

Abstract
The transmembrane GH receptor (GHR) exists at least in part as a preformed homodimer on the cell surface. Structural and biochemical studies suggest that GH binds GHR in a 1:2 stoichiometry to effect acute GHR conformational changes that trigger the activation of the receptor-associated tyrosine kinase, Janus kinase 2 (JAK2), and downstream signaling. Despite information about GHR-GHR association derived from elegant fluorescence resonance energy transfer/bioluminescence resonance energy transfer studies, an assessment of the dynamics of GH-induced GHR conformational changes has been lacking. To this end, we used a split luciferase complementation assay that allowed detection in living cells of specific ligand-independent GHR-GHR interaction. Furthermore, GH treatment acutely augmented complementation of enzyme activity between GHRs fused, respectively, to N- and C-terminal fragments of firefly luciferase. Analysis of the temporal pattern of GH-induced complementation changes, pharmacological manipulation, genetic alteration of JAK2 levels, and truncation of the GHR intracellular domain (ICD) tail suggested that GH acutely enhances proximity of the GHR homodimer partners independent of the presence of JAK2, phosphorylation of GHR-luciferase chimeras, or an intact ICD. However, subsequent reduction of complementation requires JAK2 kinase activity and the ICD tail. This conclusion is in contrast to existing models of the GHR activation process.
GH strongly influences growth and metabolism (1–4) and may affect cancer behavior and life span (5–15). GH receptor (GHR) is a single membrane-pass glycoprotein member of the type1 cytokine receptor superfamily (16) that also includes receptors for prolactin, erythropoietin (EPO), leptin, and other hormones. GH binds the cell surface GHR in its extracellular domain (ECD) and causes activation of the receptor's intracellular domain (ICD)-associated cytoplasmic tyrosine kinase, Janus kinase 2 (JAK2), to promote downstream signaling (17–20). GHR is believed to exist at least in part as a homodimer that forms independent of ligand soon after protein synthesis en route to the cell surface (21–23). GH binds GHR at 1:2 ligand-receptor stoichiometry (24, 25) and causes incompletely understood GHR conformational changes that allow associated JAK2 molecules to juxtapose, transactivate, phosphorylate receptor ICD tyrosine residues, and promote phosphorylation-dependent signaling (22, 26–28).
In addition to being a GHR signaling molecule, JAK2's interaction with GHR prevents endoplasmic reticulum-associated degradation of newly synthesized GHR, enhances cell surface GHR presentation and stability, and, if activated, hastens GH-dependent GHR endocytosis/down-regulation (18, 19, 23, 29–34). However, despite elegant structural and computational studies of GH interaction with GHR ECD (24, 35) and recent work on requirements for GHR-GHR interaction (22, 27, 36), no system has emerged to allow assessment of GHR-GHR association and ligand triggering. To this end, we developed a split luciferase complementation assay that allowed detection in living cells of specific ligand-independent GHR-GHR interaction. Furthermore, GH treatment acutely augmented the complementation of enzyme activity between GHRs fused respectively to N- and C-terminal fragments of firefly luciferase. An analysis of the temporal pattern of GH-induced complementation changes, pharmacological manipulation, genetic alteration of JAK2 levels, and truncation of the GHR ICD tail suggested that GH may acutely enhance the proximity of the GHR proximal ICD, a conclusion that contrasts with existing models of the GHR activation process.
Materials and Methods
Materials
Routine reagents were purchased from Sigma-Aldrich Corp unless otherwise noted. Restriction endonucleases were obtained from New England Biolabs. Fetal bovine serum was purchased from Atlanta Biologicals. Gentamicin sulfate, zeocin penicillin, and streptomycin were purchased from Mediatech. Recombinant human GH was kindly provided by Eli Lilly Co. B2036 was obtained from Pfizer, Inc. Recombinant G120R was produced and prepared as previously described (37). Recombinant human EPO (used at 10 U/mL) was obtained from Amgen.
Antibodies
The 4G10 monoclonal antiphosphotyrosine was purchased from Upstate Biotechnology, Inc, as was the antiphosphorylated JAK2 state-specific antibody reactive with JAK2 that is phosphorylated at residues Y1007 and Y1008. Polyclonal antiphosphorylated signal transducer and activator of transcription 5 (STAT5) was purchased from Zymed Laboratories. Polyclonal anti-STAT5 and polyclonal anti-Nluc [antiluciferase (G-19), sc-28525] were purchased from Santa Cruz Biotechnology, Inc. Polyclonal antisera against GHR (anti-GHRcyt-AL47) (38) and JAK2 (anti-JAK2AL33) (39) have been previously described, as have monoclonal anti-GHRext-mAb, anti-GHRext-mAb Fab, anti-GHRext-mAb 18.24, and anti-GHRcyt-mAb and their preparation and purification (40–44). Polyclonal anti-Cluc [antiluciferase polyclonal antibody (G7451)] was from Promega, Inc.
Cells, cell culture, and transfection
γ2A-JAK2 cells were generated by transfection of γ2A cells (45) (a gift of Dr George Stark, Cleveland Clinic, Cleveland, Ohio) with pcDNA3.1(+)/zeo-JAK2 and carried in culture, as described (32, 34). γ2A-JAK2-GHR-Nluc cells were generated by cotransfection of γ2A-JAK2 cells with pcDNA3.1(+)/zeo-GHR-Nluc and a hygromycin-encoding plasmid at a weight ratio as 20:1 and followed by hygromycin selection and single-clone amplification. γ2A-JAK2-truncated GHR (trGHR)-Nluc/trGHR-Cluc cells were generated by cotransfection of pcDNA3.1(+)/zeo-trGHR-Nluc, pcDNA3.1(+)/zeo-trGHR-Cluc, and a hygromycin-encoding plasmid at a weight ratio as 9.5:9.5:1 and followed by hygromycin selection and single-clone amplification. γ2A-JAK2-GHR-Nluc cells and γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells were maintained in 500 μg/mL hygromycin-containing γ2A-JAK2 cell medium. Transient transfection was achieved by introducing pcDNA 3.1-driven plasmids encoding GHR-luciferase chimeras (0.73 pmol for receptor-Nluc-encoding plasmid and 0.37 pmol for receptor-Cluc-encoding plasmid, as indicated, per transfection in a 6 cm2 dish), using Lipofectamine LTX Plus (Invitrogen) according to the manufacturer's instructions.
Construction of GHR, erythropoietin receptor (EPOR), estrogen receptor, and luciferase chimera expression plasmids
The human GHR cDNA was a kind gift provided by Dr R. Ross (University of Sheffield, Sheffield, UK). The full-length firefly luciferase-encoding plasmid has been described (46). The Nluc cDNA construct encoding residues 1–398 with a flexible linker (AAAGSGGGGS) on the amino terminus was created by PCR (primers available upon request) with full-length firefly luciferase as a template. This fragment was subcloned downstream of wild-type human GHR (hGHR) or hGHR truncated after residue 322, both in the context of pcDNA3.1(+)/zeo. The Cluc cDNA construct encoding firefly luciferase residues 394–550 was created in an analogous fashion and similarly subcloned downstream of wild-type or truncated hGHR. Human EPOR cDNA (pRc/CMV hEpoR) was kindly provided by Dr K. Harris (University of Alabama at Birmingham, Birmingham, AL). EPOR-Cluc and truncated EPOR-Cluc (including residues 1–301 of EPOR) were prepared by PCR in an analogous fashion as GHR-Cluc and trGHR-Cluc. pcDNA3.1/puromycin-estrogen receptor (ER) fragment (residues 130–398)-Cluc (ER-Cluc) without a linker has been described (47).
