

Abstract
We report a patient with Leigh syndrome shown to have two previously undescribed truncating mutations in the TTC19 gene. Our patient is a 4-year-old boy with global developmental delay, language regression at 13 months, and brain MRI showing T2 high-signal lesions involving the putamen, caudate body, and the brainstem, which appear to be progressing. Molecular testing showed our patient is heterozygous for two previously undescribed mutations in the TTC19 gene, c.577G>A (p.Trp186Stop) and c.964_967delGGCT (p.Gly322MetfsX8), both of which are predicted to cause loss of protein function due to either protein truncation or nonsense-mediated mRNA decay. TTC19 encodes tetratricopeptide 19 (TTC19) and is thought to be a complex III (CIII) assembly factor that is embedded on the inner mitochondrial membrane as part of two high-molecular-weight complexes, one of which coincides with CIII. The initial presentations of previously described patients with TTC19 mutations are heterogeneous and can be from childhood to adulthood. In summary, TTC19 mutations have been shown to affect CIII complex function, which results in a heterogeneous clinical phenotype including Leigh syndrome.
Introduction
TTC19 encodes tetratricopeptide 19 (TTC19) and is thought to be a CIII assembly factor that is embedded on the inner mitochondrial membrane as part of two high-molecular-weight complexes, one of which coincides with CIII. This complex transfers electrons from coenzyme Q to cytochrome c, which is critical in generating the mitochondrial electrochemical potential. TTC19 mutations were first shown to be detrimental to mitochondrial complex III (CIII) function in 2011 (Ghezzi et al. 2011).
Since that time, there have been at least five cases documented in the medical literature as having truncating mutations in the TTC19 gene (See Table 1) (Ghezzi et al. 2011; Balasubramaniam et al. 2012). The age of presentation is broad, with our patient presenting at age four with global developmental delay, language regression at 13 months of age, and brain MRI abnormalities consistent with Leigh syndrome, and another patient presenting at age 42 with subacute rapid neurological failure with death occurring 2 years later (Ghezzi et al. 2011). All mutations in the six patients currently identified are homozygous nonsense-truncating mutations. Additional biochemical testing performed in some patients has identified isolated CIII deficiency (Ghezzi et al. 2011).
Table 1.
Clinical presentation patients with TTC19 mutations
| Age of presentation (years) | Ethnic origin | Symptoms | Muscle biopsy | Functional status | Reference | |
|---|---|---|---|---|---|---|
| Patient 1 | 5 | Italian | ID, ataxia | Isolated CIII defect | Wheelchair bound (37 years) | (Ghezzi et al. 2011) |
| Patient 2 | 10 | Italian | ID, ataxia | Isolated CIII defect | Bedridden (26 years) | (Ghezzi et al. 2011) |
| Patient 3 | 5 | Italian | Psychomotor regression, ataxia | Isolated CIII defect | Bedridden (19 years) | (Ghezzi et al. 2011) |
| Patient 4 | 42 | Italian | Subacute rapid neurological failure | Isolated CIII defect | Deceased (45 years) | (Ghezzi et al. 2011) |
| Patient 5 | 8 | Iraqi | Encephalomyopathy | Isolated CIII defect | Unavailable | (Balasubramaniam et al. 2012) |
| Patient 6 | 1 | Hispanic | DD, language regression | Reduction in complex I + III (79 %), II (52 %), II + III (36 %) and IV (46 %) activities | DD, ataxia | N/A |
ID intellectual disability; DD developmental delay
Our patient is a 4-year-old Hispanic boy born to a 33-year-old gravida 7 para 3 mother with global developmental delay, language regression at 13 months, and brain MRI showing T2 high-signal lesions involving the putamen, caudate body, and the brainstem, which appear to be progressing (Fig. 1). He was considered a well infant until his parents became concerned at 7–8 months of age for developmental delay in his gross motor skills due to him not yet sitting independently. Between the age of one and two, he suffered language regression with loss of a 19-word vocabulary at 13 months of age, with subsequent MRI suggestive of mitochondrial disease, particularly Leigh syndrome. A Leigh syndrome panel was sent, which showed two novel mutations in the TTC19 gene. Additionally, biochemical analysis of the electron transport chain (ETC) in muscle showed decreased activity in complex I–IV.
Fig. 1.
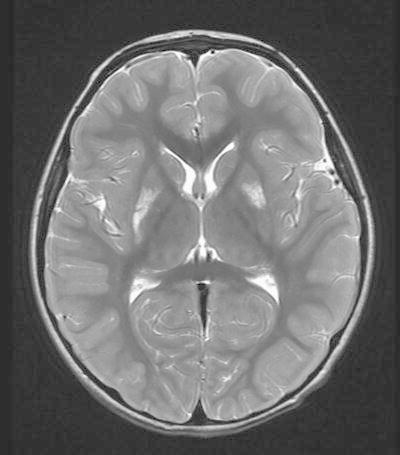
MRI of brain showing increased T2-weighted signal intensities in the caudate bodies and putamen characteristic of Leigh syndrome
Methods Used: Direct sequencing of a Leigh syndrome nuclear gene panel was performed along with a review of the electronic chart and medical literature. A commercial laboratory was used to perform the molecular analysis.
Summary of Results: Molecular testing showed our patient is heterozygous for two previously undescribed mutations in the TTC19 gene, c.577G>A (p.Trp186Stop) and c.964_967delGGCT (p.Gly322MetfsX8), both of which are predicted to cause loss of protein function due to either protein truncation or nonsense-mediated mRNA decay. The c.964_967delGGCT mutation is a four-base-pair deletion that causes a frameshift at glycine codon 322, changing it to a methionine residue and thus creating a premature stop codon at position 8 of the new reading frame. Neither of these two mutations in the TTC19 gene have previously been described in the medical literature. ETC activity on muscle demonstrated an increase in citrate synthase (1.45X mean), suggesting mitochondrial proliferation and decreased complex I + III (79 %), II (52 %), II + III (36 %) and IV (46 %) activities. We note, however, these values were not sufficiently low to fulfill minor criterion of the modified Walker criteria. We note all other patients with TTC19 mutations to date had isolated CIII deficiency identified in muscle.
Conclusions
Leigh syndrome is a subacute, necrotizing encephalomyopathy with an estimated prevalence of 1:40,000 live births. It is characterized by an initial period of normal development during the first few months of life, after which children present with progressive delay or regression in psychomotor development, nystagmus, opthalmoparesis, optic atrophy, ataxia, dysphagia, hypotonia, and dystonia. It is clear that TTC19 mutations affect mitochondrial function and therefore they should be included in nuclear gene panels for evaluation of mitochondrial dysfunction, including the Leigh syndrome phenotype. The clinical presentation of patients with TTC19 mutations is broad, with initial presentation from childhood to adulthood. Additionally, the background of patients with TTC19 mutations is varied, with one patient from consanguineous Iraqi parents, two of nonconsanguineous Italian descent and one of Hispanic descent. In summary, TTC19 mutations have been shown to affect CIII complex function, which results in a heterogeneous clinical phenotype including Leigh syndrome.
Compliance with Ethics Guidelines
Paldeep Atwal declares that he has no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Footnotes
Competing interests: None declared
Contributor Information
P. S. Atwal, Email: paldeep.atwal@gmail.com
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Balasubramaniam S, Oates S, Jolley A et al (2012) Mitochondrial complex III deficiency associated with a homozygous mutation in TTC19, Human Genetics Society of Australasia Meeting, Canberra
- Ghezzi D, Arzuffi P, Zordan M, et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43(3):259–263. doi: 10.1038/ng.761. [DOI] [PubMed] [Google Scholar]