. Cardiac echocardiogram found mild aortic valve stenosis, trivial pulmonary valve insufficiency, and normal left ventricular function. A bronchoscopy found mild glossoptosis, pharyngomalacia with posterior pharyngeal nodularity and cobble stoning, and a mild nodular appearance of the tracheal mucosa. Electromyography and nerve conduction velocity testing showed mild right median mononeuropathy at the wrist without evidence of active denervation. Cervical flexion-extension, abdomen, and chest X-rays revealed only subtle bony changes in the spine and hips. In the previous 2 years he has undergone placement of two sets of myringotomy tubes, with only one ear infection in that time. He currently wears a left hearing aid for mild hearing loss.
The patient has not undergone formal developmental testing, but at age 5.5 years his teacher reported that he had typical cognitive ability, which was evident during the last examination. None of his extended family members with MPS II had any history of cognitive impairment, and the affected individuals, who died at 18, 19, and 21 years of age, all were in or completed high school at the time of death without reports of academic problems. An MRI of the brain and spine performed at 5.5 years of age revealed some scattered foci of increased signal in the brain white matter and soft tissue thickening at the level of the foramen magnum without cord compression.
Case 6
Patient 6 is a Brazilian male with a family history of MPS, as the patient’s father has five cousins with MPS I. No prenatal genetic testing was undertaken. The child was born vaginally after 37 weeks and 5 days gestation. The physical examination revealed a scrotal hydrocele. There were no complications in the postnatal period.
At 9 weeks of age, the infant presented with wheezing, abnormal cardiac auscultation, hepatomegaly (liver palpable 2 cm below the right costal margin), and inguinal hernia. A chest X-ray revealed an increased cardiac silhouette. He was diagnosed with viral cardiomyopathy and was hospitalized. The following day, an echocardiogram revealed dilated cardiomyopathy and a ventricular ejection fraction of 25 %. The patient was transferred to the intensive care unit. Soon after, the treating physician requested enzyme activity assays in dried blood spots for alpha-L-iduronidase (MPS I), I2S (MPS II), and arylsulfatase B (MPS VI). The diagnosis of MPS II was established at 11 weeks of age, and a molecular genetic analysis revealed the p.G336E mutation. As the child’s cardiac function had continued to deteriorate, he was immediately transferred to the intensive care unit of a specialized cardiac care hospital with a diagnosis of severe congestive heart failure secondary to MPS II. A cardiac MRI performed at this hospital revealed substantial left ventricle dysfunction without myocardial fibrosis and no signs of inflammatory cardiomyopathy.
ERT with weekly infusions of 0.5 mg/kg idursulfase in 50 mL administered over 3 h and 30 min via central line was begun in the cardiac care hospital when the child was 12 weeks of age. The patient’s uGAG level at diagnosis was 27.62 mg/mmol creatinine (upper limit of normal: 35.8 mg/mmol creatinine). After five weekly infusions, the child was discharged from the cardiac care hospital due to progressive improvement in cardiac function (Table 2). Concomitant medications included captopril, furosemide, spironolactone, and carvedilol.
Table 2.
