

Abstract
Background: Nephropathic cystinosis is a rare autosomal recessive disorder caused by mutations in the CTNS gene, encoding for cystinosin, a carrier protein transporting cystine out of lysosomes. Its deficiency leads to cystine accumulation and cell damage in multiple organs, especially in the kidney. In this study, we aimed to provide the first report describing the mutational spectrum of Egyptian patients with nephropathic cystinosis and their genotype–phenotype correlation.
Methods: Fifteen Egyptian patients from 13 unrelated families with infantile nephropathic cystinosis were evaluated clinically, biochemically, and genetically. Screening for the common 57-kb deletion was performed by standard multiplex PCR, followed by direct sequencing of the ten coding exons, exon-intron interfaces, and promoter region.
Results: None of the 15 Egyptian patients had the 57-kb deletion. Twenty-seven mutant alleles and 12 pathogenic mutations were detected including six novel mutations: two frameshift (c.260_261delTT; p.F87SfsX36, c.1032delCinsTG; p.F345CfsX19), one nonsense (c.734G>A; p.W245fsX), two missense (c.1084G>A; pG362R, c.560A>G; p.K187R), and one intronic splicing mutation (IVS3+5g>t). A novel promoter region mutation (1-593-41C>T) seemed to be detected but was excluded as a pathogenic mutation by quantitative real-time PCR analysis.
Conclusions: This study could be the basis for future genetic counseling and prenatal diagnosis of patients with nephropathic cystinosis in Egyptian and surrounding populations. The screening for the 57-kb deletion is not recommended anymore outside its geographical distribution, especially in the region of the Middle East. A common Middle Eastern mutation (c.681G>A; E227E) was pointed out and discussed.
Introduction
Nephropathic cystinosis is one of the leading hereditary causes of renal Fanconi syndrome in children worldwide. It is an autosomal recessive lysosomal storage disorder caused by mutations of the CTNS gene located on chromosome 17p13 (Town et al. 1998). The CTNS gene is made up of 12 exons, the last ten encoding for cystinosin, a protein that facilitates cystine transport out of lysosomes. Defective cystinosin-mediated cystine transport leads to the accumulation and crystallization of cystine in cells of different organs, particularly the kidney, cornea, and thyroid (Nesterova and Gahl 2013). Patients with infantile nephropathic cystinosis (OMIM 219800), comprising about 95% of all cases, usually develop renal Fanconi syndrome in the first year of life and renal failure in the first decade if not treated. Other less common types are the juvenile (OMIM 219900) and the non-nephropathic ocular cystinosis (OMIM 219750) (Nesterova and Gahl 2013).
The aminothiol cysteamine is the only specific therapy for cystinosis so far. It helps in the depletion of cystine from lysosomes through different transporter mechanisms (Butler and Zatz 1984); however, it does not completely stop the progression of the disease and does not restore the lost renal function. Early diagnosis and management of nephropathic cystinosis is of utmost importance to delay renal deterioration (Gahl et al. 1990) and other systems’ affection (Kimonis et al. 1995) and, hence, improves the patient’s survival and quality of life.
The diagnosis of nephropathic cystinosis is confirmed by elevated cystine concentration in white blood cells (WBC), or even better in granulocytes (Levtchenko et al. 2004), which is the diagnostic cornerstone. Molecular analysis of the CTNS gene confirms the diagnosis and offers the advantage of prenatal diagnosis (Nesterova and Gahl 2013). Detection of corneal cystine crystals by a slit lamp examination is another confirmatory sign, but a rather late one as it is reasonably sensitive close to the second year of life (Soliman et al. 2009).
Almost 100 CTNS mutations have been reported so far in the literature (www.hgmd.cf.ac.uk). The most prevalent is the 57-kb deletion removing the first ten exons of the CTNS gene with its upstream sequence. It constitutes approximately 75% of mutated alleles in cystinotic patients from Northern European descent (Touchman et al. 2000). However, CTNS gene mutations have been rarely reported outside Europe and North America.
In this study, we evaluated CTNS mutations in 15 Egyptian pediatric patients with infantile nephropathic cystinosis and commented on their genotype–phenotype correlation. This would be the first report on the mutational spectrum of nephropathic cystinosis in Egypt and in the region of North Africa.
Patients and Methods
Patients
DNA samples of 15 Egyptian nephropathic cystinosis patients (nine males/six females, 3–16 years) from 13 different families of pure Egyptian background were obtained. Patients were recruited from the Center of Pediatric Nephrology and Transplantation (CPNT), Cairo University Children Hospitals, Cairo, Egypt, over the period from November 2010 to June 2012; however, some of them have been diagnosed and treated in the center for several years. DNA was extracted from either EDTA blood by the salting out technique or blood spots on filter paper by QIAamp, DNA mini kit (Qiagen). The study was approved by the institutional review board, and written informed consents were obtained from subjects’ parents/legal guardians.