Bioluminescence imaging
Transfected cells expressing the indicated receptor-luciferase chimeras were seeded in 96-well black wall plates (3.6 × 104 cells/well). Six hours prior to experimentation, the medium was replaced by serum-free medium, which was changed just prior to the addition of reagents and imaging to imaging medium [175 μL/well; composed of phenol red free DMEM, 1 g/L glucose, 1 mg/mL D-luciferin, 25 mM HEPES (pH 7.5), 0.1% (wt/vol) BSA] at room temperature for 10 minutes. Baseline bioluminescence signal (photons per second per square centimeter per steradian) was collected at 5-minute intervals for 20 minutes at 37°C using an IVIS 100 system (PerkinElmer; no filter; F-stop,1; field of view, level B; 5 min exposure with Bin 4 or 1 min exposure with Bin 16 in cotransient transfection experiments). GH was added in a volume of 25 μL/well to its indicated final concentration and sequential bioluminescence imaging commenced immediately thereafter at 5-minute intervals, as described above.
For the addition of antagonists, drugs, and antibodies, the same experimental procedures were used, with particular conditions as follows: B2036 (20 μg/mL) cotreatment; G120R (20 μg/mL) cotreatment; anti-GHRext-mAb(40 μg/mL), anti-GHRext-mAb Fab (13.3 μg/mL), anti-GHRext-mAb 18.24 (40 μg/mL), or anti-GHRcyt-mAb (40 μg/mL) at 30 minutes of preincubation; staurosporine (1.25 μM) at 30 minutes of preincubation; AZD1480 (20 μM) at 2 hours of preincubation; sodium orthovanadate (200 μM) at 1 hour of preincubation; and chloroquine (100 μM) at 2 hours of preincubation.
Protein extraction and immunoblotting
Cells prepared in parallel with those described above were plated in six-well plates (5.4 × 105 cells/well) and were serum starved prior to treatment with antagonists, antibodies, or drugs, as above, and stimulation with GH or EPO, as described. Detergent extraction, electrophoresis, and immunoblotting of tissue culture cells were performed as described previously (29, 44).
Statistical analysis and figure preparation
For bioluminescence complementation, each experimental condition was assessed in triplicate or quadruplicate wells of a 96-well plate. Each well was defined as a region of interest that generates a bioluminescence value expressed as total flux (photons per second). The percentage change of complementation signal was calculated by dividing the total flux value from GH- or antibody/Fab-treated wells by baseline total flux value from this same set of wells before treatment. Two-sample t tests with equal variance and pooled SE or (for concentration dependence experiments) one-way ANOVA (Statistical Analyses System, version 9.4 software) were used for statistical analysis. P < .05 was considered as significant. Immunoblots shown are in all instances representative of at least two experiments. In generating the figures, irrelevant intervening lanes from original immunoblots have been cropped for clarity of presentation. In these cases, a space is maintained in which intervening lanes were cropped. In all cases, only data from the same original blots are incorporated in the figures with consistent brightness/contrast adjustment made across each blot.
Results and Discussion
Split luciferase complementation assay reveals specific GHR-GHR interaction
The split luciferase complementation assay is a protein interaction assay with low background and high sensitivity in which one protein is molecularly fused to an N-terminal part (residues 1–398) of firefly luciferase (Nluc) and the other protein is fused to the C-terminal 157 luciferase residues (394–550) (Cluc); interaction of the Nluc- and C-luc-containing protein chimeras yields reconstituted luciferase activity from close approximation in a proper orientation (complementation) of the fragments of the enzyme, neither of which is active alone or when coexpressed with a protein known to not interact (46, 48, 49). This assay has been used to study dimerization and conformational changes of several signaling receptors, including the epidermal growth factor receptor family and others (46, 47, 50–57) but has yet to be exploited to probe the activation of a cytokine receptor system.
We first created chimeras that incorporate human GHR fused to Nluc or Cluc, human EPOR fused to Cluc, or a human ER fragment fused to Cluc (47) (Figure 1A), in each case with a flexible 10-residue linker (AAAGSGGGGS) between receptor and luciferase fragment. Each was transiently expressed in a GHR-deficient, JAK2-expressing human fibrosarcoma cell line, γ2A-JAK2 (32, 34), and specifically detected at expected Mr by anti-GHR (Figure 1B) or antiluciferase (Figure 1C) blotting, indicating each protein was intact. Endoglycosidase H digestion (not shown) verified that both the mature (cell surface) and precursor forms of GHR (31–34) were detected. Consistent with surface expression, GH induced JAK2 and STAT5 phosphorylation and EPO induced JAK2 phosphorylation, as expected, in cells transiently expressing GHR, each GHR chimera, or EPOR-Cluc (Figure 1, D and E).
Figure 1.

Specific luciferase complementation of GHR-Nluc with GHR-Cluc. A, Diagram of luciferase fragment chimeras used in this study. All constructs are described in the text and in Materials and Methods. GHR-Nluc and GHR-Cluc, full-length human GHR fused at the end of the ICD to either Nluc or Cluc, respectively, via a linker peptide; trGHR-Nluc and trGHR-Cluc, human GHR truncated after ICD residue 322 to either N-luciferase or Cluc, respectively, via a linker peptide; EPOR-Cluc, full-length human EPOR fused at the end of the ICD to Cluc via a linker peptide; truncated EPOR-Cluc, human EPOR truncated after ICD residue 301 to either Cluc via a linker peptide; ER-Cluc, human ER fragment fused to Cluc. B, GHR-Nluc and GHR-Cluc are specifically immunodetectable and exhibit expected SDS-PAGE migration. γ2A-JAK2 cells were transiently transfected to express GHR, GHR-Nluc, or GHR-Cluc or with empty vector pcDNA3.1(+)/zeo. Detergent cell extracts were resolved by SDS-PAGE and immunoblotted with anti-GHR (anti-GHRcyt-AL47). Mature forms of GHR and GHR-luciferase chimeras are indicated by an asterisk and exhibit the expected retarded migration (due to differential glycosylation) compared with precursor forms. The blot shown is representative of three such experiments. C, EPOR-Cluc and ER-Cluc are specifically immunodetectable and exhibit expected SDS-PAGE migration. γ2A-JAK2 cells were transiently transfected to express EPOR-Cluc or ER fragment (residues 130–398)-Cluc (ER-Cluc). Detergent cell extracts were resolved by SDS-PAGE and immunoblotted with antiluciferase antibody. Chimera proteins are indicated by asterisks. The blot shown is representative of three such experiments. luc, luciferase; WB, Western blot. D, GHR-Nluc and GHR-Cluc allow normal acute GH-induced signaling. γ2A-JAK2 cells were transfected to express GHR, GHR-Nluc, or GHR-Cluc or with empty vector pcDNA3.1(+)/zeo. After serum starvation for 6 hours, cells were treated with ±GH (500 ng/mL) for 10 minutes. Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated and total JAK2 and phosphorylated and total STAT5. The blot shown is representative of three such experiments. E, EPOR-Cluc allows normal acute EPO-induced signaling. γ2A-JAK2 cells were transfected to express GHR-Nluc and EPOR-Cluc. After serum starvation for 6 hours, the cells were treated with ±EPO (10 U/mL) for 10 minutes. The detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated and total JAK2. The blot shown is representative of two such experiments. F, γ2A-JAK2 cells were transiently transfected with expression plasmids encoding the indicated chimeras. Bioluminescence was determined in triplicate (inset shows actual color coded signals) and is displayed graphically as mean ± SE of total flux (photons per second × 1000). See Materials and Methods for details. The figure shown is representative of two such experiments.
