

Abstract
Mucopolysaccharidosis type VI (MPS VI, Maroteaux-Lamy syndrome) is an autosomal recessive disorder caused by the deficit of the arylsulfatase B (ARSB) enzyme, which leads to dermatan sulfate pathological storage, resulting in a wide spectrum of clinical phenotypes. To date more than 130 different mutations were reported, most of them being restricted to individual families. We here report the first study on the ARSB gene mutations in MPS VI patients of Turkish ethnogeographic origin. On the whole we analyzed 13 unrelated families recruited from 3 different Turkish clinical centers, for a total of 52 subjects, including patients, parents, and siblings. The molecular characterization of ARSB gene in these subjects lead to the identification of eight different mutations (6 missense mutations and two single-nucleotide deletions) one of which novel: c.532C>G (p.H178D). We characterized seven different genotypes, all homozygous except one. The analysis highlighted c.962T>C (p.L321P) as the most frequently detected mutation in the group of patients examined and the c.1072G>A (p.V358M) as the most frequent polymorphism. All parents and 50% of the healthy siblings analyzed carried in a heterozygous condition the mutation identified in the affected relative. The high number of homozygotes reported in this study reflects the high degree of consanguinity of the Turkish population, being the parents of most of the patients here examined, first-degree cousins. As consanguineous marriages are an integral part of the Turkish society, carriers identification accompanied by genetic counseling in families at risk is the eligible approach to minimize the effects of consanguinity in this population.
Introduction
Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI, MPS VI; MIM#253200) is an autosomal recessive lysosomal storage disorder caused by the deficit of the N-acetylgalactosamine-4-sulfatase (4-sulfatase, arylsulfatase B, ARSB, E.C.3.1.6.12) activity. This leads to dermatan and chondroitin sulfate pathological storage resulting in a wide spectrum of clinical phenotypes, the most severe of which being characterized by growth retardation, dysostosis multiplex, coarse facial features, joints stiffness, cardiac complications, respiratory difficulties, hepatosplenomegaly, hernias, corneal clouding, hearing loss, and hydrocephalus. Unlike some other storage disorders, mental development is usually normal. Death generally occurs before or during the second decade of life in patients with rapidly progressive forms, often due to cardiopulmonary complications (Valayannopoulos et al. 2010). The birth prevalence ranges between 1 in 43,261 births in the German Turkish immigrants (Baehner et al. 2005) and 1 in 1,505,160 births in Sweden (Malm et al. 2008).
The ARSB locus spans a region of 21 kb located at 5q11-q13 and contains 8 exons; the gene specifies an mRNA transcript of 6089 bp, encoding a protein of 533 amino acids with a signal peptide of 37 amino acids (Litjens et al. 1989; Modaressi et al. 1993). The 57 kDa mature form of the enzyme is formed by three disulfide-linked polypeptides of 43, 8, and 7 kDa (Kobayashi et al. 1992). To date, over 130 different mutations have been described in the ARSB gene (www.hgmd.org). About 75% of all known variations are missense or nonsense mutations, distributed all over the exons; the remaining 25% is represented by small deletions, insertions, splice-site mutations and gross deletions. Most mutations are private or restricted to a small number of subjects. Moreover, some alleles are particularly frequent in certain populations like p.R315Q in Portugal (Karageorgos et al. 2007b) and p.R152W in Russia (Voskoboeva et al. 2000). For some genetic variations a clear genotype-phenotype correlation was established (Karageorgos et al. 2007b), but in most cases this is not the rule.
Characterization of ARSB gene mutations has been reported for many populations from the American to the Australian. Although several studies on lysosomal storage disorders in the Turkish population are reported in literature, only few of them describe MPS cases, including one study reporting 3 MPS VI patients (Elcioglu et al. 2009; Emre et al. 2000; Terzioglu et al. 2002) and, recently, a paper on MPS VI that described, among others, four Maroteaux-Lamy subjects (among which two siblings) of Turkish ethnicity (Brands et al. 2013). However, no studies specifically targeted to the analysis of the MPS VI Turkish patients have been so far conducted. Therefore, this is the first report on ARSB gene mutations characterized in a group of Maroteaux-Lamy patients of Turkish ethnogeographic origin. The Turkish population is very interesting from the genetic point of view, given its social and cultural features that render consanguineous marriages very common. With the 17% of unions contracted between first-degree cousins (Koc 2008), the genetic disorders, in particular the hemoglobinopathies and phenylketonuria, represent a very important public health issue.
