

Abstract
Human and veterinary drug development addresses absorption, distribution, metabolism, elimination and toxicology (ADMET) of the Active Pharmaceutical Ingredient (API) in the target species. Metabolism is an important factor in controlling circulating plasma and target tissue API concentrations and in generating metabolites which are more easily eliminated in bile, faeces and urine. The essential purpose of xenobiotic metabolism is to convert lipid-soluble, non-polar and non-excretable chemicals into water soluble, polar molecules that are readily excreted. Xenobiotic metabolism is classified into Phase I enzymatic reactions (which add or expose reactive functional groups on xenobiotic molecules), Phase II reactions (resulting in xenobiotic conjugation with large water-soluble, polar molecules) and Phase III cellular efflux transport processes. The human–fish plasma model provides a useful approach to understanding the pharmacokinetics of APIs (e.g. diclofenac, ibuprofen and propranolol) in freshwater fish, where gill and liver metabolism of APIs have been shown to be of importance. By contrast, wildlife species with low metabolic competency may exhibit zero-order metabolic (pharmacokinetic) profiles and thus high API toxicity, as in the case of diclofenac and the dramatic decline of vulture populations across the Indian subcontinent. A similar threat looms for African Cape Griffon vultures exposed to ketoprofen and meloxicam, recent studies indicating toxicity relates to zero-order metabolism (suggesting P450 Phase I enzyme system or Phase II glucuronidation deficiencies). While all aspects of ADMET are important in toxicity evaluations, these observations demonstrate the importance of methods for predicting API comparative metabolism as a central part of environmental risk assessment.
Keywords: medicines, environment, exposure, birds, fish, invertebrates
1. Introduction
Investigations of a pharmaceutical's absorption, distribution, metabolism, elimination and toxicology (ADMET) play a central role in the pre-clinical and clinical safety assessment of human medicines [1] and potentially in environmental risk assessment (see [2]). Likewise, Active Pharmaceutical Ingredients (APIs) used in veterinary medicine are evaluated for their ADMET profile in the species of interest (e.g. poultry or ruminants) [3,4]. Metabolism of endogenous and exogenous molecules (e.g. plant toxins, pesticides and pharmaceuticals) is normally classified into Phase I enzymatic reactions (which add or expose –OH, –SH, –NH2 or –COOH functional groups on xenobiotics) and Phase II reactions (resulting in xenobiotic conjugation with large water-soluble, polar molecules). Additionally, lipophilic xenobiotics, or their metabolites, can be pumped out of cells by specific transporter proteins and this efflux pump activity is often termed Phase III metabolism [5]. For approximately 5–7% of human drugs, Phase I metabolism may be responsible for conversion of a prodrug into the API [6]. More broadly, many Phase I biotransformations of lipophilic xenobiotics are carried out by microsomal monooxygenases, located in the endoplasmic reticulum of the liver and other organs [7]. The haem protein cytochrome P450 provides the active centre of these enzymes and has huge diversity, with 37 cytochrome P450 families currently identified across many animal species [8]. It is hypothesized that the P450 superfamily has undergone repeated rounds of expansion by genome duplication, whereby approximately one and a half billion years ago, the first expansion gave rise to the P450 families primarily involved in metabolizing endogenous fatty acids, cholesterol and its derivatives (CYP4 and CYP11 families) which likely played a key role in maintaining the eukaryotic cell membrane integrity. A later expansion of the P450 family 900 Ma may have led to several endogenous steroid-synthesizing cytochrome P450 lineages (including CYP19, CYP21 and CYP27 gene families; whereby the CYP21 family later diverged to give rise to the CYP1 and CYP2 families). A final major expansion of several P450 families involved in xenobiotic metabolism (including CYP2, CYP3, CYP4 and CYP6), began about 400 Ma. This most recent expansion is thought to have been driven by first the emergence of aquatic organisms onto land, associated with eating toxic plant allelochemicals (‘animal–plant warfare’), together with exposure of terrestrial organisms to hydrocarbon-based combustion products in the atmosphere [8–12].