To examine basal (ligand independent) complementation, γ2A-JAK2 cells were transiently cotransfected with GHR-Nluc/GHR-Cluc, GHR-Nluc/EPOR-Cluc, or GHR-Nluc/ER-Cluc, treated with D-luciferin, and imaged with the IVIS-100 system (PerkinElmer) to detect bioluminescence (Figure 1F). Robust complementation was detected for GHR-Nluc/GHR-Cluc, but very little resulted for GHR-Nluc with EPOR-Cluc or GHR-Nluc with ER-Cluc. This indicates that GHR more closely associates with (likely dimerizes with) itself vs either a related cytokine receptor, EPOR, or the cytoplasmically expressed ER. Our luciferase complementation results with transient cotransfection of full-length GHR-Nluc and GHR-Cluc are consistent with elements of previous findings with fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) analysis of GHR dimerization (22), in that a specific constitutive GHR-GHR association was also seen with those techniques. However, we note that our assay measures complementation in intact cells, whereas the FRET and BRET assays previously reported were carried out in purified membrane fractions (22, 36).
Effects of GH treatment on GHR-GHR complementation
Brown et al (22) previously concluded that GH treatment does not affect the degree of GHR-GHR dimerization or exert major conformational changes in the receptor; rather, it was postulated that hormone engagement encourages rotation of dimerized receptors to activate signaling. In part, these conclusions were based on a lack of effect of GH treatment of purified membrane fractions of cells coexpressing appropriate fluorophore/enzyme-tagged GHR chimeras at 25°C on the degree of FRET or BRET signals. More recently the same group extended these studies using a combination of mutagenesis, biochemical and biophysical methods, molecular dynamics modeling, and FRET to conclude that GH induces separation of the proximal GHR ICDs and associated JAK2 molecules as part of the activation process (36). Notably, this conclusion was drawn largely from an analysis of mutant GHR orientations that resulted in constitutive activation. The time course of GH-induced conformational changes in native GHRs was not monitored.
In contrast, others have used FRET with intact cells to detect a transient GH-induced increase in signal that might relate to augmented association of GHRs within preformed dimers (58). We sought to test the effects of GH in our luciferase complementation assay, realizing that detection of such ligand-induced changes would be the most likely if surface receptor expression were optimized. We previously demonstrated that JAK2 expression enhances cell surface GHR expression, as determined by surface biotinylation and immunofluorescence microscopy and that this correlated with the proportion of GHR that achieves mature glycosylation status (30–33). Thus, we prepared γ2A-JAK2-GHR-Nluc, which stably expresses GHR-Nluc in the γ2A-JAK2 background. Anti-GHR immunoblotting (Figure 2A) revealed abundant GHR-Nluc, much of which is mature, suggesting substantial surface expression, as anticipated based on our prior studies (31, 32). Also as expected and consistent with GHR being present at the cell surface, GH treatment promoted rapid phosphorylation of GHR-Nluc, JAK2, and STAT5 (with retarded SDS-PAGE migration of the mature GHR-Nluc and STAT5, typical of robust receptor and STAT5 phosphorylation) (Figure 2A). In dose-response experiments (not shown), GH-induced (10 min) STAT5 phosphorylation was detected with 10 ng/mL GH and nearly maximal with 100–250 ng/mL, as expected based on prior stable wild-type GHR expression in these cells (23).
Figure 2.

GH specifically induces temporal changes in GHR-GHR complementation. A, GH signaling in γ2A-JAK2-GHR-Nluc cells, in which GHR-Nluc is stably expressed in the γ2A-JAK2 background. Cells were serum starved for 6 hours and stimulated ±GH (500 ng/mL) for 10 minutes. Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted with anti-pY, anti-GHRcyt-AL47, anti-p-JAK2, anti-JAK2AL33, anti-p-STAT5, and anti-STAT5. The mature form of GHR is indicated by a bracket and the precursor form is indicated by an arrowhead. Note that the position of the pY GHR-Nluc (detected by anti-pY) and the mature GHR-Nluc (both bracketed) are identical. The blot shown is representative of three such experiments. B, GH concentration-dependent changes in complementation. γ2A-JAK2-GHR-Nluc cells were transiently transfected with an expression plasmid encoding GHR-Cluc. After basal bioluminescence was determined, GH at indicated concentrations was added and bioluminescence was serially determined at 5-minute intervals over 30 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 4 per condition). For 250 ng/mL GH vs 50 ng/mL GH, the value as P < .05 at 5 minutes. For 500 ng/mL GH vs 50 ng/ml GH, the value was P < .05 at 5 minutes. For 100 ng/mL, 250 ng/mL, and 500 ng/mL GH, the value was P < .05 for 20 minutes, 25 minutes, and 30 minutes each vs 5 minutes. For 250 ng/mL and 500 ng/mL GH, the value was P < .05 for 30 minutes each vs 10 minutes. For 500 ng/mL, the value was P < .05 for 15 minutes vs 5 minutes. The figure shown is representative of three such experiments. C, GH induces sustained JAK2 phosphorylation. γ2A-JAK2-GHR-Nluc cells were transiently transfected with GHR-Cluc, serum starved for 6 hours, and then treated with 500 ng/mL GH for 0–30 minutes, as indicated. Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated and total JAK2. The blot shown is representative of three such experiments. D, GHR-specific antagonist, B2036, inhibits GH-induced JAK2 activation. γ2A-JAK2-GHR-Nluc cells were transiently transfected with GHR-Cluc, serum starved for 6 hours, and then treated ±GH (500 ng/mL, 10 min), with B2036 (20 μg/mL, 10 min) alone, or cotreated with GH plus B2036. Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated and total JAK2. The blot shown is representative of three such experiments. E, GHR-specific antagonist, B2036, inhibits GH-induced changes in GHR-GHR complementation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated with GH (500 ng/mL), B2036 (20 μg/mL), or GH plus B2036, as indicated, after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. The data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). For GH vs GH+B2036, the value was P < .05 at each time point, except at 30 minutes, 35 minutes, and 40 minutes. The figure shown is representative of three such experiments.
