

Abstract
Hunter disease (Mucopolysaccharidosis type II, MPS II) is an X-linked lysosomal storage disorder caused by deficiency of iduronate-2-sulfatase (IDS). Two main therapies have been reported for MPS II patients: enzyme-replacement therapy (ERT) and hematopoietic stem-cell transplantation (HSCT). Both treatment modalities have been shown to improve some symptoms, but the results with regard to cognitive functioning have been poor. Early initiation of therapy, i.e., before neurological symptoms have manifested, may alter cognitive outcome. The need for early identification makes Hunter disease a candidate for newborn screening (NBS). Our objective was to explore the use of a fluorometric assay that could be applicable for high-throughput analysis of IDS activity in dried blood spots (DBS). The median IDS activity in DBS samples from 1,426 newborns was 377 pmol/punch/17 h (range 78–1111). The IDS activity in one sample was repeatedly under the cutoff value (set at 20% of the median value), which would imply a recall rate of 0.07%. A sample from a clinically diagnosed MPS II individual, included in each 96-well test plate, had IDS activities well below the 20% cutoff value. Coefficients of variation in quality control samples with low, medium, and high IDS activities (190, 304, and 430 pmol/punch/17 h, respectively) were 12% to 16%. This small-scale pilot study shows that newborn screening for Hunter disease using a fluorometric assay in DBS is technically feasible with a fairly low recall rate. NBS may allow for identification of infants with Hunter disease before clinical symptoms become evident enabling early intervention.
Introduction
Hunter disease (Mucopolysaccharidosis type II, MPS II, OMIM 309900) is an X-linked lysosomal storage disorder caused by deficiency of iduronate-2-sulfatase (IDS) (Wraith et al. 2008). IDS deficiency results in accumulation of dermatan sulfate and heparan sulfate in various tissues and elevated levels in urine. Like many other lysosomal storage disorders, MPS II is clinically heterogeneous with a spectrum of disease severity ranging from childhood to adult presentation. Typical clinical features include coarse facies, dysostosis multiplex, joint stiffness/contractures, obstructive and restrictive airway disease, recurrent infections, hepatosplenomegaly, umbilical/inguinal hernias, and cardiac valve disease (Burton and Giugliani 2012). Children with severe MPS II also suffer from mental retardation, while patients with a more attenuated form usually have normal intelligence (Holt et al. 2011).
Two modes of therapeutic interventions are applied in MPS II: enzyme-replacement therapy (ERT) and bone marrow or hematopoietic stem-cell transplantation (BMT/HSCT). ERT has been shown to improve a number of symptoms (Muenzer et al. 2012; Wraith et al. 2008; Scarpa et al. 2011), but does not prevent mental retardation since the intravenously administered ERT does not cross the blood–brain barrier. HSCT has the potential advantage that transplanted cells are able to cross the blood–brain barrier and to differentiate into microglia providing an IDS enzyme source in the CNS. Unfortunately, the results of BMT/HSCT in MPS II have been variable (Vellodi et al. 1999; Krivit 2004; Guffon et al. 2009; Scarpa et al. 2011; Tanaka et al. 2012). As reported for ERT, positive effects included resolution of hepatosplenomegaly and stabilization of cardiac abnormalities, but neurological function in patients with the severe disease phenotype did not improve (Guffon et al. 2009). However, these data are limited to BMT performed in patients that already had cognitive decline. The possibility remains that early intervention by HSCT (e.g., before the age of 1 year) may change disease progression with respect to neurological symptoms. Early HSCT of patients with Hunter syndrome has not been reported to date. However, for other lysosomal storage disorders the benefits of early treatment have been shown, including Hurler disease (Boelens et al. 2013). Early intervention requires identification of patients at a very early age, before clinical features have become evident, i.e., by newborn screening (NBS) (Marsden and Levy 2010; Zhou et al. 2011). NBS has been reported for a number of lysosomal storage disorders including Pompe disease (Chien et al. 2008; Dajnoki et al. 2008), Fabry disease (Dajnoki et al. 2010), and Krabbe disease (Orsini et al. 2009), but not for MPS II. Enzymatic assays for measuring the IDS activity in dried blood spots have been described in fluorometric (Oemardien et al. 2011; Tolun et al. 2012) and in tandem mass spectrometric (Wolfe et al. 2011) platforms. In these studies small numbers of samples (<100) were used to show that the assays could be performed successfully, but to date analysis of IDS activity in large number of DBS samples has not yet been reported. Our objective was to investigate the suitability of the 4MU-based fluorometric assay for a routine diagnostic laboratory setting using microplate technology with a relatively large number of 1,426 DBS samples.