Echocardiogram findings for Patient 6
| Since start of ERT | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | −6 days | 3 days | 7 days | 14 days | 24 days | Normal range | 171 days | 293 days | Normal range | 391 days | Normal range |
| Right ventricle diameter (mm) | 8.2 | ND | 5.8 | 12 | 4.5 | <11.9 | ND | 9.3 | <13.4 | 4.8 | <15.2 |
| Diastolic septum thickness (mm) | 5.4 | ND | 4.2 | 4.5 | 5 | <4.2 | 59 | 3.7 | <5.6 | 6 | <5.7 |
| Left ventricle diastolic diameter (mm) | 37.6 | 43.7 | 41.5 | 41 | 40.6 | <21.7 | ND | 58 | <30.8 | 62.8 | <35.9 |
| Diastolic wall thickness (mm) | 4.4 | ND | 5.1 | 4.5 | 4.3 | <4.2 | ND | 3.2 | <5.4 | 4 | <6.1 |
| Systolic septum thickness (mm) | 6 | ND | 5 | 4.9 | 5.7 | NA | ND | 5.5 | NA | 7.5 | NA |
| Systolic left ventricle diameter (mm) | 34.9 | ND | 38.6 | 37 | 36.9 | NA | ND | 50.7 | NA | 54.2 | NA |
| Systolic wall thickness (mm) | 5 | ND | 6.8 | 4.1 | 6.6 | NA | ND | ND | NA | 6.8 | NA |
| Aortic diameter (mm) | 15 | ND | ND | 11.5 | ND | <11.6 | ND | 14 | <15.3 | 14 | <18.6 |
| Left atrium diameter (mm) | 17 | ND | ND | 16 | ND | <15.7 | ND | 30 | <21.2 | 30 | <23.4 |
| Left ventricle shortening fraction (%) | 7.2 | ND | 7.1 | 19 | 9.1 | 28–42 | ND | 12.6 | 28–42 | 13.7 | 28–42 |
| Left ventricle ejection fraction (%) | 12 | ND | 16 | ND | 20.3 | 56–75 | ND | 26.9 | 56–75 | 28.5 | 56–75 |
| Left ventricle ejection fraction – Simpson’s method (%) | ND | 21 | 24 | 23 | ND | NA | 23 | 26 | NA | 28 | NA |
ND not done, NA not available
The patient continues to receive ERT in the clinic via peripheral IV line and has been on treatment for about 20 months to date. No IRRs have been observed during ERT, and serum IgG and IgE antibody assays have been negative. After 5 months of ERT, the cardiologist noted a worsening of ejection fraction and recommended cardiac transplantation, which has not yet been performed. The physical examination at 1 year of age showed above-average growth [weight: 11,860 g (97th percentile), length: 80.5 cm (97th percentile), head circumference: 48.5 cm (97th percentile)], normal motor development, absence of hepatosplenomegaly, and normal joint range of motion. An echocardiogram performed when the child was 16 months of age, after approximately 13 months of ERT, showed sustained dilated cardiomyopathy, with severe impairment of left ventricle systolic function (Table 2).
Case 7
Patient 7 is an American male of Caucasian race. The patient’s mother had two brothers and a maternal first cousin with MPS II. Her maternal great-aunt had a son and a grandson with MPS II as well. All of the affected males lived into their fourth decade of life. No complications were reported during the pregnancy with Patient 7. The infant was born vaginally at 36 weeks gestation. In the postnatal period, the patient experienced respiratory distress, and continuous positive airway pressure was maintained for 48 h after the birth.
At 1 week of age a blood sample was sent for enzyme activity testing because of the family history of MPS II, and no I2S activity was detectable. Molecular genetic analysis revealed a 78-base-pair insertion in the IDS transcript that has been previously reported and has been associated with an attenuated phenotype (Rathmann et al. 1996). Weekly ERT with 0.5 mg/kg idursulfase administered via a central intravenous line over 4 h was begun when the child was 6 months of age. At baseline, an abdominal ultrasound and echocardiogram were normal. No joint restrictions were noted, although scapular flaring with right shoulder abduction was identified. A tympanogram was consistent with the presence of middle ear fluid on the right, but hearing was normal. The Mullen Scales for Early Learning and Scales of Independent Behavior-Revised, Early Development form, was used to evaluate the patient developmentally. He demonstrated skills within the average or above average in all domains without correcting for gestational age.
The child has been receiving ERT for the past 4 years with no adverse events reported. His uGAG level at baseline was 200 μg GAG/mg creatinine. After 1 year of treatment, his uGAG levels had not decreased (240.8 μg GAG/mg creatinine); no more recent uGAG data are available. At age 2 years, a formal developmental evaluation was performed, and the patient scored in the average or above average range in all domains. At the last physical examination at the age of 3 years 9 months, his growth parameters were all normal, with height and weight at the 95th percentile and head circumference at the 75th percentile. He had slightly coarse facies, with frontal bossing, a receding anterior hairline, and slight puffiness of the upper eyelids. He also exhibited tapering of the fingers with slightly reduced extension of the digits at the distal interphalangeal joint. No evidence of macroglossia, organomegaly, spine deformities, hearing loss, or hernias was seen. At the time, he was growing and developing normally. He had not had a more recent formal developmental evaluation, but he was keeping up with his peers.