Methods
Molecular analysis was performed at the laboratory of pediatrics/pediatric nephrology at UZ Leuven, K.U. Leuven. All patients were first screened for the 57-Kb deletion by a standard multiplex PCR technique using LDM1 and D17S829 primer sets as previously described (Heil et al. 2001), and then direct sequencing of the ten coding exons and exon–intron interfaces of the CTNS gene (ENSG00000040531, ENST00000046640) (www.ensembl.org) was performed (ABI 3100, Applied Biosystems). Data were analyzed using SEQUENCE Pilot (JSI Medical Systems). Promoter region sequencing was performed for patients with unidentified mutant alleles (Phornphutkul et al. 2001). Sequences of all used primers are available upon request.
Mutation prediction analysis was performed for newly detected variants using the following pathogenicity software programs: PMut, Mutation Taster, PolyPhen2, Spliceman, BDGP, and Automated Splice Site Analysis.
Total RNA was isolated from leukocytes of patient 2 (probable splicing mutation) and patient 12 (probable promoter mutation) using RNeasy MiniKit (Qiagen). Reverse transcription-PCR (RT-PCR) was performed using Superscript III Reverse-Transcriptase (Life Technologies) according to the manufacturer’s protocol. In patient 2, cDNA amplification for the CTNS gene was performed compared to a healthy control using the primers previously described [forward: 5′-CCTCTTCCAGTAACATTGAGG-3′ and reverse 5′-CGCGTGCAGGCTGAAGAAGA-3′] (AlcÄntara-Ortigoza et al. 2008) crossing the exonic boundaries and giving a product extending from exon 2 to exon 9 (722 bp in normal individuals); the resulting amplification product was directly sequenced.
In patient 12, cDNA was used for quantitative real-time PCR (qPCR) (Rotor-Gene Q, Qiagen) together with platinum Sybr Green PCR mix (Life Technologies), CTNS cDNA primers mentioned above, and primers for GAPDH as a reference gene. Three healthy control samples were assayed in the same run by qPCR. The patient and controls were assayed each in triplicate. Cycling parameters were as follows: 95°C, 30 s; 60°C, 60 s; 72°C, 60 s for 40 cycles; and then extension at 72°C for 5 min. Resulting data were expressed as percent of reduction in gene expression compared to normal controls after normalization with GAPDH using the 2-ΔΔCT method (Livak and Schmittgen 2001).
Cystine assay in WBC was recently established and performed at the Inherited Metabolic Disorder Laboratory (IMDL), Center of Social and Preventive Medicine (CSPM), Cairo University Children Hospitals. Cystine assay was performed by LC-MS/MS (Micromass, Waters), as previously described (Chabli et al. 2007), using the internal standard d,l-cystine-2,2′,3,3,3′,3′-d6 (C/D/N isotopes), L-cystine calibration curve, and a 3.0 x 50 mm, 3.5 μ particle size Xterra C18 HPLC column.
Results
Fifteen Egyptian patients with infantile nephropathic cystinosis were evaluated clinically, biochemically, and genetically. Consanguinity was reported in ten out of 13 families, and corneal cystine crystals were detectable in all patients. Age at presentation ranged from 4 to 12 months, and age at diagnosis ranged from 5 months to 9 years. Patients were already on cysteamine therapy at the time of sampling for a period ranging from 2 to 84 months, on a dosage ranging from 15 to 45 mg/kg/day. All patients were below the 3rd percentiles for weight and height in their age groups. One patient died (patient 8) at the age of 8 years before completion of the study. One patient underwent renal transplantation (patient 4) at the age of 14 years. Three patients have ESRD, and four patients have hypothyroidism and are on L-thyroxin replacement therapy. Cystine levels in WBC ranged from 1.5 to 15.3 nmol ½ cystine/mg proteins (Table 1).
Table 1.
Clinical features of Egyptian patients with nephropathic cystinosis
| Proband | Age in years | Sex | Onset of symptoms in months | Age at diagnosis in months | F.S | M.A | R | L.D | HT | SDS Weight/Hight | Creatinine (mg/dl) at diagnosis/last visit | Cysteamine (mg/kg/day) | WBC Cystine (nmol ½ cys/mg Protein) | Remarks |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | M | 6 | 20 | + | − | + | + | − | −2.8/-5.5 | 1.0/1.3 | 30 | 1.7 | |