This study was carried out on 18 MPS VI patients deriving from 13 unrelated families recruited from 3 different Turkish clinical centers. The molecular analysis of ARSB gene in these subjects lead to the characterization of 8 mutations, one of which previously undescribed.
Materials and Methods
Patients
A total of 13 unrelated Maroteaux-Lamy Turkish families, including 52 subjects (18 patients, 18 parents, and 16 healthy siblings) were recruited from the Department of Pediatric Metabolism and Nutrition-Çukurova University of Adana, the Department of Pediatric Genetics-Marmara University Hospital of Istanbul, and the Diyarbakır Children’s Hospital-Department of Pediatrics of Diyarbakır, and evaluated in this study. All families belong to the Turkish ethnic group. Patients are offspring of consanguineous marriages between first-degree cousins (11 families), second-degree cousins (1 family), or “distant cousins” (1 family). Clinical evaluations were performed in the Turkish clinical centers. Arylsulfatase B enzyme activity assays were performed in leukocytes by 3 different laboratories: Willink Biochemical Genetics Laboratory-Manchester (United Kingdom), Gazi University Medical Faculty Pediatric Metabolism Laboratory-Ankara (Turkey), and Sahlgrenska’s University Laboratory-Molndal (Sweden). ARSB molecular analysis was carried out at the Laboratory of Diagnosis and Therapy of Lysosomal Disorders, University of Padova (Italy). Informed consent for genetic analysis was obtained for all patients and their relatives included in the study. For a better comprehension, patients phenotype was classified in the two principal forms of rapidly progressing, or severe, and slowly progressing, or mild, according to a previously published description (Valayannopoulos et al. 2010).
ARSB Mutation Analysis
Genomic DNA was extracted from peripheral blood leukocytes or from skin fibroblasts using the commercial QIAmp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany). ARSB exons and their flanking regions were PCR-amplified using appropriate intronic primers as previously described (Zanetti et al. 2009). Sequence variations were confirmed by sequencing duplicate PCR products in both directions. Obtained sequences were compared to the genomic reference sequence NC_000005.9.
Sequence Variations Nomenclature
All sequence variations were described according to the mutation nomenclature [(den Dunnen and Antonarakis 2001) HGVS, http://www.hgvs.org/mutnomen].
Analysis of the New Missense Sequence Variation
In order to establish if the new non-synonymous variation was a polymorphism or a mutation, an in silico prediction of the pathogenicity was obtained through the software PONP (Pathogenic-or-Not-Pipeline) which integrates five tools (PhD-SNP, SIFT, PolyPhen-2, SNAP, I-Mutant) to predict the probability that variations may affect protein function and may consequently be disease-related (Olatubosun et al. 2012). Interrogation of dbSNPs [http://www.ncbi.nlm.nih.gov/projects/SNP/] and 1000 genomes [http://www.1000genomes.org/] databases for alleles and genotype frequency was also performed.
Results
The molecular characterization of the ARSB gene mutations in the 18 Turkish Maroteaux-Lamy patients allowed the identification of eight point mutations: six missense mutations and two single-nucleotide deletions. One sequence variation had never been described before: c.532C>G (p.H178D). Five out of eight mutations are located in exon 5.
On the whole, we observed seven different genotypes of which only one is a composite heterozygote while the others are homozygotes. Nine individuals (including one couple of twins and two of brothers) carried the genotype c.[962T>C]+[962T>C]; 3 subjects (including 2 siblings) revealed the genotype c.[ 1036delG]+[ 1036delG].
Interestingly, patients P3, P6, P11, and P12 come from the same city and all of them except patient P3 carry the genotype c.[962T>C]+[962T>C]. Also patient P15 and the two brothers identified as P13 and P14 come from the same town and present with the same mutation c.1036delG.
Most of the patients analyzed in this study did not present a significant residual ARSB enzymatic activity; five out of the seven patients in whom such activity was found showed less than 1% of the lower level normally given as control value; in one patient we measured an activity of 2.81% and in another one 8.23% of the control levels.
Genotype-Phenotype Correlation
Patients’ phenotypes ranged from mild to severe and are reported in Table 1.
Table 1.