Much data exist on the metabolism of pharmaceuticals and other xenobiotics by the liver microsomes of mammals, birds and other species, with rates of microsomal oxidative metabolism determined across a range of vertebrates [3,13,14]. For example, Abass et al. [15] studied the metabolism of the insecticide benfuracarb by hepatic microsomes taken from seven mammalian species to investigate species-specific metabolic pathways. Benfuracarb is metabolized via sulfur-oxidation and nitrogen–sulfur bond cleavage (producing carbofuran which is further metabolized). Clearance rates for the seven species ranged from 1.4 (monkey) to 3.5 (rat), these differences being due to variability in CYP enzyme expression [15]. Among herbivorous and omnivorous mammals, there is a clear inverse correlation between the microsomal monooxygenase activity and body weight [16,17]. When hepatic monooxygenase activities are expressed in terms of body weight, much higher values are found in small rodents than in large mammals. This observation is consistent with the concept of a coevolutionary arms race between plants and herbivorous animals. In this context, small mammals need to consume more food per unit body weight than do large ones in order to maintain body temperature due to their high surface area to volume ratios. In contrast to the mammalian species studied by Walker and co-workers, the carnivorous (piscivorous or raptorial) species showed distinctly lower microsomal monooxygenase activities than did herbivorous or omnivorous birds (an observation also explicable in terms of ‘animal–plant arms race’ theory). Predatory mammals (e.g. cats) and birds (e.g. raptors) eat very little, if any, plant material and therefore do not incur major pressure to drive the evolution of enzymes to metabolize plant toxins [17–20]. Interestingly, zebrafish (a widely used model in pharmaceutical research) show a dramatic increase in Phase I and II enzyme activity at the juvenile life stage in association with being fed plant-based diets [21].
In contrast to terrestrial vertebrates, Phase I enzyme activities in fish are generally lower and there is only a weak correlation with body weight (whereas individual avian species show a correlation between body weight and hepatic microsomal monooxygenase activity across species) [13]. For fish, this has been explained on the grounds that they can excrete as well as take up (see [22]) many xenobiotics by diffusion across gills into the large volume of ambient water and it has been argued that there has not been a strong pressure for the evolution of highly active detoxification enzymes as seen in mammals [14,23]. A similar situation is thought to apply to aquatic invertebrates [24–26]. Nonetheless, as molecular and biochemical methods have advanced, there is growing evidence of both Phase I and II enzyme activity in fish [21,27,28] and recent studies have addressed how dietary and trophic variables may affect enzyme activity in fish [29]. There are also a growing number of studies on the metabolism of pharmaceuticals in fish [30–40] and to a far lesser extent invertebrates [41]. Veterinary pharmaceuticals have also been studied from a comparative metabolism perspective [42,43]. Table 1 summarizes Phase I pathways of pharmaceutical and xenobiotic metabolism in mammals and other vertebrates, adapted from Parkinson and Ogu & Maxa [44,45] and updated with examples from the DrugBank online database (http://www.drugbank.ca/) established by Wishart et al. [46].
Table 1.
Summary of vertebrate metabolic pathways with examples of pharmaceutical and xenobiotic substrates and inhibitors.
| enzyme | localization | substrate | inhibitor |
|---|---|---|---|
| Phase I—hydrolysis reactions | |||
| esterase | microsomes and cytosol | trandolapril | tamoxifen |
| peptidase | lysosomes | — | alogliptin |
| epoxide hydrolase | microsomes and cytosol | diazepam | valproate |
| Phase I—reduction reactions | |||
| azo- and nitro-reduction | microsomes and cytosol | prontosil | clofibrate |
| carbonyl reduction | microsomes and cytosol | loxoprofen | befunolol |
| disulfide reduction | cytosol | captopril | — |
| sulfoxide reduction | cytosol | — | dimethylsulfoxide |
| quinone reduction | microsomes and cytosol | trenimon | warfarin |
| reductive dehalogenation | microsomes | chloramphenicol | — |