Case 8
Patient 8 is an American male of Caucasian race with a positive family history for MPS II, as his mother’s uncle (his maternal great-uncle) has an attenuated form of the disease and is currently alive at the age of 42 years. No complications were noted during the pregnancy. A prenatal ultrasound was significant for the fetus being large for gestational age, but otherwise was unremarkable. The infant was born at 38 weeks’ gestation. He was transferred to the NICU due to respiratory distress and was discharged after 6 days. There was no documentation of any abnormalities in the immediate postnatal period.
At 2 months of age, the patient was hospitalized for 1 week for the treatment of bronchiolitis. At 5.5 months of age, the patient’s grandmother noticed a lump on his back; subsequent evaluation by orthopedics and a skeletal survey revealed a thoracolumbar gibbus with the apex at L2 (Fig. 1e) as well as bilateral forefoot adductus. The astute pediatric orthopedist also noted macrocephaly; a saddle-shaped nose; and small, thick ears, which triggered his suspicion of a metabolic disorder. Upon questioning the mother about a family history, he referred the patient to medical genetics for a possible diagnosis of MPS II.
On examination in the metabolic genetics clinic at 6 months of age, the patient showed mild coarse facies; small, thickened ears; hepatomegaly; and bilateral syndactyly of the second and third toes, in addition to the gibbus and bilateral foot adduction. The mother reported frequent upper respiratory infections, snoring, and recurrent episodes of acute otitis media. A uGAG analysis revealed a level of 106.4 mg/mmol creatinine (normal range: 6.7–16.5 mg/mmol creatinine). A molecular genetic analysis revealed a small, 4-base-pair duplication (c.1362–1365dup) in the IDS gene. Findings on echocardiogram and MRI of the brain were normal.
ERT was initiated when the patient was 6.5 months of age. Initially an entire 6 mg vial of idursulfase was infused in order not to waste medication, yielding a dose of 0.66 mg/kg weekly (an off-label dosage). As the patient grew, the dose was adjusted to 0.5 mg/kg weekly. At the time of last follow-up at 4 years of age, he had not experienced any adverse events to ERT. His uGAG levels had fallen from 112.4 mg/mmol creatinine (normal: 6.2–13.6 mg/mmol creatinine) at baseline to 19.4 mg/mmol creatinine. Overall, the patient has done well on ERT. His liver size normalized, and echocardiogram results have remained normal. Myringotomy tubes were placed concurrently with a tonsillectomy and adenoidectomy at age 19 months, and this procedure was repeated at age 3.5 years. The gibbus deformity progressed during his second year of life, but has stabilized since 3 years of age. He developed slight contractures of the joints in the upper extremities by 3.5 years of age. A bilateral carpal tunnel decompression surgery was performed at 3.5 years of age. A formal neurodevelopmental evaluation was conducted at the age of 3 years 8 months using the Capute Scales. The patient’s score on the Cognitive Adaptive Test was the age equivalent of 30 months. Given his chronological age, that yields a visual-motor problem solving quotient of 68. On the Clinical Linguistic and Auditory Milestone Scale, his age equivalent was 33 months, which is a language quotient of 75.
Discussion
We have presented a case series of eight patients with MPS II whose treatment with idursulfase was begun under the age of 1 year. The age at initiation of treatment was between 10 days and 6.5 months, and the duration of treatment was between 6 weeks and 5.5 years. No new safety concerns were identified and none of the patients experienced an IRR. Our experience here suggests that the development of disease manifestations occurs rapidly after birth, or perhaps even begins prenatally. All of the patients showed improvements and/or stabilization of some somatic manifestations while on treatment.