Materials and Methods
DBS Samples
Anonymous DBS samples, residues from routine newborn screening, were obtained from the National Institute for Public Health and the Environment (RIVM). To obtain a representative selection of the Dutch population, random samples were taken from all Dutch provinces in numbers proportional to the number of newborns per province. DBS were thoroughly air dried and stored at 4°C in sealed plastic bags. The samples were 2–3 years old at the time the enzyme assays were performed. Disease-positive DBS were obtained from a clinically diagnosed MPS II individual. Quality control DBS, manufactured to contain low (QC low), medium (QC medium), and high (QC high) lysosomal enzyme activities, were provided by Dr. Hui Zhou from the Centers for Disease Control and Prevention, Atlanta, Georgia (De Jesus et al. 2009).
This study was approved by the Erasmus University Medical Center Institutional Review Board.
Enzyme Assay
Iduronate-2-sulfatase (IDS) activities were determined essen-tially as described previously (Voznyi et al. 2001; Civallero et al. 2006; Oemardien et al. 2011), but using 96-well microplates instead of reaction vials. Briefly, a 3.2 mm DBS punch was placed in a microplate well and incubated for 17 h with 40 μL reaction mixture (1.25 mmol/L 4-methylumbelliferryl-α-l-iduronate 2-sulfate (Moscerdam, Oegstgeest, The Netherlands), 10 mmol/L lead acetate, 100 mmol/L sodium acetate pH 5), and 20 μL of a solution containing 1 μg/mL recombinant human α-l-iduronidase (Tolun et al. 2012) (R&D Systems, commercial preparation diluted in water containing 0.2% BSA). Following 17-h incubation at 37°C, the plates were placed on ice and protein precipitation was performed by addition of 30 μL 16% trichloroacetic acid. After 10 min, the plates were centrifuged for 5 min at 1,500g and 4°C. Subsequently, 60 μL supernatant from each well was transferred to the corresponding well of a 96-well Optiplate (Perkin Elmer) using a Microlab Star liquid handling workstation (Hamilton), and 200 μL sodium carbonate buffer (pH 10.7) containing 0.25% Triton X-100 was added to enhance the fluorescence of the reaction product 4-methylumbelliferone (4MU).
Following mixing, fluorescence intensity was measured with a fluorometer (Varioskan, Thermo Electron Corporation) at an excitation wavelength of 365 nm and an emission wavelength of 448 nm. Each 96-wel plate included 90 unknown samples, two blanks (blank filter paper punch), one MPS II patient sample, and one of the three different QC samples. The last two wells were used for a calibrator (60 μL of 12.5 μmol/L 4-methylumbelliferone), which was added after the 17-h incubation and included in the subsequent sample processing steps. IDS activities were calculated by subtracting the blank value and converting fluorescence readings to pmol 4MU per 3.2 mm punch per 17 hours (pmol/punch/17 h).
Results and Discussion
IDS activity was determined in 1,426 DBS from newborns. The median activity was 377 pmol/punch/17 h (Fig. 1) (range 78–1111, average 388, SD 118). For each 96-well plate, specimens with an activity less than 20% of the plate median value (in total 7 of the 1,426 samples; 0.5%) were retested using a second punch from the original blood spot card. Six of the seven originally positive samples had normal IDS activity in the second testing. The false-positive outcomes must have been caused by erroneous sample or liquid handling. Inclusion of a second enzyme assay in the first testing could have allowed discriminating between these two possibilities, but would have required blood-spot extraction and sample splitting before substrate addition. The IDS activity in one of the seven originally positive samples remained below the cutoff value upon retesting. Since the NBS cards used in our study were de-identified and not traceable to a person, we could not confirm the low IDS activity in a fresh blood sample from this newborn. A disease-positive control sample (i.e., from an MPS II individual) was blindly loaded at different positions in each of the 17 microplates that were used to analyze the 1,426 samples. All these MPS II samples had IDS activity well below the 20% cutoff value (Fig. 2; mean activity −21 pmol/punch/17 h). The negative value is due to the method we used to calculate the activity, which includes subtraction of the blank.