Genotypes and clinical phenotypes encountered in the 13 MPS VI families included in the study. Arylsulfatase B activity at diagnosis is reported; the assay was performed in three different laboratories as reported in the materials and methods section: (a) Turkey, (b) the United Kingdom, (c) Sweden. Enzyme activities are expressed as percentage of activity with respect to the lowest limit of the normal range (range of values detected in a healthy control population) or to the value of the normal control (contemporarily examined). F family, pt patient, n.d. not defined
| F | Pt | c.DNA alteration | Amino acid substitution | Reference | Polymorphisms | Arylsulfatase B activity | Phenotype |
|---|---|---|---|---|---|---|---|
| 1 | P1 | c.[160G>A]+ [1057T>A] | p. [D54N]+ [W353R] | Karageorgos et al. 2007b (HGMD: CM074027; HGMD: CM070023) | c.[1072G>A]+ [1072G>A] | 0%b | Severe |
| 2 | P2 | c.[532C>G]+ [532C>G] | p.[H178D]+ [H178D] | Previously undescribed | c. [1191G>A] + [1191G>A] | 2.81%a | Mild |
| 3 | P3 | c.[903C>G]+ [903C>G] | p.[N301K]+ [N301K] | Brands et al. 2013 | c.[1151G>A]+ [1151G>A] | 0%b | Moderate |
| 4 | P4 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | Isbrandt et al. 1994 (HGMD: CM940120) | c.[1072G>A]+ [1072G>A] | 0%b | Severe |
| P5 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 0.35%b | n.d. | |
| 5 | P6 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 0%b | Severe |
| 6 | P7 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 0%b | Mild-moderate |
| P8 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 0%b | Mild-moderate | |
| 7 | P9 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 8.23%c | Moderate |
| P10 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | n.a. | Moderate | |
| 8 | P11 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 0.37%b | Moderate |
| 9 | P12 | c.[962T>C]+ [962T>C] | p.[L321P ]+[L321P] | As above | c.[1072G>A]+ [1072G>A] | 0.07%b | Mild |
| 10 | P13 | c.[1036delG]+ [1036delG] | --- | Karageorgos et al. 2007 (HGMD: CD075326) | c.[1072G>A]+ [1072G>A] | 0%a | Severe |
| P14 | c.[1036delG]+ [1036delG] | --- | As above | c.[1072G>A]+ [1072G>A] | 0%a | Mild | |
| 11 | P15 | c.[1036delG]+ [1036delG] | --- | As above | c.[1072G>A]+ [1072G>A] | 0.07%b | Mild |
| 12 | P16 | c.[1079T>C]+ [1079T>C] | p.[L360P ]+[L360P] | Voskoboeva et al. 2000 (HGMD: CM003998) | c.[1151G>A]+ [1151G>A] | 0%b | Moderate |
| P17 | c.[1079T>C]+ [1079T>C] | p.[L360P ]+[L360P] | As above | c.[1151G>A]+ [1151G>A] | 0.75%b | Moderate | |
| 13 | P18 | c. [1577delC]+ [1577delC] | --- | Isbrandt et al. 1994 (HGMD: CD941599) | --- | 0%b | Severe |
Laboratory in which was performed the ARSB activity assay:
aGazi University Medical Faculty Pediatric Metabolism Laboratory-Ankara (Turkey)
bWillink Biochemical Genetics Laboratory-Manchester (United Kingdom)
cSahlgrenska’s University Laboratory-Molndal (Sweden)
The mutation c.962T>C (p.L321P) was first described in homozygosis in a patient with an intermediate phenotype, who was born from a consanguineous marriage (Isbrandt et al. 1994). Then it was reported, again in homozygosis, in one patient, with a non-specified phenotype (Karageorgos et al. 2007b).
In this study it was detected in a homozygous state in six families including nine patients, among which two couples of siblings and one of twins. The phenotypes of these nine cases range from severe to mild, while the arylsulfatase B activity was undetectable for all patients except for patient P9 showing a very low activity and a moderate phenotype.
The c.1036delG mutation leads to the production of a 176 amino acids shorter protein with respect to the wild-type ARSB. It was first reported in a homozygote German patient with a very high level of urinary GAG (699 mg/ml creatinine) who showed symptoms of a rapidly progressing disease (Karageorgos et al. 2007b).
In our analysis, the deletion was detected in the two siblings P13 and P14 and in patient P15. One of the two siblings presents with a severe pathology, while the other one, in ERT since 10 months of age, seems to carry an attenuated form of the disease. The third subject shows mild symptoms.
The genetic variation c.1079T>C (p.L360P) was detected for the first time in an attenuated phenotype (Voskoboeva et al. 2000).
In this study, this variation was diagnosed in two siblings (P16 and P17) both presenting with moderate symptoms.
The deletion c.1577delC was previously reported in a severe homozygote patient, son of consanguineous parents (Isbrandt et al. 1994).
Our patient (P18) carrying this mutation had a severe phenotype and died at 8 years of age for post-surgery complications.