The use of ERT in very young patients with MPS II has generated much recent interest, both in the safety profile in this age group and in the ability of ERT to modify the natural history of the disease in younger versus older patients. The pivotal phase II/III trial for idursulfase in MPS II only enrolled patients over the age of 5 years due to the need for patients to participate in the forced vital capacity and 6-min walk tests that made up the composite primary endpoint (Muenzer et al. 2006). Reassuring results from a recent open-label clinical trial of idursulfase in 28 patients aged 1.4–7.5 years indicated no new safety concerns in this population, with improvements seen in liver size and uGAG levels (Giugliani et al. 2013). In addition, there are non-controlled patient registry data suggesting that the safety profile of idursulfase is similar in younger and older patients in clinical practice. A retrospective study using data from the Hunter Outcome Survey (HOS), a voluntary patient registry for patients with MPS II regardless of treatment status (Wraith et al. 2008), evaluated the safety and effectiveness of idursulfase in 124 patients younger than 6 years and compared these findings with those seen in 289 older patients (Muenzer et al. 2011a). The mean age at start of ERT in the younger group was 3.6 ± 1.6 years. There were no new safety concerns in the younger group as compared with the older group. In our experience of using ERT in eight patients under 1 year of age, no new safety concerns were seen and none of the patients experienced IRRs after a treatment duration of between 6 weeks and 5 years. This is encouraging given that the majority of IRRs appear to occur during the first 3 months of therapy (Burton and Whiteman 2011).
The ability of ERT to change the natural history of the disease when begun in very young patients has not been systematically evaluated to date. This is in part because identifying and diagnosing very young patients without a family history of the disease generally requires newborn screening, which has not yet been generally instituted for MPS II (Nakamura et al. 2011). In the HOS study of patients under 6 years of age, ERT effectiveness as assayed by reductions in liver size and uGAG levels was found to be similar between both the younger and older groups of patients, but the clinical significance in altering the long-term disease course could not be evaluated using the available registry data (Muenzer et al. 2011a). There are sibling-pair case study data for MPS I, II, and VI suggesting that the initiation of ERT in infancy results in a better long-term clinical outcome (Gabrielli et al. 2010; McGill et al. 2010; Tajima et al 2013; Tylki-Szymanska et al. 2012). The most current recommendations from the Hunter Syndrome European Expert Council state that the relationship between progressive GAG storage and clinical manifestations in MPS II provides a strong argument for the initiation of ERT as early as possible following diagnosis (Scarpa et al. 2011). Indeed, our experience with these eight patients shows that disease manifestations are present from a few days or weeks of age, or even prenatally in some cases, upon careful examination and imaging. Six out of eight patients displayed organomegaly, five out of eight displayed mild coarse facies, and three out of eight had an umbilical and/or inguinal hernia. Among the five patients who underwent a skeletal survey before starting ERT, four had spinal malformations (kyphosis or gibbus), as seen in Fig. 1a–e.
In this report, we provide evidence that early ERT has resulted in somatic improvements among our patients, although two of the eight patients (Patient 1 and Patient 3) only received ERT for a short time before HSCT. For Patient 1, ERT plus HSCT was tried based on an extrapolation of the results seen with MPS I (de Ru et al. 2011); such an approach is not the standard of care for MPS II. Early marked decrease in uGAGs was seen in Patients 1 and 3, but the short duration of treatment makes interpretations of longer-term ERT effectiveness impossible for these patients. Among the patients who did not undergo HSCT, Patient 2 has been treated for 2 years; during that time, no new disease manifestations have been seen, and the mild frontal bossing and pectus excavatum which were noted at birth remained stable. His hepatomegaly improved on treatment. Patient 4 has been treated for about 3 years to date. His uGAG levels have significantly improved, he has no respiratory complaints, and his spinal malformation and facial features have remained stable. His hearing loss has progressed, and he has minor joint stiffness in his elbows and wrists, although he has no trouble with ambulation. His parents subjectively report that he is doing much better than his older affected brother at the same age. Patient 5 has been receiving ERT for approximately 5.5 years and has experienced improvements in uGAG level and hepatomegaly. His signs and symptoms include very mild joint restrictions, mild aortic stenosis, mononeuropathy of the right median nerve, subtle bony changes in the spine and hips, umbilical hernia (repaired at 5 years of age), mild hearing loss in the left ear, and otitis media leading to the placement of ventilation tubes. He is a healthy appearing child who is active and doing very well in school. Other affected maternal relatives were reported to have somatic manifestations that led to death at the ages of 13, 18, 19, and 21 years. The patient’s mother reported that the other affected family members had obvious short stature at that same age, although their records are not available.