We transfected GHR-Cluc into γ2A-JAK2-GHR-Nluc cells, yielding specific basal luciferase complementation, as expected. To track GHR-GHR complementation, we adopted a 96-well format protocol of GH treatment at 37°C and serial bioluminescence measurements, using in vivo imaging system imaging. Interestingly, GH induced substantial concentration-dependent changes in complementation (Figure 2B), acutely augmenting bioluminescence by approximately 40%–60% over basal at 500 ng/mL with a reproducible time-dependent pattern thereafter (Figures 2B, 2E, 3D, and 4B). The pattern of GH-induced complementation change is summarized as follows: 1) acute increase (0–5 min; upstroke); 2) rapid partial decline (5–10 min; downstroke); and 3) more moderate decline (10–40 min) to near baseline. Of note, GH-induced phosphorylation of GHR-luciferase chimeras (not shown), JAK2 (Figure 2C), and STAT5 (not shown) was detected in these cells within 5 minutes and persisted for at least 30 minutes. Thus, the temporal pattern of GH-induced changes in GHR-Nluc/GHR-Cluc complementation may reflect the postengagement conformational changes in GHRs that are involved in signaling.
Figure 3.

Effect of antagonistic GHR antibodies on GHR-GHR complementation. A, Diagram of regions of GHR that interact with each monoclonal antibody (or Fab) used in this study, GH, or the GH antagonist, B2036. The regions indicated are subdomains 1 and 2 of the ECD and the ICD. B, GHR-specific monoclonal antibodies or a Fab fragment specifically inhibit GH-induced phosphorylation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc cells were serum starved, preincubated with anti-GHRext-mAb (40 μg/mL), anti-GHRext-mAb-18.24 (40 μg/mL), anti-GHRext-mAb-Fab (13.3 μg/mL), or anti-GHRcyt-mAb (40 μg/mL) (a control mAb against the GHR ICD) for 30 minutes, and then treated ±GH (500 ng/mL, 10 min). Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated GHR-luciferase chimeras (with anti-pY) and total GHR-luciferase chimeras and phosphorylated and total JAK2. The blot shown is representative of three such experiments. C, Treatment with GHR-specific monoclonal antibodies or a Fab fragment specifically augments basal (non-GH dependent) GHR-GHR complementation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated with vehicle, anti-GHRext-mAb (40 μg/mL), anti-GHRext-mAb-18.24 (40 μg/mL), anti-GHRext-mAb-Fab (13.3 μg/mL), or anti-GHRcyt-mAb (40 μg/mL, a control mAb against the GHR ICD) for 30 minutes, after which bioluminescence was determined. Data are displayed graphically as mean ± SE total flux (photons per second × 1000). *, P < .05 compared with vehicle treatment. The figure shown is representative of three such experiments. D, GHR-specific monoclonal antibodies or Fab fragment specifically inhibit GH-induced changes in GHR-GHR complementation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated with GH (500 ng/mL) or GH plus the indicated antibodies (40 μg/mL) or Fab fragment (13.3 μg/mL) after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). Baseline signal is normalized for each condition. For GH vs GH+anti-GHRext-mAb/anti-GHRext-mAb 18.24/anti-GHRext-mAb Fab, the value was P < .05 at each time point; for GH vs GH+anti-GHRcyt-mAb, the value was P > .05 at each time point. The figure shown is representative of three such experiments.
Figure 4.

JAK2 activity is not required for GH-induced augmentation of GHR-GHR complementation but contributes to the subsequent decline in complementation. A, Staurosporine inhibits and orthovanadate enhances acute GH-induced signaling in γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc. γ2A-JAK2-GHR-Nluc cells were transiently transfected with GHR-Cluc, serum starved for 6 hours, and treated ±GH (500 ng/mL, 10 min) alone or after pretreatment with either staurosporine (1.25 μM, 30 min), or sodium orthovanadate (200 μM, 1 h). Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated GHR (with anti-pY) and total GHR and phosphorylated and total JAK2. The blot shown is representative of three such experiments. B, Staurosporine prevents and vanadate enhances the decline in GH-induced GHR-GHR complementation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated with GH (500 ng/mL) alone or in the presence of either staurosporine (1.25 μM) or vanadate (200 μM), as indicated, after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). For GH vs GH+staurosporine, the value was P < .05 at each time point; for GH vs GH+vanadate, the value was P < .05 at each time point. The figure shown is representative of three such experiments. C, JAK-specific kinase inhibitor, AZD1480, mimics the effect of staurosporine on GH-induced GHR-GHR complementation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated with GH (500 ng/mL) in the presence of either staurosporine (1.25 μM) or AZD1480 (20 μM), as indicated, after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). The figure shown is representative of two such experiments. D, AZD1480 inhibits GH-induced GHR-luciferase chimera phosphorylation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated ±GH (500 ng/mL, 10 min) alone or after pretreatment with either staurosporine (1.25 μM, 30 min) or AZD1480 (20 μM, 2 h), as indicated. Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated GHR-luciferase chimeras (with anti-pY) and total GHR-luciferase chimeras. The blot shown is representative of two such experiments.
B2036 is a GH mutant GHR-specific antagonist that binds GHR with high affinity but cannot induce conformational changes required for signaling (59); thus, B2036 itself does not activate GHR signaling but prevents receptor activation by GH. As expected, B2036 did not activate JAK2 in γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc and strongly inhibited GH-induced JAK2 activation (Figure 2D). Importantly, B2036 did not itself alter basal GHR-GHR complementation but completely inhibited GH-induced complementation changes (Figure 2E), verifying that the GH-induced complementation changes are closely linked to GHR activation. [In data not shown, the same effects were observed using a separate GH antagonist, hGH-G120R (G120R) (26, 60).]
These data suggest a relationship between GHR-Nluc/GHR-Cluc complementation and GHR signaling. To further study this relationship, we used our antagonist GHR monoclonal antibodies, anti-GHRext-mAb and anti-GHRext-mAb-18.24, that prevent GH-induced GHR signaling but are not themselves able to activate GHR; these characteristics are shared by a monovalent Fab fragment of anti-GHRext-mAb (43, 44). In contrast to GH and B2036, which interact mainly with GHR ECD subdomain 1 residues, anti-GHRext-mAb and anti-GHRext-mAb-18.24 have similar, but nonidentical, discontinuous epitopes within ECD subdomain 2 including the interface between GHR monomers within the GHR dimer (see Figure 3A). This is consistent with findings (43) that anti-GHRext-mAb does not significantly impair GH binding and suggests anti-GHRext-mAb and anti-GHRmAb 18.24 block GH signaling by preventing GH-induced conformational changes that allow signaling and/or by inducing unproductive conformational changes within GHR dimers that prevent subsequent GH-induced signaling. Indeed, neither monoclonal antibody (mAb) induced signaling and both blocked GH-induced phosphorylation in γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc (Figure 3B). Yet, unlike B2036, both antibodies and the Fab fragment, but not a control mAb (anti-GHRcyt-mAb), themselves markedly enhanced basal (no GH) GHR-Nluc/GHR-Cluc complementation that plateaued after 30–40 minutes at 140%–160% increase over basal (Figure 3C). However, GH-induced complementation changes were specifically blocked by pretreatment with anti-GHRext-mAb, anti-GHRext-mAb 18.24, and anti-GHRext-mAb Fab (Figure 3D). These data suggest that complementation, presumably reflecting GHR dimer partner conformational changes, can occur without activation of signaling but that both its pattern and degree differ from that induced by GH and associated with signaling. Furthermore, the degree of complementation induced by the antibodies may reflect a maximum that cannot be exceeded by hormone treatment (see also below in Figure 4, B and C).