Fig. 1.
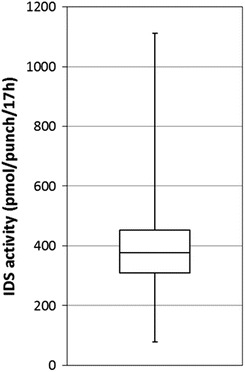
Iduronate-2-sulfatase activities in anonymous newborn screening filter cards ( n = 1426). The box represents those results within the 25th to 75th percentiles, the error bars show the range of enzyme activities and the horizontal line the median
Fig. 2.
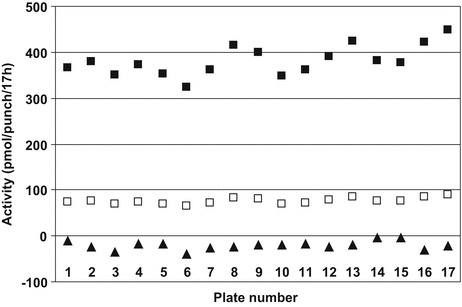
Iduronate-2-sulfatase activities in anonymous newborn screening filter cards ( n = 1426) and DBS obtained from an established MPS II patient. Seventeen microplates were required to test all samples. On the horizontal axis, the microplate number is indicated. On the vertical axis, the following IDS activities are depicted: the median IDS activity calculated for 90 newborn screening DBS samples included in each plate (■), the value corresponding to 20% of the plate median activity (□, used as the cutoff value to identify samples that were analyzed a second time), and the value obtained for an MPS II sample included in each plate (▲)
Intra-assay variation was 11% (mean value of two different samples). QC samples provided by the CDC had the following mean IDS activities in pmol/punch/17 h: QC low 190 (SD 23; n = 5), QC medium 304 (SD 41; n = 6), and QC high 430 (SD 69; n = 5). Inter-assay variation coefficients calculated from these values were 12% to 16%. The QC low and QC high specimen clearly had distinct values, but a relatively high activity was recorded for the QC low specimen. In contrast, other lysosomal enzymes, such as α-glucosidase, α-galactosidase, and α-l-iduronidase, had very low activities in the QC low sample compared to QC medium and QC high in our laboratory (results to be published elsewhere), as well as other laboratories (De Jesus et al. 2009). We do not have an obvious explanation for high IDS activity in the QC low sample, but this may be accounted for by the DBS preparation procedure, which includes mixing of several blood fractions, including serum, and the notion that considerable IDS activity is present in plasma. IDS activity was stable for at least 2 months in DBS stored at 4°C. After 2 months of storage, IDS activities in the three different QC samples were 86% to 103% of the original value confirming previous results (Tolun et al. 2012).
Importantly, in our study only one infant would be recalled for confirmation in a second sample. Based on this result, the recall rate would be 0.07%. Although this number remains to be confirmed by larger studies, it is comparable to the results obtained in studies for other lysosomal storage disorders: Pompe disease (0.039%) (Dajnoki et al. 2008) and Krabbe disease (0.06%) (Orsini et al. 2009). A higher recall rate (0.82%) was reported by Chien et al. for Pompe NBS in Taiwan, but this likely results from a common α-glucosidase variant with low enzyme activity present in the Asian population (Chien et al. 2008). After several more years’ experience with implementation of NBS for Pompe in Taiwan, the screening algorithm has been recalculated to achieve a recall rate of 0.009% (41 of 473,738 infants, calculated by us on the basis of the study by Chiang et al. (2012)). Our study did not allow estimation of a false-negative rate.