Patient 6 is somewhat unusual in that there was no family history of the disease. He presented at 9 weeks of age with dilated cardiomyopathy and a reduced ventricular ejection fraction, and his cardiac function deteriorated rapidly over the course of the next 3 weeks. After five infusions of ERT, a progressive improvement in cardiac function was seen, and the boy was discharged from the hospital. He has received treatment for the past 20 months; however, his ejection fraction worsened after 5 months on ERT and he is now a candidate for cardiac transplantation. The cardiologist has speculated that the patient may have had both viral myocarditis and cardiac disease due to MPS II in the neonatal period. Interestingly, Patient 2 also displayed very early cardiac signs with a mildly dilated left ventricle at 1 month of age. Cardiac involvement is a well-documented, characteristic feature of MPS II, reported for about two-thirds of patients in HOS (Kampmann et al. 2011). Because cardiologists may be unaware of the signs and symptoms of the MPS, it would be worthwhile to include these disorders in listings of the differential diagnoses for cardiomyopathy in the young child.
Patient 7 began treatment at the age of 6 months and has been receiving ERT for about 4 years. Currently at age 4.5 years, his disease manifestations are limited to slightly coarse facies and tapering of the fingers with slightly reduced extension of the digits at the distal interphalangeal joint. Patient 8, who in this case series initiated ERT the latest, at 6.5 months, experienced improvements in uGAG level and normalization of liver size on treatment. His echocardiogram results have remained normal. However, some disease features have shown progression during the 3.5 years that he has been on treatment, including hearing loss, airway disease, slight joint contractures of the upper extremities, and carpal tunnel syndrome. In addition, his gibbus deformity progressed for about a year before stabilizing.
In summary, in our experience the use of ERT in MPS II patients under 1 year of age did not produce any new safety concerns, and all of our patients showed improvements and/or stabilization of some somatic manifestations while on treatment. We note with interest that, in our cohort of early-treated patients, the effectiveness of ERT does not appear to have decreased over time, even after 3–5 years of treatment in some cases. In addition, in some of the cases, caregivers made comparisons with older affected siblings or other affected family members and subjectively reported that the early treated patients experienced a less severe clinical course, consistent with other case reports (Tajima et al. 2013; Tylki-Szymanska et al. 2012). Unfortunately, the limited nature of the medical records available for other family members precluded a more rigorous comparison of phenotypes. Long-term follow-up of these and other MPS II patients who began therapy within the first months of life will provide valuable information on the ability of ERT to possibly prevent or delay the development of certain disease manifestations. Such efforts would be greatly helped by the implementation of newborn screening programs for this disorder (Nakamura et al. 2011).
Acknowledgments
We would like to thank all the patients and families for their participation. Editorial assistance to the authors was provided by Jillian Lokere, MS, of The Curry Rockefeller Group, LLC, Tarrytown, New York, and was funded by Shire. The authors received no payment for their work, and they confirm independence from the funding source.
Abbreviations
- CT
Computed tomography
- ERT
Enzyme replacement therapy
- GAG
Glycosaminoglycan
- HSCT
Hematopoietic stem cell transplantation
- I2S
Iduronate-2-sulfatase
- IRR
Infusion-related reaction
- IV
Intravenous
- LSD
Lysosomal storage disorder
- MPS
Mucopolysaccharidosis
- MRI
Magnetic resonance imaging
- uGAG
Urinary glycosaminoglycan
Synopsis
In a case series of eight MPS II patients treated with idursulfase enzyme replacement therapy started under 1 year of age, treatment was well tolerated and produced some somatic improvements, with no new safety concerns seen.
Compliance with Ethics Guidelines
Individual Contributions
The planning, writing, and content decisions for the manuscript were performed by all the authors equally.
Financial Disclosure
Editorial assistance to the authors was provided by Jillian Lokere, MS, of The Curry Rockefeller Group, LLC, Tarrytown, New York and was funded by Shire. The sponsor played no role in the writing of this report. The authors received no payment for their work. The authors confirm independence from the funding source.