The presence of JAK2- and GH-induced kinase activity affects the pattern of GH-induced GHR-GHR complementation
To better understand the determinants of GH-induced GHR-GHR complementation, we examined factors that impact GHR signaling without affecting GH engagement per se. Staurosporine is a kinase inhibitor that prevents GH-induced JAK2 activation and GHR-luciferase chimera tyrosine phosphorylation (references 40, 62, and 63 and Figure 4A). Conversely, sodium orthovanadate (vanadate), a protein tyrosine phosphatase inhibitor, potentiates GH-induced JAK2 and GHR-luciferase chimera phosphorylation (Figure 4A). Both staurosporine and vanadate treatments notably affected GH-induced GHR-GHR complementation (Figure 4B), whereas neither affected basal complementation (not shown). Vanadate blunted (but did not prevent) the GH-induced complementation upstroke but dramatically hastened and augmented the decline to below baseline levels. In contrast, staurosporine substantially increased the upstroke (by more than 100% above basal) and prevented the rapid downstroke and decline over 40 minutes. The effect of staurosporine on GH-induced GHR-GHR complementation was mimicked by the JAK-specific kinase inhibitor, AZD1480 (64, 65) (Figure 4, C and D), suggesting the effect was via inhibition of JAK2. These data indicate that neither tyrosine kinase nor protein tyrosine phosphatase activity is required for the acute complementation upstroke, but GH-induced JAK2 activity promotes the downstroke and return to baseline.
To further pursue this, we isolated a stable clone in which GHR-Nluc was expressed without JAK2 in JAK2-deficient γ2A cells. As expected, GH-induced GHR-luciferase chimera and STAT5 phosphorylation were absent in these cells without immunodetectable JAK2 (Figure 5A). Transfection of GHR-Cluc into γ2A-GHR-Nluc cells enabled robust (∼250% increase above basal) GH-induced acute GHR-GHR complementation upstroke (Figure 5B), but, like staurosporine and AZD1480 treatments, no reduction in complementation ensued over the next 35 minutes in the cells lacking JAK2 expression, consistent with the idea that GH-induced JAK2 activity and/or phosphorylation of JAK2 substrates both blunt the acute increase in complementation and promote its subsequent decline.
Figure 5.
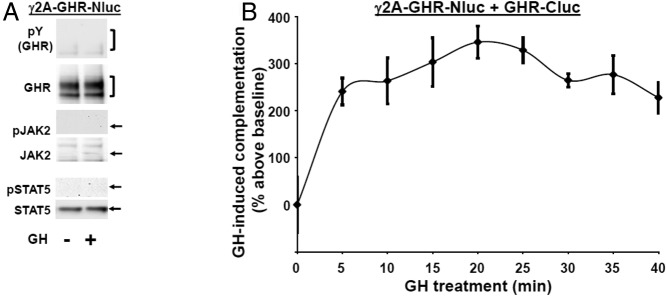
GH augments acute GHR-GHR complementation but fails to cause a subsequent decline in complementation in JAK2-deficient cells. A, JAK2-deficient cells lack GH-induced phosphorylation of GHR-Nluc, JAK2, and STAT5. Serum-starved JAK2-deficient γ2A-GHR-Nluc cells were treated ±GH (500 ng/mL, 10 min). Detergent cells extracts were resolved by SDS-PAGE and sequentially immunoblotted with anti-pY, anti-GHRcyt-AL47, anti-p-JAK2, anti-JAK2AL33, anti-p-STAT5, and anti-STAT5. GHR-Nluc is indicated by a bracket. The positions of JAK2, pJAK2, STAT5, and pSTAT5 are indicated by arrows. Note the absence of phosphorylated GHR-Nluc, JAK2, or STAT5 and the lack of JAK2. The blot shown is representative of two such experiments. B, JAK2 deficiency prevents the decline in GH-induced GHR-GHR complementation. γ2A-GHR-Nluc cells transiently expressing GHR-Cluc were treated with GH (500 ng/mL) after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). The figure shown is representative of three such experiments.
A parsimonious interpretation may be that GH acutely induces the C-terminal tails of predimerized ICDs to move toward each other but that JAK2 activity promotes rapid and steady splaying of the chimera tails from each other. However, the decline in complementation could also reflect, in principle, GH- and tyrosine phosphorylation-dependent GHR-luciferase chimera down-regulation/degradation (29, 32, 66). To test this, we expressed GHR-Cluc in γ2A-JAK2-GHR-Nluc cells and treated cells with GH in the absence or presence of chloroquine, which blocks GH-dependent GHR down-regulation in γ2A-JAK2-GHR cells (32). The profile of GH-induced GHR-GHR complementation varied only slightly in the presence of chloroquine, manifesting only a modest lessening of the decline during the 20- to 40-minute period of GH exposure (Figure 6). Thus, GH-induced GHR-luciferase chimera down-regulation contributes little to the complementation decline. Another possible interpretation for the decline in complementation may be a phosphorylation-dependent reduction in luciferase activity. Such a finding has not been reported but cannot be entirely ruled out.
Figure 6.
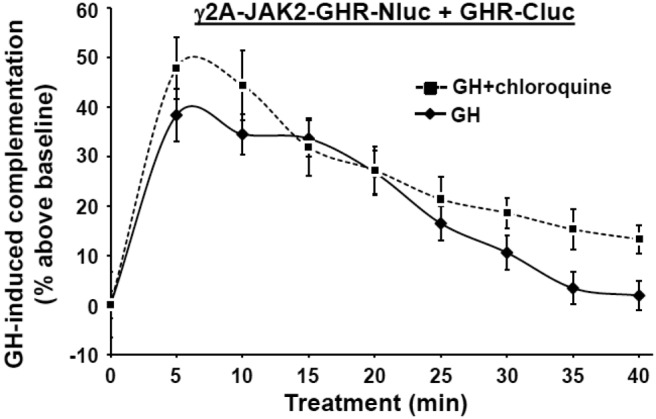
Chloroquine only modestly affects GH-induced GHR-GHR complementation. γ2A-JAK2-GHR-Nluc cells transiently expressing GHR-Cluc were treated with GH (500 ng/mL) alone or in the presence of chloroquine (100 μM), as indicated, after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). A value of P < .05 was attained at 30 minutes, 35 minutes, and 40 minutes. The figure shown is representative of three such experiments.
GH-induced complementation of truncated GHR-luciferase chimera dimers
The experiments in Figures 1–6 use chimeras incorporating full-length GHRs fused at their distal ICD tails to luciferase fragments that report proximity of the distal tails of GHR dimers. However, JAK2 associates with GHR in proline-rich Box1 element within the proximal ICD (17, 18, 20). Recent theory regarding the mechanism of GH-induced JAK2 activation suggests that GH acutely induces GHR proximal ICDs to move apart (35, 36) rather than together; however, experimental evidence for this is lacking in terms of dynamic assessment of GH-induced changes in living cells. We thus asked how GH-induced complementation changes for full-length GHR-luciferase chimeras compare with conformational changes in the proximal ICD in which JAK2 associates by preparing chimeras in which Nluc or Cluc fragments are fused to GHRs truncated at ICD residue 322; these chimeras [trGHR-Nluc and trGHR-Cluc (diagrammed in Figure 1A)] include only 51 of the 350 ICD residues and retain the Box1 element and thus JAK2 association. Coexpression of trGHR-Nluc with trGHR-Cluc specifically complemented (Figure 7A).