The mutation c.903C>G (p.N301K) was recently described for the first time in homozygosis condition, in a Turkish patient with a rapidly progressing phenotype, child of consanguineous parents. Through the in vitro expression of the mutated protein they evidenced a slightly reduced amount of the 66 kDa ARSB precursor, while they did not detect the 46 kDa mature form of the enzyme (Brands et al. 2013).
In this study, the p.N301K mutation was diagnosed in homozygosis in patient P3 who presented with moderate symptoms.
The only compound heterozygote detected in our analysis is patient P1 carrying the genotype c.[160G>A]+ [1057T>A] (p.[D54N]+[W353R]) and a severe form of the disease. Both mutations were previously described (Karageorgos et al. 2007b): the p.D54N was detected in a homozygote and in a compound heterozygote patients both of Portuguese origin; this mutation, directly affecting the ARSB catalytic site, might result in an early-onset and rapidly progressing pathology. The p.W353R mutation was found in a compound heterozygote with a non-specified phenotype.
The novel variation diagnosed in this study is the c.532C>G (p.H178D), detected in a homozygous condition in patient P2, showing mild symptoms. This undescribed nucleotide substitution is not reported in both dbSNPs and 1,000 genomes databases. Moreover, in silico prediction of its pathogenicity through PONP integrated tools revealed that these variations belong to the “pathogenic” Predicted class with a Probability of pathogenicity of 0.72 (standard error 0.1 e–1) confirming that the variation could not be considered a polymorphism but a disease-causing mutation.
The c.1072G>A (p.V358M) is the most frequent polymorphism encountered in the group of patients analyzed: 13 patients out of 18 were homozygotes for this variant. The polymorphism c.1151G>A (p.S384N) was detected in homozygosis in 3 patients, while only one patient was homozygote for the polymorphism c.1191G>A (p.P397P). Patient P18 did not carry anyone of the polymorphisms mentioned above. No patients carried the quite common polymorphism c.1126G>A (p.V376M).
Family Studies
Both parents of each patient were genetically characterized for 11 families out of 13; of the two remaining families, for one only the mother sample was available, for the other one no parents’ samples were collected due to social problems of the family. All parents analyzed carried the child’s mutation in heterozygous condition. We also performed carrier analysis in 16 healthy siblings: eight of them resulted carrier of the mutation detected in the affected relative.
Three families presented a family history reporting at least one previous child dead for worsening of MPS VI clinical conditions; two families had a history reporting at least one miscarriage; one family reported stillbirth siblings.
A pedigree was reconstructed for the families of patient P6 and of patients P13-P14 (Fig. 1a and b, respectively). Among the families evaluated in this study, these two presented with the highest number of affected children and/or miscarriages or termination of pregnancy events in their clinical history. The high degree of consanguinity is clearly shown in the two pedigrees reported, being the parents first-degree cousins.
Fig. 1.
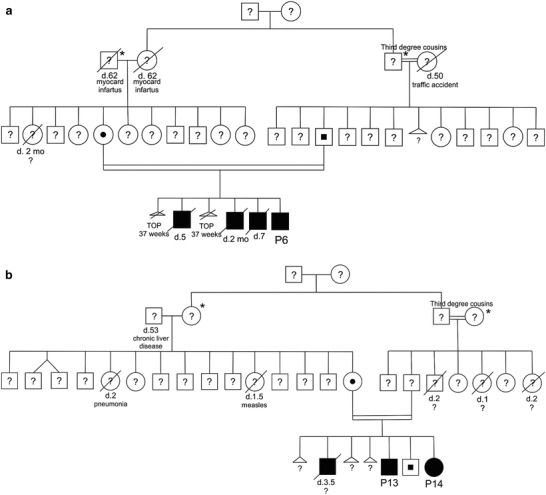
Pedigrees of patient P6 (a) and patients P13-P14 families (b). Subjects identified with * and # are first-degree cousins. Molecular characterization of ARSB gene was performed only for the probands P6, P13, and P14, their parents and siblings. For all other relatives reported in the pedigrees, molecular status of ARSB gene was not investigated. TOP termination of pregnancy
Discussion
In this study, we report the results of the molecular analysis of the ARSB gene in a group of Turkish MPS VI patients. Turkish population is a very young heterogeneous population composed mainly by the Turkish ethnic group but also by other minor communities (Kurdish, Arabic, Greek, Circassian, Georgian, Armenian, and Jewish). Data from the 2003 Turkey Demographic and Health Survey indicates that Turkey is a country with a high level of consanguinity, as other Muslim populations of North Africa, Middle East, South and Central Asia. At present consanguineous marriages account for 22% of all unions, 76% of which being contracted between first-degree cousins. However, no later than 4 decades ago the percentage assessed around 27, the gradual reduction being due to the modernization of the Turkish society (Koc 2008). This social habit is motivated by cultural, religious but also economical reasons. The highest prevalence of consanguinity is found in the less developed regions of the country, in families with a low socioeconomic status in which women, but also their husbands, tend to have lower level of education.