| Phase I—oxidation reactions | |||
| alcohol dehydrogenase | cytosol | ethanol | fomepizole |
| aldehyde dehydrogenase | mitochondria and cytosol | acetaldehyde | disulfiram |
| aldehyde oxidase | cytosol | aldehyde | raloxifene |
| xanthine oxidase | cytosol | xanthine | allopurinol |
| monoamine oxidase | mitochondria | monoamine | moclobemide |
| diamine oxidase | cytosol | diamine | phenformin |
| prostaglandin H synthase | microsomes | arachidonic acid | ibuprofen |
| flavin-monooxygenases | microsomes | riboflavin | nitric oxide |
| cytochrome P450: | microsomes | — | — |
| CYP1A1 | microsomes | 7-ethoyxyresorufin | galangin |
| CYP1A2 | microsomes | clozapine propranolol | cimetidine citalopram |
| CYP2C19 | microsomes | citalopram diazepam | fluoxetine ketoconazole |
| CYP2C9 | microsomes | diclofenac ibuprofen | fluconazole fluoxetine |
| CYP2D6 | microsomes | metoprolol tramadol | fluoxetine sertraline |
| CYP2E1 | microsomes | acetaminophen ethanol | disulfiram water cress |
| CYP3A4 | microsomes | carbamazepine simvastatin | flavonoids ketoconazole |
| Phase II—enzyme reactions | |||
| glucuronide conjugation | microsomes | Phase I metabolites | valproic acid |
| sulfate conjugation | cytostol | Phase I metabolites | harmol |
| glutathione conjugation | microsomes and cytosol | Phase I metabolites | tannic acid |
| amino acid conjugation | microsome | Phase I metabolites | kinetin |
| acetylation | mitochondria and cytosol | Phase I metabolites | garcinol |
| methylation | microsomes and cytosol | Phase I metabolites | 5-A-2′deoxycytidine |
2. In vitro and in silico methods to understand comparative metabolism
In vitro systems are widely used for the investigation of xenobiotic metabolism in mammals [1], birds [47] and fish [38,40]. Systems include: (i) whole liver tissue slices which retain an accurate, structural framework of the liver; (ii) whole isolated hepatocytes where the endoplasmic-reticulum-bound and cytosolic enzymes are present but the structural integrity of liver network lost; (iii) after centrifugation at 9000g, the S9 fraction supernatant from liver (or other tissue) homogenate containing both cytosolic (predominantly Phase II) and microsomal (predominantly Phase I) enzymes; and (iv) microsomes comprising endoplasmic-reticulum-bound enzymes that have been separated from cytosolic enzymes (P450 enzymes are concentrated in this subcellular fraction). These methods are routinely used to determine the rate and extent of metabolism and mass-spectroscopic analysis of specific metabolites. Results for clearance rates obtained from in vitro metabolism experiments can then be extrapolated to the in vivo situation using scaling factors (e.g. number of hepatocytes per liver; weight of microsomal protein per gram of liver, etc.). Allometric methods can also be used to scale in vitro results between different species (used in drug development for scaling from pre-clinical species to man). Where such values are known for wildlife species, this may allow for approximations between different species [16,17] and form a basis for models to aid in environmental risk assessment using fish [31,48,49], invertebrates [41] and plants [50].
Novel in silico tools may also be useful to predict metabolism, this approach tending to focus on the semi-quantitative prediction of potential metabolites and identification of the specific enzymes responsible for the metabolism. Prediction of metabolic rates of drug metabolism remains a key challenge, especially with regard to identification of potential metabolites (which may be associated with specific toxicities) and identification of the enzymes responsible (combined with knowledge of different enzyme expression in different species). Kirchmair et al. [51] provide an overview of in silico tools for predicting key factors associated with metabolism (including sites of metabolism within a molecule; potential metabolites; cytochrome P450 (CYP) binding affinity/inhibition and prediction of CYP induction). Table 2 shows a representative software tool for each of these categories, however, many other tools are available [51].
Table 2.
Representative examples of computational tools for predicting factors associated with mammalian metabolism (programs may have additional capabilities).