Figure 7.

Basal and GH-augmented trGHR-trGHR luciferase complementation. A, Specific luciferase complementation of trGHR-Nluc with trGHR-Cluc. γ2A-JAK2 cells were transiently transfected with expression plasmids encoding the indicated chimeras. Bioluminescence was determined in triplicate (inset shows actual color coded signals) and is displayed graphically as mean ± SE total flux (photons per second × 1000). See Materials and Methods for details. The figure shown is representative of two such experiments. B, GH signaling in γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells, in which trGHR-Nluc and trGHR-Cluc are stably coexpressed in the γ2A-JAK2 background. Cells were serum starved for 6 hours and stimulated ±GH (500 ng/mL) for 10 minutes. Detergent cells extracts were resolved by SDS-PAGE and sequentially immunoblotted with anti-Nluc, anti-Cluc, anti-p-JAK2, and anti-JAK2AL33. The blot shown is representative of two such experiments. C, GH concentration-dependent changes in complementation. γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells were treated with GH at the indicated concentrations after basal bioluminescence was determined. Bioluminescence was thereafter serially determined at 5 minutes intervals over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). For each time point, a value of P < .05 was attained for signals produced by each GH concentration with the exception of the following: 20 minutes, 25 minutes, 30 minutes, 35 minutes, and 40 minutes, 250 ng/mL GH vs 500 ng/mL GH; and 40 minutes, 100 ng/mL GH vs 250 ng/mL. The figure shown is representative of two such experiments. D, GHR-specific antagonist, B2036, inhibits GH-induced JAK2 activation via ICD-truncated GHR chimeras. γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells were serum starved for 6 hours and then treated ±GH (500 ng/mL, 10 min), with B2036 (20 μg/mL, 10 min) alone, or cotreated with GH plus B2036. Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated and total JAK2. The blot shown is representative of two such experiments. E, GHR-specific antagonist, B2036, inhibits GH-induced changes in trGHR-trGHR complementation. γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells were treated with GH (500 ng/mL), B2036 (20 μg/mL), or GH plus B2036, as indicated, after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 25 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). GH vs GH+B2036: P < .05 at each time point. The figure shown is representative of two such experiments.
To examine GH's impact on trGHR-Nluc/trGHR-Cluc complementation, we prepared γ2A-JAK2-trGHR-Nluc/trGHR-Cluc, in which both truncated GHR chimeras are stably coexpressed in γ2A-JAK2 cells. Immunoblotting verified expression of each chimera at the expected Mr and that GH induced acute JAK2 tyrosine phosphorylation, as expected (Figure 7B). We tested effects of GH on trGHR-trGHR complementation in combined concentration and time-course experiments (Figure 7C). GH dose dependently promoted acutely increased complementation of up to nearly 40% above basal within the first few minutes of treatment, similar to the effect of GH on full-length GHR-luciferase chimera complementation observed in Figure 2B, indicating that truncated, predimerized GHR-luciferase chimeras experience an acute GH-induced further approximation of the attached luciferase fragments, which in these chimeras reside in the proximal ICD near its JAK2-associating region. Furthermore, as observed with full-length GHR-luciferase chimeras pretreated with staurosporine or expressed in the absence of JAK2, this GH-induced abrupt increase in complementation did not subsequently decrease. Indeed, complementation dose dependently further increased, reaching a sustained maximum with 500 ng/mL GH. Further evidence of the specificity of augmented trGHR/trGHR complementation is that B2036 alone had no effect but completely blocked GH's effect on complementation of the truncated GHR-luciferase chimeras (Figure 7, D and E). This suggests GH engages both the full-length and truncated GHR-luciferase chimeras similarly, as we previously found for nonluciferase chimerized GHRs (18). Also consistent with findings for GHR-luciferase chimeras, basal (non-GH dependent) complementation was specifically increased (without downstream signaling) by anti-GHRext-mAb, anti-GHRext-mAb 18.24, and anti-GHRext-mAb Fab but not the control anti-GHRcyt-mAb (Figure 8, A and B) and GH-induced trGHR-luciferase chimera complementation is blocked by the two specific mAbs and the Fab (Figure 8C).
Figure 8.

Effect of antagonistic GHR antibodies on trGHR-trGHR complementation. A, Treatment with GHR-specific monoclonal antibodies or a Fab fragment specifically augments basal (non-GH dependent) trGHR-trGHR complementation. γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells Cluc were treated with vehicle, anti-GHRext-mAb (40 μg/mL), anti-GHRext-mAb-18.24 (40 μg/mL), anti-GHRext-mAb-Fab (13.3 μg/mL), or anti-GHRcyt-mAb (40 μg/mL, a control mAb against the GHR ICD) for 30 minutes, after which bioluminescence was determined. Data are displayed graphically as mean ± SE total flux (photons per second × 10000). *, P < .05 compared with vehicle treatment. The figure shown is representative of two such experiments. B, GHR-specific monoclonal antibodies or a Fab fragment blocks GH-induced JAK2 phosphorylation via ICD truncated GHR-luciferase chimeras. γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells were serum starved, preincubated with anti-GHRext-mAb (40 μg/mL), anti-GHRext-mAb-18.24 (40 μg/mL), anti-GHRext-mAb-Fab (13.3 μg/mL), or anti-GHRcyt-mAb (40 μg/mL) (a control mAb against the GHR ICD) for 30 minutes and then treated ±GH (500 ng/mL, 10 min). Detergent cell extracts were resolved by SDS-PAGE and sequentially immunoblotted to detect phosphorylated and total JAK2. The blot shown is representative of two such experiments. C, GHR-specific monoclonal antibodies or Fab fragment specifically inhibit GH-induced changes in trGHR-trGHR complementation. γ2A-JAK2-trGHR-Nluc/trGHR-Cluc cells were treated with GH (500 ng/mL) or GH plus the indicated antibodies (40 μg/mL) or Fab fragment (13.3 μg/mL), after basal bioluminescence was determined. Bioluminescence was measured serially thereafter over 40 minutes. Data are expressed as mean ± SE of GH-induced signal as a percentage above baseline signal (n = 3 per condition). For GH vs GH+anti-GHRext-mAb/anti-GHRext-mAb 18.24/anti-GHRext-mAb Fab, a value of P < .05 was attained at each time point; for GH vs GH+anti-GHRcyt-mAb, a value of P > .05 was attained at each time point. Note that the data for GH-only treatment are the same as those for GH (500 ng/mL) shown in Figure 7C because they are from the same experiment. The figure shown is representative of three such experiments.