Turkish population represents a challenge for genetic studies as its high rate of consanguineous marriages could be a contributing factor to the high incidence of some rare autosomal recessive diseases registered in the country; in particular, hereditary blood disorders (hemoglobinopathies and thalassemia) and phenylketonuria are the genetic pathologies presenting the highest frequencies (Tuncbilek and Ozguc 2007). On the other side, groups of the Turkish population might also represent a rare opportunity to evaluate specific pathological alleles in homozygous condition and in a quite conserved genomic background.
Until now, analyses of ARSB gene mutations have been reported for many world populations including the North (Isbrandt et al. 1994; Jin et al. 1992; Karageorgos et al. 2007a; Simonaro and Schuchman 1995) and the South American (Karageorgos et al. 2007a; Petry et al. 2003, 2005), the Western European (Arlt et al. 1994; Isbrandt et al. 1994, 1996; Karageorgos et al. 2007a; Villani et al. 1998, 1999), the Eastern European (Isbrandt et al. 1994; Jurecka et al. 2012; Voskoboeva et al. 1994, 2000), the Asiatic (Dou et al. 2006; Wicker et al. 1991; Wu et al. 2000), and the Australian populations (Litjens et al. 1992, 1996; Wicker et al. 1991). Three studies have been published describing few cases of mucopolysaccharidoses in the Turkish population (Elcioglu et al. 2009; Emre et al. 2000; Terzioglu et al. 2002). Among these, Emre and colleagues characterized biochemically and genetically a group of MPS patients including three MPS VI cases (Emre et al. 2002). Recently, Brands described four MPS VI patients of Turkish ethnogeographic origin (Brands et al. 2013). Hence this is the first study reporting the molecular characterization of the ARSB mutations in a quite large group of Maroteaux-Lamy cases from Turkey. Eighteen patients coming from 13 unrelated families were genetically characterized. Since all MPS VI subjects analyzed have consanguineous parents (in most cases first-degree cousins), we evidenced, as expected, a lower degree of genetic heterogeneity with respect to what is generally described for other populations, confirming the effect of the social habits mentioned above. In addition, founder effects might be postulated, given the small number of different mutations detected in the group analyzed, for a gene as ARSB, which shows in most populations a wide variety of private mutations. Also the high frequency of homozygous genotypes (12 out of 13 families) detected in this study reflects the above-mentioned aspects.
To date more than 130 different mutations have been described in the literature for the ARSB gene confirming the high genetic heterogeneity of MPS VI, a feature shared with all the other MPSs. This large number of different mutations, which are often restricted to single families or are novel, renders the analysis of genotype-phenotype correlation quite difficult in the MPS VI disease, although this might result easier in homozygous subjects carrying the same mutation. An attempt to establish a genotype-phenotype correlation was therefore made for the patients characterized in this study, considering the high rate of consanguinity and hence a likely similar genetic background which might help in establishing such a relationship. However the picture we revealed was not so unambiguous. For example, clinical evaluation of the 8 subjects homozygote for the p.L321P mutation showed a continuum of phenotypes from the mild-slowly to the severe-rapidly progressing form of the disease. Also, for the c.1036delG mutation we observed both a mild phenotype and a severe phenotype. These findings allow to hypothesize the existence of other genetic or epigenetic factors which could be potentially important in the phenotype determination.
The most frequent polymorphisms detected in the Maroteaux-Lamy population analyzed are the p.V358M and p.S384N. In particular the p.V358M, when present, was found in homozygosis, suggesting a co-segregation with the disease-causing mutations. In fact, all patients carrying the mutations p.L321P or c.1036delG, presented also the polymorphism p.V358M. The allele frequency was 36.1%, slightly higher than that (32%) found in the population previously analyzed (Karageorgos et al. 2007b). Also the frequency of the allele p.S384N is higher with respect to the frequency detected by Karageorgos (16.6% versus 6.3%), although in that paper it was erroneously identified as pathological mutation. On the contrary, the polymorphism p.P397P was found in the 30.5% of the alleles here examined, less than half of the frequency (74.3%) calculated on the cases previously reported by the cited author (Karageorgos et al. 2007b).