| 2 | 5 | F | 8 | 14 | + | + | + | − | − | −1.7/−2.8 | 0.3/1.2 | 30 | 3.8 | |
| 3 | 3 | M | 9 | 22 | + | − | + | − | − | −2.9/−3.6 | 0.5/0.7 | 30 | 5.1 | |
| 4 | 15 | F | 12 | 108 | − | − | - | + | − | −3.7/−8.2 | 4.8/0.7 | 25 | 4.0 | RTX one year ago and on GH |
| 5a | 16 | F | 6 | 60 | + | + | + | − | + | −4.5/−7.6 | 2.7/6.1 | 25 | 4.9 | On regular HD |
| 5b | 6 | M | 5 | 6 | + | − | + | − | − | −2.3/−5.0 | 0.7/0.6 | 30 | 10.6 | |
| 6 | 7.5 | M | 5 | 9 | + | − | + | - | − | −3.5/−6.8 | 0.6/0.9 | 30 | 1.5 | |
| 7 | 5 | M | 6 | 14 | + | + | + | − | − | −2.9/−5.4 | 1.7/4.9 | 40 | 9.9 | On regular HD and prepared for RTX |
| 8 | 8 | M | 6 | 9 | + | + | + | - | - | −2.7/−5.4 | 0.8/5.6 | – | – | Deceased |
| 9 | 4 | M | 7 | 15 | + | − | + | + | − | −2.7/−3.7 | 0.4/0.3 | 35 | 4.7 | |
| 10 | 4.5 | F | 10 | 30 | + | − | − | − | − | – | – | 30 | – | |
| 11a | 11 | F | 5 | 30 | + | + | + | − | + | −2.7/−5.0 | 0.7/5.6 | 15 | – | On regular HD |
| 11b | 6.5 | F | 4 | 5 | + | + | + | − | + | −2.9/−8.5 | 0.3/1.5 | 45 | – | |
| 12 | 3.5 | M | 8 | 14 | + | + | + | − | − | −4.1/−3.9 | 0.5/0.6 | 45 | 15.4 | |
| 13 | 4 | M | 6 | 36 | + | + | + | + | + | −4.0/−5.9 | 1.4/1.7 | 45 | 7.5 |
F.S Fanconi syndrome, GH Growth hormone, HD Hemodialysis, HT Hypothyroidism, L.D Limb deformity, M.A Metabolic acidosis, R Rickets, RTX Renal transplantation, SDS Standardized score, Families 5 and 11 have 2 siblings each (a) and (b)
None of the 15 Egyptian patients had the common European 57-Kb deletion. Twelve pathogenic mutations were identified representing 27 discovered mutant alleles (Table 2). Ten out of 15 patients had homozygous mutations. Six previously reported CTNS mutations were detected in our study: c.829dup; p.T277NfsX19 (Besouw et al. 2012), c.922G>A; G308R (Shotelersuk et al. 1998), 809_811delCCT; p.S270del (Attard et al. 1999), c.15G>A; p.W5X (Kalatzis et al. 2002), c.681G>A; E227E (Aldahmesh et al. 2009), and c.1015G>A; p.G339R (Shotelersuk et al. 1998). All these mutations were previously associated with infantile nephropathic cystinosis in various populations. Figure 1 presents the worldwide geographical distribution of the previously reported CTNS mutations detected in Egyptian patients.
Table 2.
Genotype of Egyptian patients with nephropathic cystinosis
| Proband | Mutant allele1 | Mutant allele2 | Location | Protein effects | Consequence of mutation | Reference |
|---|---|---|---|---|---|---|
| 1 | c.829dup | c.829dup | Exon 10 | p.T277NfsX19 | Truncated protein at AA296 | (Besouw et al 2012) |
| 2 | IVS3+5g>t | IVS3+5g>t | Intron 3 | – | Skipping of exon 3 | This study |
| 3 | c.922G>A | c.922G>A | Exon 11 | G308R | AA change at TM6 | (Shotelersuk et al 1998) |
| 4 | 809_811del | 809_811del | Exon 10 | p.S270del | AA deleted from TM5 | (Attard et al 1999) |
| 5a,5b | c.829dup | c.829dup | Exon 10 | p.T277NfsX19 | Truncated protein at AA296 | (Besouw et al 2012) |
| 6 | c.15G>A | c.15G>A | Exon 3 | p.W5X | Truncated protein at AA5 | (Kalatzis et al 2002) |
| 7 | c.681G>A | c.681G>A | Exon 9 | E227E | Alternative splicing | (Aldahmesh et al 2009) |
| 8 | 260_261delTT | c.560A>G | Exons 6 and 8 | p.F87SfsX36, p.K187R | Truncated protein at AA123, Alternative splicing | This study |
| 9 | c.1015G>A | c.1015G>A | Exon 12 | p.G339R | AA change at TM7 | (Shotelersuk et al 1998) |
| 10 | c.1084G>A | ND | Exon 12 | pG362R | AA change at cytosolic LTM | This study |
| 11a,11b | c.734G>A | c.1032delCinsTG | Exons 10 and 12 | p.W245fsX, p.F345CfsX19 | Truncated protein at AA245, Truncated protein at AA364 | This study |
| 12 | ND | ND | – | – | – | – |
| 13 | c.829dup | c.829dup | Exon 10 | p.T277NfsX19 | Truncated protein at AA296 | (Besouw et al 2012) |
AA, Amino acid; LTM, Lysosomal targeting motif; ND, Not detected; TM, Transmembrane domain; Families 5 and 11 have 2 siblings each (a) and (b).
Fig. 1.