These findings obtained with cells that express only truncated GHR chimeras bear important implications. They echo previous findings (22, 23) in indicating that GHR predimerization does not require most the receptor ICD, but more compellingly, they suggest that GH acutely causes enhanced proximity of the proximal ICD. Whether this reflects critical features related to the activation mechanism of the associated JAK2 molecules is a worthwhile area for further exploration that uses the luciferase complementation methods herein. In comparison with the existing data with FRET and BRET methods, the reliable and dissectible temporal nature of GH-dependent complementation we observe with our methods should provide a useful scaffold for such studies. As with each of these methods, a caveat to be addressed is the possibility that the detection of enhanced proximity may also reflect higher order (oligomeric rather than dimeric) complex formation. Thus, companion biochemical and biophysical techniques (61) may be useful in these efforts. We also note that our findings of GH-induced enhanced complementation could, in principle, reflect a more favorable approximation of luciferase fragments resulting from the proximal ICDs transiently separating and rotating in response to GH. Further studies are required to determine whether such a conclusion may better harmonize our findings with recent models of GHR activation (35, 36).
Acknowledgments
We acknowledge the expert assistance of Sharon Samuel and helpful discussion with members of the Frank laboratory.
This work was supported by National Institutes of Health Grants DK58259 and DK46395 (both to S.J.F.) and a Veterans Affairs Merit Review award (to S.J.F.).
Parts of this work were presented at the 95th Annual Meeting of The Endocrine Society, San Francisco, CA, 2013, and the 96th Annual Meeting of The Endocrine Society, Chicago, IL, 2014.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BRET
- bioluminescence resonance energy transfer
- ECD
- extracellular domain
- EPO
- erythropoietin
- EPOR
- EPO receptor
- ER
- estrogen receptor
- FRET
- fluorescence resonance energy transfer
- GHR
- GH receptor
- hGHR
- human GHR
- ICD
- intracellular domain
- JAK2
- Janus kinase 2
- mAb
- monoclonal antibody
- STAT5
- signal transducer and activator of transcription 5
- trGHR
- truncated GHR.
References
- 1. Isaksson OG, Eden S, Jansson JO. Mode of action of pituitary growth hormone on target cells. Annu Rev Physiol. 1985;47:483–499 [DOI] [PubMed] [Google Scholar]
- 2. Kaplan S. Hormonal regulation of growth and metabolic effects of growth hormone. In: Kostyo J, Goodman HM, eds. Handbook of Physiology. New York: Oxford University Press; 1999:129–143 [Google Scholar]
- 3. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30:152–177 [DOI] [PubMed] [Google Scholar]
- 4. Waters MJ, Hoang HN, Fairlie DP, Pelekanos RA, Brown RJ. New insights into growth hormone action. J Mol Endocrinol. 2006;36:1–7 [DOI] [PubMed] [Google Scholar]
- 5. Wang Z, Luque RM, Kineman RD, et al. Disruption of growth hormone signaling retards prostate carcinogenesis in the probasin/TAg rat. Endocrinology. 2008;149:1366–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang X, Mehta RG, Lantvit DD, et al. Inhibition of estrogen-independent mammary carcinogenesis by disruption of growth hormone signaling. Carcinogenesis. 2007;28:143–150 [DOI] [PubMed] [Google Scholar]
- 7. Wang Z, Prins GS, Coschigano KT, et al. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology. 2005;146:5188–5196 [DOI] [PubMed] [Google Scholar]
- 8. Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology. 2003;144:3799–3810 [DOI] [PubMed] [Google Scholar]
- 9. Bonkowski MS, Rocha JS, Masternak MM, Al Regaiey KA, Bartke A. Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc Natl Acad Sci USA. 2006;103:7901–7905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ikeno Y, Hubbard GB, Lee S, et al. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J Gerontol A Biol Sci Med Sci. 2009;64:522–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swanson SM, Unterman TG. The growth hormone-deficient Spontaneous Dwarf rat is resistant to chemically induced mammary carcinogenesis. Carcinogenesis. 2002;23:977–982 [DOI] [PubMed] [Google Scholar]
- 13. Shen Q, Lantvit DD, Lin Q, et al. Advanced rat mammary cancers are growth hormone dependent. Endocrinology. 2007;148:4536–4544 [DOI] [PubMed] [Google Scholar]
- 14. Ramsey MM, Ingram RL, Cashion AB, et al. Growth hormone-deficient dwarf animals are resistant to dimethylbenzanthracine (DMBA)-induced mammary carcinogenesis. Endocrinology. 2002;143:4139–4142 [DOI] [PubMed] [Google Scholar]
- 15. Steuerman R, Shevah O, Laron Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur J Endocrinol. 2011;164:485–489 [DOI] [PubMed] [Google Scholar]
- 16. Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci USA. 1990;87:6934–6938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Argetsinger LS, Campbell GS, Yang X, et al. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell. 1993;74:237–244 [DOI] [PubMed] [Google Scholar]
- 18. Frank SJ, Gilliland G, Kraft AS, Arnold CS. Interaction of the growth hormone receptor cytoplasmic domain with the JAK2 tyrosine kinase. Endocrinology. 1994;135:2228–2239 [DOI] [PubMed] [Google Scholar]
- 19. Frank SJ, Yi W, Zhao Y, et al. Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor. J Biol Chem. 1995;270:14776–14785 [DOI] [PubMed] [Google Scholar]
- 20. Vanderkuur JA, Wang X, Zhang L, et al. Domains of the growth hormone receptor required for association and activation of JAK2 tyrosine kinase. J Biol Chem. 1994;269:21709–21717 [PubMed] [Google Scholar]
- 21. Gent J, van Kerkhof P, Roza M, Bu G, Strous GJ. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc Natl Acad Sci USA. 2002;99:9858–9863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown RJ, Adams JJ, Pelekanos RA, et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol. 2005;12:814–821 [DOI] [PubMed] [Google Scholar]
- 23. Yang N, Wang X, Jiang J, Frank SJ. Role of the growth hormone (GH) receptor transmembrane domain in receptor predimerization and GH-induced activation. Mol Endocrinol. 2007;21:1642–1655 [DOI] [PubMed] [Google Scholar]
- 24. de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science. 1992;255:306–312 [DOI] [PubMed] [Google Scholar]
- 25. Cunningham BC, Ultsch M, De Vos AM, Mulkerrin MG, Clauser KR, Wells JA. Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science. 1991;254:821–825 [DOI] [PubMed] [Google Scholar]
- 26. Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA. Rational design of potent antagonists to the human growth hormone receptor. Science. 1992;256:1677–1680 [DOI] [PubMed] [Google Scholar]
- 27. Rowlinson SW, Yoshizato H, Barclay JL, et al. An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway. Nat Cell Biol. 2008;10:740–747 [DOI] [PubMed] [Google Scholar]
- 28. Frank SJ. Receptor dimerization in GH and erythropoietin action—it takes two to tango, but how? Endocrinology. 2002;143:2–10 [DOI] [PubMed] [Google Scholar]
- 29. Deng L, Jiang J, Frank SJ. Growth hormone-induced JAK2 signaling and GH receptor down-regulation: role of GH receptor intracellular domain tyrosine residues. Endocrinology. 2012;153:2311–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He K, Wang X, Jiang J, et al. Janus kinase 2 determinants for growth hormone receptor association, surface assembly, and signaling. Mol Endocrinol. 2003;17:2211–2227 [DOI] [PubMed] [Google Scholar]