The fluorometry-based method is easily implemented in a routine diagnostic laboratory, but has as disadvantage that the 4MU-based enzyme assay used to determine IDS activity is rather expensive due to the high substrate cost (5 US$ per sample). Downscaling the assay volume, for instance, by employing digital microfluidics (DMF) as recently described (Sista et al. 2011), might reduce the costs, although the cost structure using disposable cartridges is not yet clear.
Conclusion
This small-scale pilot study shows that newborn screening for Hunter disease using a fluorometric assay in DBS is feasible and has a fairly low recall rate. The assay is ideally performed overnight, requires 1–2-h hands-on time after punching, and gives a result within 24 h of sample receipt. NBS may allow for identification of infants with Hunter disease before clinical symptoms become evident enabling early intervention by, e.g., ERT or HSCT.
Acknowledgments
This research was funded through Top Institute Pharma, Leiden, the Netherlands as part of project T6-208-1, “Sustainable orphan drug development through registries and monitoring”. The project was financially supported by Genzyme Corporation, the Dutch Health Care Insurance Board (College voor Zorgverzekeringen), Shire Corporation, the Dutch Steering Committee on Orphan Drugs, Erasmus MC University Medical Center, Utrecht University Medical Center, and the Academic Medical Center at the University of Amsterdam. The project steering committee included representatives of the Dutch Association for Neuromuscular Diseases and the Netherlands patients’ association for metabolic disorders VKS [Volwassenen en kinderen met stofwisselingsziekten].
The National Institute for Public Health and the Environment (RIVM) is gratefully acknowledged for donating newborn screening cards. The authors thank Dr Hui Zhou (Centers for Disease Control and Prevention, Atlanta, Georgia) for providing QC DBS.
Abbreviations
- 4MU
4-methylumbelliferrone
- BMT
Bone marrow transplantation
- DBS
Dried blood spot
- ERT
Enzyme-replacement therapy
- HSCT
Hematopoietic stem-cell transplantation
- IDS
Iduronate-2-sulfatase
- MPS
Mucopolysaccharidosis
- NBS
Newborn screening
Conflict of Interest
Salaries of GJGR, DAG, and SSW were funded in part by a grant through Top Institute Pharma, which was financially supported by Genzyme Corporation, the Dutch Health Care Insurance Board (College voor Zorgverzekeringen), Shire Corporation, the Dutch Steering Committee on Orphan Drugs, Erasmus MC University Medical Center, Utrecht University Medical Center, and the Academic Medical Center at the University of Amsterdam. The corporate sponsors of this research played no role in the design of the study, review, and interpretation of data, or preparation or approval of the manuscript.
AMB, JvdB, ATvdP, LHE, and AJReuser declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
This study was approved by the Erasmus University Medical Center Institutional Review Board.
Details of the Contributions of Individual Authors
G. Ruijter, A. van der Ploeg, S. Weinreich, and A. Reuser contributed to the planning, conduct, and reporting of the work described in the article.
D. Goudriaan, A. Boer, J van den Bosch, and L. Elvers contributed to the conduct and reporting of the work described in the article.