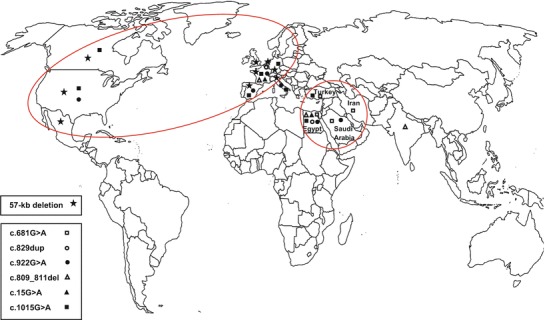
Worldwide geographical distribution of 57-kb deletion and previously reported CTNS mutations detected in the Egyptian population. Oval: geographical distribution of 57-kb deletion. Circle: geographical distribution of the Middle Eastern mutation c.681G>A
Newly detected CTNS variants include two frameshift mutations (c.260_261del; p.F87SfsX36 and c.1032delCinsTG; p.F345CfsX19), one nonsense mutation (c.734G>A; p.W245fsX), two missense mutations (c.1084G>A; pG362R and c.560A>G; p.K187R), and one intronic splicing mutation (IVS3+5g>t) (Fig. 2). A novel promoter region variant was also detected in a homozygous state in patient 12 (1-593-41C>T) corresponding to the important specificity protein-1 (Sp-1) binding motif (GGCGGCG) that was reported to extend from 1-593-46 to 1-593-40 in the CTNS promoter area (Phornphutkul et al. 2001) (Fig. 2).
Fig. 2.
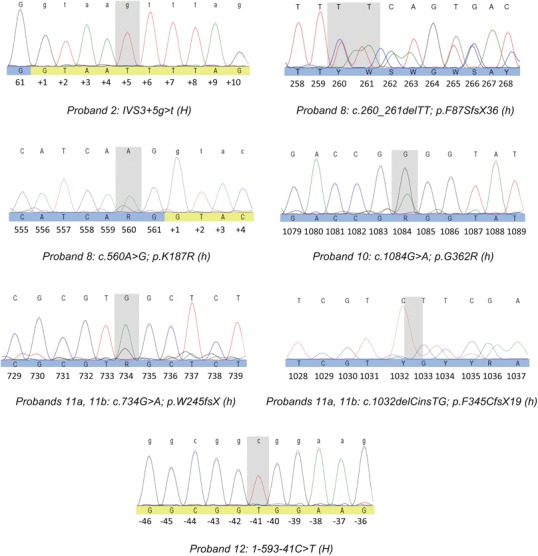
Newly discovered CTNS variants in Egyptian patients. H homozygous, h heterozygous
The newly detected variants were tested with different predictive software programs. Table 3 provides a summary for all prediction results. The frameshift and the nonsense variants led to truncated proteins and were easily predicted pathogenic. c.1084G>A in patient 10 replacing glycine with arginine at position 362, which is a highly conserved residue among different species, was also detected as being pathogenic. The other missense variant (c.560A>G) in patient 8 replacing lysine with arginine at position 187 was expected to be neutral based on the amino acid change; however, this mutation occurs at the second base before exon–intron 8 interface (Fig. 2) and could be an exonic splicing mutation as expected by three splicing evaluation software programs. The intronic mutation (IVS35g>t) in patient 2 was more strongly suspected to have a splicing damaging effect during transcription (Table 3).
Table 3.
Predicted effects of new CTNS variants
| Variant | Prediction | ||||||
|---|---|---|---|---|---|---|---|
| Deletions or insertions | |||||||
| c.260-261delTT; p.F87SfsX36 | Frame shift | ||||||
| c.1032delCinsTG; p.F345CfsX19 | Frame shift | ||||||
| Nonsense | |||||||
| c.734G>A; p.W245fsX | Nonsense | ||||||
| Missense | PMut (AA change) | Mutation taster (AA change/splice) | PolyPhen2 (AA change) | ||||
| NN output | Reliability | Result | Probability | Result | Score | Result | |
| c.1084G>A; p.G362R | 0.8395 | 6 | Pathogenic | 0.999 | Pathogenic | 0.647 | Possibly damaging |
| c.560A>Ga; p.K187R | 0.0145 | 9 | Neutral | 0.952 | Pathogenic | 0.002 | Benign |
| Probable splicing variants | Spliceman (splice) | BDGP (splice) | Automated splice site analysis (splice) | ||||
| Ranking | Result | Score | Result | Final/initial binding | Result | ||
| IVS3+5g>t | 0.85 | Pathogenic | 1.0>0.67 | Pathogenic | 6.6% | Pathogenic | |
| c.560A>Ga | 0.55 | Pathogenic | 1.0>0.99 | Neutral | 20.4% | Pathogenic | |
AA Amino acid
PMut: http://mmb2.pcb.ub.es:8080/PMut/; Mutation taster: www.mutationtaster.org/; PolyPhen2: genetics.bwh.harvard.edu/pph2/; Spliceman: fairbrother.biomed.brown.edu/spliceman/; BDGP: www.fruitfly.org/seq_tools/splice.html; Automated splice site analysis: https://splice.uwo.ca/
aA missense mutation and a probable splicing mutation
The splicing mutation (IVS3+5g>t) was confirmed by demonstrating the skipping of exon 3 evidenced by sequence analysis after RT-PCR (Fig. 3). On the other hand, the promoter region mutation in patient 12 (1-593-41C>T) led to a modest reduction (27 ± 6.5%) in CTNS RNA gene expression after normalization to GAPDH and comparison with three healthy controls. To our regret, RNA samples from patient 8 (c.560A>G) or his parents were unobtainable.
Fig. 3.