- 31. He K, Loesch K, Cowan JW, et al. Janus kinase 2 enhances the stability of the mature growth hormone receptor. Endocrinology. 2005;146:4755–4765 [DOI] [PubMed] [Google Scholar]
- 32. Deng L, He K, Wang X, et al. Determinants of growth hormone receptor down-regulation. Mol Endocrinol. 2007;21:1537–1551 [DOI] [PubMed] [Google Scholar]
- 33. Loesch K, Deng L, Wang X, He K, Jiang J, Frank SJ. Endoplasmic reticulum-associated degradation of growth hormone receptor in Janus kinase 2-deficient cells. Endocrinology. 2007;148:5955–5965 [DOI] [PubMed] [Google Scholar]
- 34. Loesch K, Deng L, Cowan JW, et al. JAK2 influences growth hormone receptor metalloproteolysis. Endocrinology. 2006;147:2839–2849 [DOI] [PubMed] [Google Scholar]
- 35. Pang X, Zhou HX. A common model for cytokine receptor activation: combined scissor-like rotation and self-rotation of receptor dimer induced by class I cytokine. PLoS Comput Biol. 2012;8:e1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brooks AJ, Dai W, O'Mara ML, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344:1249783. [DOI] [PubMed] [Google Scholar]
- 37. Yang N, Langenheim JF, Wang X, Jiang J, Chen WY, Frank SJ. Activation of growth hormone receptors by growth hormone and growth hormone antagonist dimers: insights into receptor triggering. Mol Endocrinol. 2008;22:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, Guan R, Jiang J, et al. Growth hormone (GH)-induced dimerization inhibits phorbol ester-stimulated GH receptor proteolysis. J Biol Chem. 2001;276:24565–24573 [DOI] [PubMed] [Google Scholar]
- 39. Jiang J, Liang L, Kim SO, Zhang Y, Mandler R, Frank SJ. Growth hormone-dependent tyrosine phosphorylation of a GH receptor-associated high molecular weight protein immunologically related to JAK2. Biochem Biophys Res Commun. 1998;253:774–779 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y, Jiang J, Kopchick JJ, Frank SJ. Disulfide linkage of growth hormone (GH) receptors (GHR) reflects GH-induced GHR dimerization. Association of JAK2 with the GHR is enhanced by receptor dimerization. J Biol Chem. 1999;274:33072–33084 [DOI] [PubMed] [Google Scholar]
- 41. Alele J, Jiang J, Goldsmith JF, Yang X, et al. Blockade of growth hormone receptor shedding by a metalloprotease inhibitor. Endocrinology. 1998;139:1927–1935 [DOI] [PubMed] [Google Scholar]
- 42. Kim SO, Jiang J, Yi W, Feng GS, Frank SJ. Involvement of the Src homology 2-containing tyrosine phosphatase SHP-2 in growth hormone signaling. J Biol Chem. 1998;273:2344–2354 [DOI] [PubMed] [Google Scholar]
- 43. Jiang J, Wang X, He K, et al. A conformationally-sensitive GHR [growth hormone (GH) receptor] antibody: impact on GH signaling and GHR proteolysis. Mol Endocrinol. 2004;18:2981–2996 [DOI] [PubMed] [Google Scholar]
- 44. Jiang J, Wan Y, Wang X, et al. Inhibitory GH receptor extracellular domain monoclonal antibodies: three-dimensional epitope mapping. Endocrinology. 2011;152:4777–4788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kohlhuber F, Rogers NC, Watling D, et al. A JAK1/JAK2 chimera can sustain α and γ interferon responses. Mol Cell Biol. 1997;17:695–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Paulmurugan R, Gambhir SS. Firefly luciferase enzyme fragment complementation for imaging in cells and living animals. Anal Chem. 2005;77:1295–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paulmurugan R, Gambhir SS. An intramolecular folding sensor for imaging estrogen receptor-ligand interactions. Proc Natl Acad Sci USA. 2006;103:15883–15888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA. 2004;101:12288–12293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Villalobos V, Naik S, Piwnica-Worms D. Detection of protein-protein interactions in live cells and animals with split firefly luciferase protein fragment complementation. Methods Mol Biol. 2008;439:339–352 [DOI] [PubMed] [Google Scholar]
- 50. Yang KS, Ilagan MX, Piwnica-Worms D, Pike LJ. Luciferase fragment complementation imaging of conformational changes in the EGF receptor. J Biol Chem. 2009;284(12):7474–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Macdonald-Obermann JL, Adak S, Landgraf R, Piwnica-Worms D, Pike LJ. Dynamic analysis of the EGF receptor-ErbB2-ErbB3 network by luciferase fragment complementation imaging. J Biol Chem. 2013;288(42):30773–30784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Macdonald-Obermann JL, Piwnica-Worms D, Pike LJ. Mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc Natl Acad Sci USA. 2012;109:137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paulmurugan R, Massoud TF, Huang J, Gambhir SS. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004;64:2113–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paulmurugan R, Umezawa Y, Gambhir SS. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc Natl Acad Sci USA. 2002;99:15608–15613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Paulmurugan R, Tamrazi A, Massoud TF, Katzenellenbogen JA, Gambhir SS. In vitro and in vivo molecular imaging of estrogen receptor α and β homo- and heterodimerization: exploration of new modes of receptor regulation. Mol Endocrinol. 2011;25:2029–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Defour JP, Itaya M, Gryshkova V, et al. Tryptophan at the transmembrane-cytosolic junction modulates thrombopoietin receptor dimerization and activation. Proc Natl Acad Sci USA. 2013;110:2540–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luker KE, Gupta M, Steele JM, Foerster BR, Luker GD. Imaging ligand-dependent activation of CXCR7. Neoplasia. 2009;11:1022–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Biener-Ramanujan E, Ramanujan VK, Herman B, Gertler A. Spatio-temporal kinetics of growth hormone receptor signaling in single cells using FRET microscopy. Growth Horm IGF Res. 2006;16:247–257 [DOI] [PubMed] [Google Scholar]
- 59. Goffin V, Bernichtein S, Carriere O, Bennett WF, Kopchick JJ, Kelly PA. The human growth hormone antagonist B2036 does not interact with the prolactin receptor. Endocrinology. 1999;140:3853–3856 [DOI] [PubMed] [Google Scholar]
- 60. Kopchick JJ, Parkinson C, Stevens EC, Trainer PJ. Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev. 2002;23:623–646 [DOI] [PubMed] [Google Scholar]
- 61. Sedek M, van der Velden LM, Strous GJ. Multimeric growth hormone receptor complexes serve as signaling platforms. J Biol Chem. 2014;289:65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang Y, Kim S-O, Yang N, Jiang J, Frank SJ. Physical and functional interaction of GH and IGF-1 signaling elements. Mol Endocrinol. 2004;18:1471–1485 [DOI] [PubMed] [Google Scholar]
- 63. Frank SJ, Gilliland G, Van Epps C. Treatment of IM-9 cells with human growth hormone (GH) promotes rapid disulfide linkage of the GH receptor. Endocrinology. 1994;135:148–156 [DOI] [PubMed] [Google Scholar]
- 64. Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McFarland BC, Ma JY, Langford CP, et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10:2384–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Frank SJ, Fuchs SY. Modulation of growth hormone receptor abundance and function: roles for the ubiquitin-proteasome system. Biochim Biophys Acta. 2008;1782:785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]