Footnotes
Competing interests: None declared
Contributor Information
G J G Ruijter, Email: g.ruijter@erasmusmc.nl.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Boelens JJ, Aldenhoven M, Purtill D, et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood. 2013;121:3981–3987. doi: 10.1182/blood-2012-09-455238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton BK, Giugliani R. Diagnosing Hunter syndrome in pediatric practice: practical considerations and common pitfalls. Eur J Pediatr. 2012;171:631–639. doi: 10.1007/s00431-012-1703-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S-C, Hwu W-L, Lee N-C, Hsu L-W, Chien Y-H. Algorithm for Pompe disease newborn screening: results from the Taiwan screening program. Mol Gen Metab. 2012;106:281–286. doi: 10.1016/j.ymgme.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Chien Y-H, Chiang S-C, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122:e39–45. doi: 10.1542/peds.2007-2222. [DOI] [PubMed] [Google Scholar]
- Civallero G, Michelin K, de Mari J, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta. 2006;372:98–102. doi: 10.1016/j.cca.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Dajnoki A, Mühl A, Fekete G, et al. Newborn screening for Pompe disease by measuring acid alpha-glucosidase activity using tandem mass spectrometry. Clin Chem. 2008;54:1624–1629. doi: 10.1373/clinchem.2008.107722. [DOI] [PubMed] [Google Scholar]
- Dajnoki A, Fekete G, Keutzer J, et al. Newborn screening for Fabry disease by measuring GLA activity using tandem mass spectrometry. Clin Chim Acta. 2010;411:1428–1431. doi: 10.1016/j.cca.2010.03.009. [DOI] [PubMed] [Google Scholar]
- De Jesus VR, Zhang XK, Keutzer J, et al. Development and Evaluation of Quality Control Dried Blood Spot Materials in Newborn Screening for Lysosomal Storage Disorders. Clin Chem. 2009;55:158–164. doi: 10.1373/clinchem.2008.111864. [DOI] [PubMed] [Google Scholar]
- Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. 2009;154:733–737. doi: 10.1016/j.jpeds.2008.11.041. [DOI] [PubMed] [Google Scholar]
- Holt JB, Poe MD, Escolar ML. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics. 2011;127:e1258–1265. doi: 10.1542/peds.2010-1274. [DOI] [PubMed] [Google Scholar]
- Krivit W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases. Springer Semin Immunopathol. 2004;26:119–132. doi: 10.1007/s00281-004-0166-2. [DOI] [PubMed] [Google Scholar]
- Marsden D, Levy H. Newborn screening of lysosomal storage disorders. Clin Chem. 2010;56:1071–1079. doi: 10.1373/clinchem.2009.141622. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Bodamer O, Burton B, et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr. 2012;171:181–188. doi: 10.1007/s00431-011-1606-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oemardien LF, Boer AM, Ruijter GJG, et al. Hemoglobin precipitation greatly improves 4-methylumbelliferone-based diagnostic assays for lysosomal storage diseases in dried blood spots. Mol Gen Metab. 2011;102:44–48. doi: 10.1016/j.ymgme.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Orsini JJ, Morrissey MA, Slavin LN, et al. Implementation of newborn screening for Krabbe disease: population study and cutoff determination. Clin Biochem. 2009;42:877–884. doi: 10.1016/j.clinbiochem.2009.01.022. [DOI] [PubMed] [Google Scholar]
- Scarpa M, Almássy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi: 10.1186/1750-1172-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sista R, Eckhardt AE, Wang T, Séllos-Moura M, Pamula VK. Rapid, single-step assay for Hunter syndrome in dried blood spots using digital microfluidics. Clin Chim Acta. 2011;412:1895–1897. doi: 10.1016/j.cca.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Okuyama T, Suzuki Y, et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: a nationwide survey in Japan. Mol Gen Metab. 2012;107:513–520. doi: 10.1016/j.ymgme.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Tolun AA, Graham C, Shi Q, et al. A novel fluorometric enzyme analysis method for Hunter syndrome using dried blood spots. Mol Gen Metab. 2012;105:519–521. doi: 10.1016/j.ymgme.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Vellodi A, Young E, Cooper A, Lidchi V, Winchester B, Wraith JE. Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis. 1999;22:638–648. doi: 10.1023/A:1005525931994. [DOI] [PubMed] [Google Scholar]
- Voznyi YV, Keulemans JL, van Diggelen OP. A fluorimetric enzyme assay for the diagnosis of MPS II (Hunter disease) J Inherit Metab Dis. 2001;24:675–680. doi: 10.1023/A:1012763026526. [DOI] [PubMed] [Google Scholar]
- Wolfe BJ, Blanchard S, Sadilek M, Scott CR, Turecek F, Gelb MH. Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: application to screening newborns for mucopolysaccharidosis II (Hunter Syndrome) Anal Chem. 2011;83:1152–1156. doi: 10.1021/ac102777s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–277. doi: 10.1007/s00431-007-0635-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Fernhoff P, Vogt RF. Newborn Bloodspot Screening for Lysosomal Storage Disorders. J Pediatr. 2011;159:7–13. doi: 10.1016/j.jpeds.2011.02.026. [DOI] [PubMed] [Google Scholar]