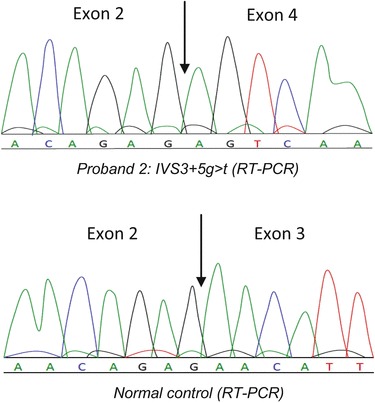
RT-PCR/sequence analysis of the mutation (IVS3+5g>t) of patient 2 in which skipping of exon 3 of the CTNS gene is evident compared to a healthy individual
Discussion
We here present the first report on the mutational spectrum of Egyptian patients with nephropathic cystinosis. Thirteen unrelated families and 15 patients representing a wide geographical distribution within Egypt were screened for the CTNS gene mutations.
Most of the Egyptian patients had a severe phenotype as evident in Table 1, but this phenotypic severity could be attributed to many factors. The CTNS genotype is definitely an important one (Attard et al. 1999); however, delayed diagnosis in many of our patients, underdosage due to financial or logistical problems, and differences in response to cysteamine therapy may all be implicated (Soliman et al. 2013).
In our series of patients, growth retardation was a striking feature in almost all patients; however, more severe cases were usually associated with truncating mutations (Table 2). The recent introduction of growth hormone in the therapeutic panel of Egyptian nephropathic cystinosis patients hopefully will improve the overall growth pattern. Although all therapeutic measures were taken to oppose the effects of hypocalcemia and hypophosphatemia, still most of our patients are complaining of rickets and some already show skeletal deformities at relatively young ages. The only patient who underwent renal transplantation (patient 4) is doing well with normally functioning graft and is showing increased annual height velocity post transplantation. Not surprisingly though, she is still suffering remarkable growth retardation and residual genu valgum given the significantly late diagnosis in this particular patient (at 9 years of age) and the consequent therapeutic delay.
The detection of six novel mutations and six mutations previously reported in 13 families denotes the marked genetic heterogeneity of Egyptian patients with nephropathic cystinosis, which is different from many populations in which a single mutation may constitute over 50% of causative mutations. Also the high rate of homozygous mutations in our study (67%) sheds light on the gravity of the role of consanguineous marriage in elevating the incidence of nephropathic cystinosis and other autosomal recessive disorders in Egypt and in other Arab countries with similar and even higher rates of consanguineous marriages (Tadmouri et al. 2009).
The novel mutations found in this study include the frameshift mutation (c.260_261delTT; p.F87SfsX36) detected in patient 8, converting phenylalanine at position 87 to serine and resulting in a truncated protein at AA 123, completely abolishing the protein function. Likewise, the other novel frameshift mutations detected in sisters 11a and 11b (c.1032delCinsTG; p.F345CfsX19) resulted in the conversion of phenylalanine to cysteine at position 345 and a truncated protein at AA 364, disrupting the seventh and final transmembrane domain of cystinosin (Attard et al. 1999). The nonsense mutation (c.734G>A; p.W245fsX) also detected in patients 11a and 11b led to the conversion of tryptophan at position 245 into a stop codon (TAG), thus immediately stopping the protein translation.
The missense mutation (c.1084G>A; pG362R) detected in patient 10 is the first reported mutation in the lysosomal targeting motif of cystinosin (GYDQL) extending from AA 362 to 366. This mutation replaced the highly conserved glycine at position 362 at the C-terminus of the protein with the basic amino acid arginine. This charge alteration is expected to disrupt the cytosolic lysosomal targeting motif (Attard et al. 1999). Although previous intentional site-directed mutagenesis of C-terminus lysosomal targeting motif led to the redirection of most of cystinosin to the plasma membrane, still partial localization to lysosomes occurred, denoting the presence of a second lysosomal targeting signal, which was identified in the third cytoplasmic loop (YFPQA) extending from AA 281 to 285 (Cherqui et al. 2001). This could be the reason of the relatively milder phenotype of patient 10 (no metabolic acidosis, rickets, limb deformity, or hypothyroidism) (Table1).
The other missense mutation was detected in patient 8 (c.560A>G; p.K187R), and although exonic, it is highly likely to cause alternative splicing, as it was detected as being pathogenic by three different splicing software programs: Mutation Taster, Spliceman, and Automated Splice Site Analysis (Table 3). Unfortunately, the patient with this mutation died recently at the age of 8 years from intracranial hemorrhage complicating hemodialysis before we could confirm the splicing nature of his mutation.
The intronic splice site mutation (IVS3+5g>t) detected in patient 2 led to the skipping of exon 3 after RT-PCR/sequencing analysis as the similar splicing mutation at the same position (IVS3+5g>a), which was also demonstrated to lead to exon 3 skipping in a Mexican patient with nephropathic cystinosis (AlcÄntara-Ortigoza et al. 2008), confirming the importance of the guanine base at the IVS+5 position for CTNS exon 3 transcription.