Most of the patients analyzed in this study did not present a significant residual ARSB enzymatic activity; five out of the seven patients in whom such activity was found showed less than 1% of the lower level normally given as control value; in 1 patient we measured an activity of 2.81% and in another 8.23% of the control levels. All these seven patients showed a mild to moderate phenotype. None of the patients with a severe-defined phenotype reported residual enzymatic activity, however also for some of the mild/moderate patients we registered null ARSB activity. Therefore, from our data we might infer that the presence of a residual enzymatic activity usually correlates with a mild/moderate phenotype, but a total lack of activity is not always associated with a severe phenotype/prognosis.
In conclusion, analysis of the Turkish population has confirmed for MPS VI similar influence of the cultural habits on the incidence of inherited diseases, already denounced for hemoglobinopathies and phenylketonuria, as an important public health issue (Koc 2008). Similar issues are taken into consideration in other countries such as Israel, characterized by a high level of consanguineous marriages due to the existence of few separate ethnic groups (Bach et al. 2007) and in which counseling measures adopted in recent years have significantly reduced the prevalence of genetic disorders in the population.
In particular, measures addressed to identify in the population the most frequent genetic diseases and the set up of screening programs for them would help the individuation of carriers; more restricted screenings as well as prenatal diagnosis may also be conducted within at-risk families. All this should allow a possible reduction of the incidence of MPS VI as well as other recessive genetic diseases.
One-Sentence Take-Home Message
First ARSB molecular analysis extensive report of MPS VI Turkish patients.
Details of the Contributions of Individual Authors
AZ: conception and design of the study, performing of molecular analysis, analysis and interpretation of data, writing and critical revision of the manuscript
NO: conception and design of the study, clinical data collection, and critical revision of the manuscript
NE: clinical data collection and critical revision of the manuscript
MNO: clinical data collection and critical revision of the manuscript
DK: clinical data collection and critical revision of the manuscript
EL: cells culture and critical revision of the manuscript
MS: conception and design of the study and critical revision of the manuscript
RT: conception and design of the study, writing and critical revision of the manuscript, final approval of the manuscript
Name of One Author Who Serves as Guarantor
Rosella Tomanin
Details of Funding
This study was partly funded by the Brains for Brain (B4B) Foundation (www.brains4brain.eu). The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Details of Ethics Approval
Ethics approval was not required to perform the described studies.
A Patient Consent Statement
Informed consent for genetic analyses in patients and relatives was obtained from all subjects involved in the study, including parents or tutors.
Conflict of Interest
Maurizio Scarpa has received research grants and honoraria and travel support for speaking engagements from Actelion, Shire HGT, Genzyme Corporation, and BioMarin.
Alessandra Zanetti, Neslihan Önenli-Mungan, Nursel Elcioglu, Mehmet Nuri Özbek, Deniz Kör, Elisabetta Lenzini, and Rosella Tomanin declare no conflicts of interest.
Footnotes
Competing interests: None declared
The first two authors contributed equally to this work
Contributor Information
Rosella Tomanin, Email: rosella.tomanin@unipd.it.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Arlt G, Brooks DA, Isbrandt D, Hopwood JJ, Bielicki J, Bradford TM, Bindloss-Petherbridge CA, von Figura K, Peters C. Juvenile form of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) A C-terminal extension causes instability but increases catalytic efficiency of arylsulfatase B. J Biol Chem. 1994;269(13):9638–9643. [PubMed] [Google Scholar]