Conflict of Interest Statements
Christina Lampe, M.D., has received grants and/or speaker honoraria and/or research grants from BioMarin Pharmaceutical, and Shire. Andrea Atherton, MS, GCG, has received travel and educational grants and/or speaker honoraria and/or has participated on advisory boards from BioMarin Pharmaceutical, Genzyme Corporation, and Shire. Barbara Burton, M.D., has received funding for the conduct of clinical trials from BioMarin Pharmaceutical, Genzyme Corporation, Shire, and Synageva BioPharma, and consulting fees and/or honoraria from BioMarin Pharmaceutical, Genzyme Corporation, Hyperion Therapeutics, and Shire. Maria Descartes, M.D., has nothing to declare. Roberto Giugliani, M.D., Ph.D., has received travel grants and/or speaker honoraria and/or investigator fees from Actelion Pharmaceuticals, Amicus Therapeutics, BioMarin Pharmaceutical, Genzyme Corporation, and Shire. Dafne D.G. Horovitz, M.D., has received educational travel grants and/or speaker honoraria from BioMarin Pharmaceutical, Genzyme Corporation, and Shire. Sandra Obikawa Kyosen, M.D., has received travel grants from BioMarin Pharmaceutical and Genzyme Corporation. Tatiana S.P.C. Magalhães, M.D., has received travel grants from BioMarin Pharmaceutical, Genzyme Corporation, and Shire. Ana Maria Martins, M.D., has received travel grants and/or speaker honoraria and/or investigator fees from Actelion Pharmaceuticals, BioMarin Pharmaceutical, Genzyme Corporation, and Shire. Nancy J. Mendelsohn, M.D. has received financial reimbursement for travel expenses and honoraria from BioMarin Pharmaceutical and Shire; she has provided consulting support to Genzyme Corporation, and is also engaged in ongoing research projects with BioMarin Pharmaceutical, Genzyme Corporation, and Shire. Joseph Muenzer, M.D., Ph.D., has received travel expense reimbursement and honoraria for speaking from BioMarin Pharmaceutical, Genzyme Corporation, and Shire. He has served on advisory boards and has been a principal investigator for MPS I and MPS II enzyme replacement clinical trials for BioMarin Pharmaceutical, Genzyme Corporation, and Shire. He is currently the principal investigator for a phase I/II intrathecal enzyme replacement clinical trial for the severe form of MPS II sponsored by Shire. Laurie D. Smith, M.D., Ph.D., has served on advisory boards for Shire.
Informed Consent for Identifying Information
Consent to publish identifying information and facial images was obtained from all parents/guardians of patients for whom identifying information is included in this report.
Footnotes
Competing interests: None declared
Authorship Statement: All authors other than the first author contributed equally to this work. Authors are listed in alphabetical order after the first author.
Contributor Information
Christina Lampe, Email: christina_lampe@gmx.de.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Baldo G, Matte U, Artigalas O, et al. Placenta analysis of prenatally diagnosed patients reveals early GAG storage in mucopolysaccharidoses II and VI. Mol Genet Metab. 2011;103:197–198. doi: 10.1016/j.ymgme.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Burton BK, Whiteman DA. Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: a perspective from the Hunter Outcome Survey (HOS) Mol Genet Metab. 2011;103:113–120. doi: 10.1016/j.ymgme.2011.02.018. [DOI] [PubMed] [Google Scholar]
- de Ru MH, Boelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55. doi: 10.1186/1750-1172-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froissart R, Maire I, Millat G, et al. Identification of iduronate sulfatase gene alterations in 70 unrelated Hunter patients. Clin Genet. 1998;53:362–368. doi: 10.1111/j.1399-0004.1998.tb02746.x. [DOI] [PubMed] [Google Scholar]
- Gabrielli O, Clarke LA, Bruni S, Coppa GV. Enzyme-replacement therapy in a 5-month-old boy with attenuated presymptomatic MPS I: 5-year follow-up. Pediatrics. 2010;125:e183–e187. doi: 10.1542/peds.2009-1728. [DOI] [PubMed] [Google Scholar]
- Giugliani R, Hwu P, Tylki-Szymanska A, Whiteman DAH, Pano A. A multicenter, open-label study evaluating safety and clinical outcomes in children (1.4–7.5 years) with Hunter syndrome receiving idursulfase enzyme replacement therapy. Genet Med. 2013 doi: 10.1038/gim.2013.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Almassy Z, Beck M, et al. Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS) J Inherit Metab Dis. 2009;32:534–543. doi: 10.1007/s10545-009-1119-7. [DOI] [PubMed] [Google Scholar]