Promoter region mutations reported in the CTNS gene are rare. Only three different mutations were reported in eight patients (Phornphutkul et al. 2001, Mason et al. 2003). The most commonly reported is the mutation causing infantile type (1-593-42G>C) which was discovered in six patients, one from the USA and five from Italy. The other two reported mutations (1-593-50G>T and 1-593-50insT) resulted in ocular cystinotic phenotypes. All three mutations were located at or near a highly critical region in the promoter area corresponding to the Sp-1 binding motif essential for gene transcription (Phornphutkul et al. 2001). We here report the fourth promoter region mutation at the Sp-1 binding motif in the CTNS gene (1-593-41C>T) detected in patient 12 in our study in a homozygous state. qPCR analysis of this patient’s RNA sample showed only a modest reduction in CTNS gene expression when compared to three control individuals (27 ± 6.5 %). This indicates that this variant is not the mutation responsible for the patient phenotype (Table 1); however, reducing CTNS gene expression in the first place is also indicative of the importance of the Sp-1 binding motif for CTNS gene transcription. Caution during the interpretation of promoter region mutations is highly recommended.
In addition to the 13 Egyptian cystinotic families, the absence of the 57-kb deletion has been previously reported in the region of the Middle East in 13 families from Saudi Arabia (Aldahmesh et al. 2009), 10 from Turkey (Topaloglu et al. 2012), and 24 from southwestern Iran (Shahkarami et al. 2013). A single study in the Far East (Thailand) also reported the absence of the 57-kb mutant allele in six patients of Thai and Cambodian origins (Yeetong et al. 2012). Apparently, this common mutation is restricted to the Northern European/American populations and, to a lesser extent, to countries of possible genetic contact as in Italy (Mason et al. 2003) and Mexico (AlcÄntara-Ortigoza et al. 2008). This supports the theory that this founder mutation originated very recently during human evolution, perhaps less than 2,000 years ago somewhere in Northern Europe (Kalatzis and Antignac 2002); so it has not got the chance to spread to remote ethnicities. Based on these observations, we do not recommend anymore the routine screening for the 57-kb deletion before CTNS sequencing in populations outside its geographical distribution, at least in the region of the Middle East.
The most common mutation detected in Egyptian patients is c.829dup; p.T277NfsX19, discovered in a homozygous state in four patients belonging to three unrelated families (6/26 or 23.1% of Egyptian familial mutant alleles). This mutation is a frameshift mutation starting at codon 277, leading eventually to a truncated protein at amino acid 296. It was associated with increased creatinine levels in two Egyptian unrelated patients (1 and 13) at the time of diagnosis (2 and 3 years, respectively), and the latter patient is already complaining of hypothyroidism at 4 years (Table 1). This mutation has been reported only once before in a heterozygous state in a European patient (Besouw et al. 2012). It is apparently a hot spot in Egypt and could be the focus for further investigations. Apart from this mutation, all other detected CTNS mutations in Egyptian patients were only present in a single family.
In similar studies in the Middle East, only one mutation was detected in all populations. This mutation was the exonic splice site mutation c.681G>A; p.E227E, completely abolishing the adjacent donor site at exon 9 and replacing it with a cryptic donor site (Aldahmesh et al. 2009). It comprises 39.5% of Iranian (Shahkarami et al. 2013), 20% of Turkish (Topaloglu et al. 2012), and 15.4% of Saudi familial mutant alleles (Aldahmesh et al. 2009). This mutation was not detected previously in European or American populations (a pure Middle Eastern mutation), and it was present in the Egyptian population in our study in a homozygous state in a single patient (7.7% of familial mutant alleles); so its prevalence decreases gradually upon heading to the west (Fig. 1). This could suggest that the origin of this founder mutation is Iran or perhaps a place further to the east.
Other previously reported CTNS mutations detected in our study include c.15G>A; p.W5X, which is a nonsense mutation leading to truncated protein at AA 5 with complete loss of function. c.922G>A; p.G308R and c.1015G>A; p.G339R are two missense mutations that were tested on the level of cystinosin protein carrier function and led to absent carrier capability (Kalatzis et al. 2004). Likewise, the small deletion c.809_811del; p.S270del was tested functionally and resulted in almost complete loss of function (Kalatzis et al. 2004). The most widely distributed among the six previously reported mutations in Egyptian patients was c.1015G>A; p.G339R, as it was reported in Turkey (Topaloglu et al. 2012), Italy (Mason et al. 2003), Germany (Kiehntopf et al. 2002), France (Attard et al. 1999), Spain (Macías-Vidal et al. 2009), the USA (Shotelersuk et al. 1998), and Canada (Rupar et al. 2001), followed by c.922G>A; p.G308R reported in Saudi Arabia (Aldahmesh et al. 2009), Italy (Mason et al. 2003), France (Attard et al. 1999), Spain (Macías-Vidal et al. 2009), and the USA (Shotelersuk et al. 1998). The mutation c.809_811del; p.S270del was also relatively widespread, as it was reported in France (Attard et al. 1999) and India (Tang et al. 2009) (Fig. 1).
The most common Egyptian mutation (c.829dup; p.T277NfsX19) was completely absent in other studies from the Middle East. Likewise, the most common mutation in the Saudi population (1013 T>G; L338R), representing 34.6 % of familial mutant alleles, was not detected in the three surrounding populations, and apart from the founder mutation (c.681G>A; E227E), there were no other mutations detected in common among the Saudi, Turkish, and Iranian patients. This is quite remarkable considering the long history of commercial relations, invasions, and genetic contact between these four close countries over the last few thousand years.