- Bach G, Zeigler M, Zlotogora J. Prevention of lysosomal storage disorders in Israel. Mol Genet Metab. 2007;90(4):353–357. doi: 10.1016/j.ymgme.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlschutter A, Kampmann C, Beck M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28(6):1011–1017. doi: 10.1007/s10545-005-0112-z. [DOI] [PubMed] [Google Scholar]
- Brands MM, Hoogeveen-Westerveld M, Kroos MA, Nobel W, Ruijter GJ, Ozkan L, Plug I, Grinberg D, Vilageliu L, Halley DJ, et al. Mucopolysaccharidosis type VI phenotypes-genotypes and antibody response to galsulfase. Orphanet J Rare Dis. 2013;8(1):51. doi: 10.1186/1750-1172-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109(1):121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- Dou W, Peng C, Zheng J, Sheng HZ, Gu X, Chen J, Zhang W, Huang S. Two novel mutations of the arylsulfatase B gene in a Chinese MPS VI child undergoing bone marrow transplantation therapy. Clin Chim Acta. 2006;374(1–2):171–172. doi: 10.1016/j.cca.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Elcioglu NH, Pawlik B, Colak B, Beck M, Wollnik B. A novel loss-of-function mutation in the GNS gene causes Sanfilippo syndrome type D. Genet Couns. 2009;20(2):133–139. [PubMed] [Google Scholar]
- Emre S, Topcu M, Terzioglu M, Renda Y. Arylsulfatase A pseudodeficiency incidence in Turkey. Turk J Pediatr. 2000;42(2):115–117. [PubMed] [Google Scholar]
- Emre S, Terzioglu M, Coskun T, Tokath A, Ozalp I, Muller V, Hopwood J. Biochemical and molecular analysis of mucopolysaccharidoses in Turkey. Turk J Pediatr. 2002;44(1):13–17. [PubMed] [Google Scholar]
- Isbrandt D, Hopwood JJ, von Figura K, Peters C. Two novel frameshift mutations causing premature stop codons in a patient with the severe form of Maroteaux-Lamy syndrome. Hum Mutat. 1996;7(4):361–363. doi: 10.1002/(SICI)1098-1004(1996)7:4<361::AID-HUMU12>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Isbrandt D, Arlt G, Brooks DA, Hopwood JJ, von Figura K, Peters C. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): Six unique arylsulfatase B gene alleles causing variable disease phenotypes. Am J Hum Genet. 1994;54(3):454–463. [PMC free article] [PubMed] [Google Scholar]
- Jin WD, Jackson CE, Desnick RJ, Schuchman EH. Mucopolysaccharidosis type VI: Identification of three mutations in the arylsulfatase B gene of patients with the severe and mild phenotypes provides molecular evidence for genetic heterogeneity. Am J Hum Genet. 1992;50(4):795–800. [PMC free article] [PubMed] [Google Scholar]
- Jurecka A, Piotrowska E, Cimbalistiene L, Gusina N, Sobczynska A, Czartoryska B, Czerska K, Ounap K, Wegrzyn G, Tylki-Szymanska A. Molecular analysis of mucopolysaccharidosis type VI in Poland, Belarus, Lithuania and Estonia. Mol Genet Metab. 2012;105(2):237–243. doi: 10.1016/j.ymgme.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Karageorgos L, Brooks DA, Harmatz P, Ketteridge D, Pollard A, Melville EL, Parkinson-Lawrence E, Clements PR, Hopwood JJ. Mutational analysis of mucopolysaccharidosis type VI patients undergoing a phase II trial of enzyme replacement therapy. Mol Genet Metab. 2007;90(2):164–170. doi: 10.1016/j.ymgme.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Karageorgos L, Brooks DA, Pollard A, Melville EL, Hein LK, Clements PR, Ketteridge D, Swiedler SJ, Beck M, Giugliani R, et al. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat. 2007;28(9):897–903. doi: 10.1002/humu.20534. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Honke K, Jin T, Gasa S, Miyazaki T, Makita A. Components and proteolytic processing sites of arylsulfatase B from human placenta. Biochim Biophys Acta. 1992;1159(3):243–247. doi: 10.1016/0167-4838(92)90051-E. [DOI] [PubMed] [Google Scholar]
- Koc I. Prevalence and sociodemographic correlates of consanguineous marriages in Turkey. J Biosoc Sci. 2008;40(1):137–148. doi: 10.1017/S002193200700226X. [DOI] [PubMed] [Google Scholar]
- Litjens T, Brooks DA, Peters C, Gibson GJ, Hopwood JJ. Identification, expression, and biochemical characterization of N-acetylgalactosamine-4-sulfatase mutations and relationship with clinical phenotype in MPS-VI patients. Am J Hum Genet. 1996;58(6):1127–1134. [PMC free article] [PubMed] [Google Scholar]
- Litjens T, Morris CP, Robertson EF, Peters C, von Figura K, Hopwood JJ. An N-acetylgalactosamine-4-sulfatase mutation (delta G238) results in a severe Maroteaux-Lamy phenotype. Hum Mutat. 1992;1(5):397–402. doi: 10.1002/humu.1380010509. [DOI] [PubMed] [Google Scholar]