- Kampmann C, Beck M, Morin I, Loehr JP. Prevalence and characterization of cardiac involvement in Hunter syndrome. J Pediatr. 2011;159(327–331):e322. doi: 10.1016/j.jpeds.2011.01.054. [DOI] [PubMed] [Google Scholar]
- Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome) Pediatrics. 2008;121:e377–e386. doi: 10.1542/peds.2007-1350. [DOI] [PubMed] [Google Scholar]
- McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age–a sibling control study. Clin Genet. 2010;77:492–498. doi: 10.1111/j.1399-0004.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome) Genet Med. 2006;8:465–473. doi: 10.1097/01.gim.0000232477.37660.fb. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Beck M, Eng CM, et al. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124:e1228–e1239. doi: 10.1542/peds.2008-0999. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Beck M, Giugliani R, et al. Idursulfase treatment of Hunter syndrome in children younger than 6 years: results from the Hunter Outcome Survey. Genet Med. 2011;13:102–109. doi: 10.1097/GIM.0b013e318206786f. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Bodamer O, Burton B, et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr. 2012;171:181–188. doi: 10.1007/s00431-011-1606-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Hattori K, Endo F. Newborn screening for lysosomal storage disorders. Am J Med Genet C Semin Med Genet. 2011;157:63–71. doi: 10.1002/ajmg.c.30291. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- Rathmann M, Bunge S, Beck M, Kresse H, Tylki-Szymanska A, Gal A. Mucopolysaccharidosis type II (Hunter syndrome): mutation “hot spots” in the iduronate-2-sulfatase gene. Am J Hum Genet. 1996;59:1202–1209. [PMC free article] [PubMed] [Google Scholar]
- Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi: 10.1186/1750-1172-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Frenking G, Jones SA, Roberts J, Beck M, Wraith JE. Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis. 2011;34:203–208. doi: 10.1007/s10545-010-9215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shire Human Genetic Therapies (2013) Elaprase® (idursulfase) solution for intravenous infusion [prescribing information]. Shire Human Genetic Therapies, Inc., Lexington, MA
- Tajima G, Sakura N, Kosuga M, Okuyama T, Kobayashi M. Effects of idursulfase enzyme replacement therapy for mucopolysaccharidosis type II when started in early infancy: comparison in two siblings. Mol Genet Metab. 2013;108:172–177. doi: 10.1016/j.ymgme.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Tomatsu S, Gutierrez MA, Ishimaru T, et al. Heparan sulfate levels in mucopolysaccharidoses and mucolipidoses. J Inherit Metab Dis. 2005;28:743–757. doi: 10.1007/s10545-005-0069-y. [DOI] [PubMed] [Google Scholar]
- Tylki-Szymanska A, Jurecka A, Zuber Z, Rozdzynska A, Marucha J, Czartoryska B. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: a 3-year follow-up. Acta Paediatr. 2012;101:e42–e47. doi: 10.1111/j.1651-2227.2011.02385.x. [DOI] [PubMed] [Google Scholar]
- Vafiadaki E, Cooper A, Heptinstall LE, Hatton CE, Thornley M, Wraith JE. Mutation analysis in 57 unrelated patients with MPS II (Hunter’s disease) Arch Dis Child. 1998;79:237–241. doi: 10.1136/adc.79.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the Hunter Outcome Survey. Genet Med. 2008;10:508–516. doi: 10.1097/GIM.0b013e31817701e6. [DOI] [PubMed] [Google Scholar]
- Young ID, Harper PS. The natural history of the severe form of Hunter’s syndrome: a study based on 52 cases. Dev Med Child Neurol. 1983;25:481–489. doi: 10.1111/j.1469-8749.1983.tb13794.x. [DOI] [PubMed] [Google Scholar]
- Young ID, Harper PS, Archer IM, Newcombe RG. A clinical and genetic study of Hunter’s syndrome. 1. Heterogeneity. J Med Genet. 1982;19:401–407. doi: 10.1136/jmg.19.6.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ID, Harper PS, Newcombe RG, Archer IM. A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe forms. J Med Genet. 1982;19:408–411. doi: 10.1136/jmg.19.6.408. [DOI] [PMC free article] [PubMed] [Google Scholar]