The newly detected mutations in our study may not be restricted to the Egyptian population, as the cystinotic genotypes of most Arab and all African populations are still largely obscure. However, our study helps in the understanding of the genetic basis of this orphan disease and its correlation with phenotypic features in this part of the world where consanguinity is the rule, not the exception. It also paves the way for future family counseling and prenatal diagnosis in the region.
Acknowledgments
We would like to thank our patients and their parents for their kind collaboration. This work was supported by a grant from the Cystinosis Research Network (CRN) to Neveen A Soliman and Elena Levtchenko in 2012. Elena Levtchenko was supported by the Fund for Scientific Research, Flanders (F.W.O. Vlaanderen), grant 1801110 N.
Synopsis
This study summarizes the Egyptian experience in the diagnosis and management of patients with nephropathic cystinosis over the past few years and reports six novel and six recurrent mutations in the CTNS gene.
Compliance with Ethics Guidelines
Conflict of Interest
Neveen A. Soliman, Mohamed A. Elmonem, Lambertus van den Heuvel, Rehab H. Abdel Hamid, Mohamed Gamal, Inge Bongaers, Sandrine Marie, and Elena Levtchenko declare that they have no conflict of interest regarding the contents of the current study.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consents were obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
Neveen A Soliman was involved in the conception and design of the study, recruitment and management of patients, interpretation of data, and drafting the manuscript.
Mohamed A Elmonem was involved in the conception and design of the study, biochemical and genetic analysis of patients, interpretation of data, and drafting the manuscript.
Lambertus van den Heuvel was involved in the conception and design of the study, genetic analysis of patients, interpretation of data, and revising the manuscript critically for intellectual content.
Rehab H. Abdel Hamid was involved in the design of the study, recruitment and management of patients, interpretation of data, and revising the manuscript critically for intellectual content.
Mohamed Gamal was involved in the design of the study, recruitment and management of patients, interpretation of data, and revising the manuscript critically for intellectual content.
Inge Bongaers was involved in the design of the study, genetic analysis of patients, interpretation of data, and revising the manuscript critically for intellectual content.
Sandrine Marie was involved in the design of the study, biochemical analysis of patients, interpretation of data, and revising the manuscript critically for intellectual content.
Elena Levtchenko was involved in the conception and design of the study, genetic analysis of patients, interpretation of data, and revising the manuscript critically for intellectual content.
Footnotes
Competing interests: None declared
Neveen A. Soliman and Mohamed A. Elmonem contributed equally to this study
Contributor Information
Mohamed A. Elmonem, Email: mohamed.abdelmonem@kasralainy.edu.eg
Collaborators: Johannes Zschocke and K Michael Gibson
References
- AlcÄntara-Ortigoza MA, Belmont-Martínez L, Vela-Amieva M, GonzÄlez-Del Angel A. Analysis of the CTNS gene in nephropathic cystinosis Mexican patients: report of four novel mutations and identification of a false positive 57-kb deletion genotype with LDM-2/exon 4 multiplex PCR assay. Genet Test. 2008;12:409–414. doi: 10.1089/gte.2008.0014. [DOI] [PubMed] [Google Scholar]
- Aldahmesh MA, Humeidan A, Almojalli HA, et al. Characterization of CTNS mutations in Arab patients with cystinosis. Ophthalmic Genet. 2009;30:185–189. doi: 10.3109/13816810903200953. [DOI] [PubMed] [Google Scholar]
- Attard M, Jean G, et al. Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum Mol Genet. 1999;8:2507–2514. doi: 10.1093/hmg/8.13.2507. [DOI] [PubMed] [Google Scholar]
- Besouw MT, Van Dyck M, Francois I, Van Hoyweghen E, Levtchenko EN. Detailed studies of growth hormone secretion in cystinosis patients. Pediatr Nephrol. 2012;27:2123–2127. doi: 10.1007/s00467-012-2213-x. [DOI] [PubMed] [Google Scholar]
- Butler JD, Zatz M. Pantethine and cysteamine deplete cystine from cystinotic fibroblasts via efflux of cysteamine-cysteine mixed disulfide. J Clin Invest. 1984;74:411–416. doi: 10.1172/JCI111436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabli A, Aupetit J, Raehm M, Ricquier D, Chadefaux-Vekemans B. Measurement of cystine in granulocytes using liquid chromatography-tandem mass spectrometry. Clin Biochem. 2007;40:692–698. doi: 10.1016/j.clinbiochem.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Cherqui S, Kalatzis V, Trugnan G, Antignac C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J Biol Chem. 2001;20(276):13314–13321. doi: 10.1074/jbc.M010562200. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Schneider JA, Schulman JD, Thoene JG, Reed GF. Predicted reciprocal serum creatinine at age 10 years as a measure of renal function in children with nephropathic cystinosis treated with oral cysteamine. Pediatr Nephrol. 1990;4:129–135. doi: 10.1007/BF00858823. [DOI] [PubMed] [Google Scholar]