- Litjens T, Baker EG, Beckmann KR, Morris CP, Hopwood JJ, Callen DF. Chromosomal localization of ARSB, the gene for human N-acetylgalactosamine-4-sulphatase. Hum Genet. 1989;82(1):67–68. doi: 10.1007/BF00288275. [DOI] [PubMed] [Google Scholar]
- Malm G, von Dobeln U, Naess K, Ringden O. Progress on lysosomal diseases yields hope for cure and improvement. Lakartidningen. 2008;105(51–52):3731–3735. [PubMed] [Google Scholar]
- Modaressi S, Rupp K, von Figura K, Peters C. Structure of the human arylsulfatase B gene. Biol Chem Hoppe Seyler. 1993;374(5):327–335. doi: 10.1515/bchm3.1993.374.1-6.327. [DOI] [PubMed] [Google Scholar]
- Olatubosun A, Valiaho J, Harkonen J, Thusberg J, Vihinen M. PON-P: Integrated predictor for pathogenicity of missense variants. Hum Mutat Aug. 2012;33(8):1166–1174. doi: 10.1002/humu.22102. [DOI] [PubMed] [Google Scholar]
- Petry MF, Dieter T, Burin M, Giugliani R, Leistner S. Identification of a novel mutation in the ARSB gene that is frequent among Brazilian MPSVI patients. Genet Test. 2003;7(4):347–349. doi: 10.1089/109065703322783743. [DOI] [PubMed] [Google Scholar]
- Petry MF, Nonemacher K, Sebben JC, Schwartz IV, Azevedo AC, Burin MG, de Rezende AR, Kim CA, Giugliani R, Leistner-Segal S. Mucopolysaccharidosis type VI: Identification of novel mutations on the arylsulphatase B gene in South American patients. J Inherit Metab Dis. 2005;28(6):1027–1034. doi: 10.1007/s10545-005-0020-2. [DOI] [PubMed] [Google Scholar]
- Simonaro CM, Schuchman EH. N-acetylgalactosamine-4-sulfatase: identification of four new mutations within the conserved sulfatase region causing mucopolysaccharidosis type VI. Biochim Biophys Acta. 1995;1272(3):129–132. doi: 10.1016/0925-4439(95)00070-4. [DOI] [PubMed] [Google Scholar]
- Terzioglu M, Tokatli A, Coskun T, Emre S. Molecular analysis of Turkish mucopolysaccharidosis IVA (Morquio A) patients: identification of novel mutations in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene. Hum Mutat. 2002;20(6):477–478. doi: 10.1002/humu.9088. [DOI] [PubMed] [Google Scholar]
- Tuncbilek E, Ozguc M. Application of medical genetics in Turkey. Turk J Pediatr. 2007;49(4):353–359. [PubMed] [Google Scholar]
- Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi: 10.1186/1750-1172-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani GR, Balzano N, Di Natale P (1998) Two novel mutations of the arylsulfatase B gene in two Italian patients with severe form of mucopolysaccharidosis. Mutations in brief no. 127. online. Hum Mutat 11(5):410 [DOI] [PubMed]
- Villani GR, Balzano N, Vitale D, Saviano M, Pavone V, Di Natale P. Maroteaux-Lamy syndrome: five novel mutations and their structural localization. Biochim Biophys Acta. 1999;1453(2):185–192. doi: 10.1016/S0925-4439(98)00099-4. [DOI] [PubMed] [Google Scholar]
- Voskoboeva E, Isbrandt D, von Figura K, Krasnopolskaya X, Peters C. Four novel mutant alleles of the arylsulfatase B gene in two patients with intermediate form of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) Hum Genet. 1994;93(3):259–264. doi: 10.1007/BF00212019. [DOI] [PubMed] [Google Scholar]
- Voskoboeva EI, Krasnopol'skaia KD, Peters K, von Figura K. Identification of mutations in the arylsulfatase B gene in Russian mucopolysaccharidosis type VI patients. Genetika. 2000;36(6):837–843. [PubMed] [Google Scholar]
- Wicker G, Prill V, Brooks D, Gibson G, Hopwood J, von Figura K, Peters C. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). an intermediate clinical phenotype caused by substitution of valine for glycine at position 137 of arylsulfatase B. J Biol Chem. 1991;266(32):21386–21391. [PubMed] [Google Scholar]
- Wu JY, Yang CF, Lee CC, Chang JG, Tsai FJ. A novel mutation (Q239R) identified in a Taiwan Chinese patient with type VI mucopolysaccharidosis (Maroteaux-Lamy syndrome) Hum Mutat. 2000;15(4):389–390. doi: 10.1002/(SICI)1098-1004(200004)15:4<389::AID-HUMU31>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Zanetti A, Ferraresi E, Picci L, Filocamo M, Parini R, Rosano C, Tomanin R, Scarpa M. Segregation analysis in a family at risk for the Maroteaux-Lamy syndrome conclusively reveals c.1151G>A (p.S384N) as to be a polymorphism. Eur J Hum Genet. 2009;17(9):1160–1164. doi: 10.1038/ejhg.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]