- Heil SG, Levtchenko E, Monnens LA, Trijbels FJ, Van der Put NM, Blom HJ. The molecular basis of Dutch infantile nephropathic cystinosis. Nephron. 2001;89:50–55. doi: 10.1159/000046043. [DOI] [PubMed] [Google Scholar]
- Kalatzis V, Antignac C. Cystinosis: from gene to disease. Nephrol Dial Transplant. 2002;17:1883–1886. doi: 10.1093/ndt/17.11.1883. [DOI] [PubMed] [Google Scholar]
- Kalatzis V, Cohen-Solal L, Cordier B, et al. Identification of 14 novel CTNS mutations and characterization of seven splice site mutations associated with cystinosis. Hum Mutat. 2002;20:439–446. doi: 10.1002/humu.10141. [DOI] [PubMed] [Google Scholar]
- Kalatzis V, Nevo N, Cherqui S, Gasnier B, Antignac C. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet. 2004;13:1361–1371. doi: 10.1093/hmg/ddh152. [DOI] [PubMed] [Google Scholar]
- Kiehntopf M, Schickel J, Gönne B, et al. Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis. Hum Mutat. 2002;20:237–244. doi: 10.1002/humu.9063. [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Troendle J, Rose SR, Yang ML, Markello TC, Gahl WA. Effects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosis. J Clin Endocrinol Metab. 1995;80:3257–3261. doi: 10.1210/jcem.80.11.7593434. [DOI] [PubMed] [Google Scholar]
- Levtchenko E, de Graaf-Hess A, Wilmer M, van den Heuvel L, Monnens L, Blom H. Comparison of cystine determination in mixed leukocytes vs PMN leukocytes for diagnosis of cystinosis and monitoring of cysteamine therapy. Clin Chem. 2004;50:1686–1688. doi: 10.1373/clinchem.2004.031872. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Macías-Vidal J, Rodés M, HernÄndez-Pérez JM, Vilaseca MA, Coll MJ. Analysis of the CTNS gene in 32 cystinosis patients from Spain. Clin Genet. 2009;76:486–489. doi: 10.1111/j.1399-0004.2009.01222.x. [DOI] [PubMed] [Google Scholar]
- Mason S, Pepe G, Dall'Amico R, et al. Mutational spectrum of the CTNS gene in Italy. Eur J Hum Genet. 2003;11:503–508. doi: 10.1038/sj.ejhg.5200993. [DOI] [PubMed] [Google Scholar]
- Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol. 2013;28:51–59. doi: 10.1007/s00467-012-2242-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phornphutkul C, Anikster Y, Huizing M, et al. The promoter of a lysosomal membrane transporter gene, CTNS, binds Sp-1, shares sequences with the promoter of an adjacent gene, CARKL, and causes cystinosis if mutated in a critical region. Am J Hum Genet. 2001;69:712–721. doi: 10.1086/323484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupar CA, Matsell D, Surry S, Siu V. A G339R mutation in the CTNS gene is a common cause of nephropathic cystinosis in the south western Ontario Amish Mennonite population. J Med Genet. 2001;38:615–616. doi: 10.1136/jmg.38.9.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahkarami S, Galehdari H, Ahmadzadeh A, Babaahmadi M, Pedram M. The first Molecular genetics analysis of individuals suffering from nephropatic cystinosis in the Southwestern Iran. Nefrologia. 2013;33:308–315. doi: 10.3265/Nefrologia.pre2012.Sep.11558. [DOI] [PubMed] [Google Scholar]
- Shotelersuk V, Larson D, et al. CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet. 1998;63:1352–1362. doi: 10.1086/302118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman NA, El-Baroudy R, Rizk A, Bazaraa H, Younan A. Nephropathic cystinosis in children: an overlooked disease. Saudi J Kidney Dis Transpl. 2009;20:436–442. [PubMed] [Google Scholar]
- Soliman NA, Bazaraa HM, Abdel Hamid RH, Badawi N. Nephropathic cystinosis in a developing country: the Egyptian experience. Saudi J Kidney Dis Transpl. 2013;24:147–149. doi: 10.4103/1319-2442.106315. [DOI] [PubMed] [Google Scholar]
- Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;8:6–17. doi: 10.1186/1742-4755-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Danda S, Zoleikhaeian M, Simon M, Huang T. An Indian boy with nephropathic cystinosis: a case report and molecular analysis of CTNS mutation. Genet Test Mol Biomarkers. 2009;13:435–438. doi: 10.1089/gtmb.2008.0156. [DOI] [PubMed] [Google Scholar]
- Topaloglu R, Vilboux T, Coskun T, et al. Genetic basis of cystinosis in Turkish patients: a single-center experience. Pediatr Nephrol. 2012;27:115–121. doi: 10.1007/s00467-011-1942-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchman JW, Anikster Y, Dietrich NL, et al. The genomic region encompassing the nephropathic cystinosis gene (CTNS): complete sequencing of a 200-kb segment and discovery of a novel gene within the common cystinosis-causing deletion. Genome Res. 2000;10:165–173. doi: 10.1101/gr.10.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- Yeetong P, Tongkobpetch S, Kingwatanakul P, et al. Two novel CTNS mutations in cystinosis patients in Thailand. Gene. 2012;499:323–325. doi: 10.1016/j.gene.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]