| factor predicted | software | summary of method | website or key citation |
|---|---|---|---|
| (i) site of metabolism | Metaprint2D | predicts sites of Phase I metabolism in dog, human and rat through data-mining and statistical analysis of published metabolic transformations | http://www-metaprint2d.ch.cam.ac.uk/metaprint2d |
| (ii) potential metabolites | Meteor Nexus | uses expert knowledge rules for metabolism to predict metabolites which are presented in metabolic trees | http://www.lhasalimited.org/products/meteor-nexus.htm |
| (iii) CYP binding affinity/inhibition | isoCYP | predicts the predominant human cytochrome P450 isoform by which a compound is metabolized | http://www.molecular-networks.com/products/isocyp |
| (iv) CYP induction | VirtualToxLab | predicts binding affinities to Aryl hydrocarbon receptor (and other targets) using flexible docking and quantitative structure–activity relationships | http://www.biograf.ch/index.php?id=projects&subid=virtualtoxlab |
In silico tools have a number of potential advantages and provide complementary techniques to in vitro methods. One area where information from both fields can be combined to build improved predictions is in physiologically based pharmacokinetic (PBPK) modelling. In this method, an organism is divided into a sequence of physiological compartments (e.g. brain, liver, lungs, etc.). The models integrate compound-specific data (e.g. physico-chemical properties, such as log P, pKa or solubility; these values may be measured or predicted using in silico techniques) and species- (or even subject-) specific data (e.g. physiological factors such as body or organ weights, volumes or blood flow rates). Subject to validation, these models are potentially of high value in predicting concentration-time profiles for pharmaceuticals in wildlife species [31,36,48]. Understanding inter-species differences in metabolism is essential for reliable PBPK models, especially in non-mammalian species. For example, Ohyama et al. [47] studied methoxychlor (MXC) metabolism in rat, mouse, Japanese quail and rainbow trout using liver slices. Each species showed differences in metabolism, considered due to the substrate specificity of CYP450s involved. MXC was metabolized to bis-OH-MXC which was then glucuronidated (with only rats producing the bis-OH-MXC 4 O-sulfate 4-O-glucuronide). In mice and Japanese quail, mono-OH-MXC (and glucuronide conjugate) were the main metabolites and little bis-OH-MXC glucuronide was formed (dechlorinated mono-OH-MXC glucuronide was found only in mice). Rainbow trout liver slices formed similar amounts of both metabolites. In conclusion, rat and trout liver slices were able to metabolize both MXC and mono-OH MXC, whereas only MXC could be metabolized in mouse and Japanese quail [47].
3. In vivo approaches in studying comparative metabolism
The overall effect a xenobiotic has on any organism is ultimately the result of its intrinsic activity and its concentration at the target site. Concentration at a given target site is determined by the ADMET properties of the compound. The history of studying the time course and concentration of xenobiotics at different sites within the body has been developed predominantly within the pharmaceutical industry, with respect to drug effects on humans. However, the techniques are applicable to diverse chemical space and across diverse species. In vivo measurements determining the pharmacokinetic profiles of xenobiotics in environmental species are largely unavailable, hence extrapolation and predictive models (combining in silico and in vitro methods) become essential tools in determining organ-level concentrations [52]. Metabolism is one of the key factors to consider when modelling the time course of a xenobiotic within an organism, not only as it can determine the overall period of exposure, but also because the metabolite(s), rather than the parent drug, may be responsible for the toxic effect [1,53]. In the non-mammalian area, where much less is known about metabolic profiles of drugs in animals, in vivo experiments still have a major role to play to derive reliable environmental risk assessments (for case studies with freshwater fish, see [35,36,54]) and also in wildlife forensic studies (see following case study on birds).
4. Case study: vulture toxicity to non-steroidal anti-inflammatory drug (a process of zero-order metabolism)
The dramatic impact of diclofenac (a non-steroidal anti-inflammatory drug or ‘NSAID’) on Asian vulture populations represents one of the most serious ecological catastrophes of recent times. In just over a decade, diclofenac has been responsible for the deaths of millions of vultures of the Asian white-backed (Gyps bengalensis), long-billed (Gyps indicus), slender-billed (Gyps tenuirostris), Egyptian (Neophron percnopterus) and red-headed (Sarcogyps calvus) species across the Indian subcontinent [55,56] (also see [57]). In addition to the scale of the toxicity, the exposure route to the product was probably highly unconventional as these birds were inadvertently being poisoned by the oral route even though diclofenac was only available as an injectable cattle formulation. Whereas previous veterinary medicines and pesticides had caused their negative effects by ending up in the water, soil or general environment of the species affected, these vultures were being exposed to this product as residues in the meat of the dead cattle carcasses upon which they fed. This unique mode of exposure was linked to cultural and religious practices in the region, whereby sick and old cattle were routinely treated in a palliative manner with diclofenac, a cheap and effective NSAID. The net effect of this practice was an unfortunate high occurrence of diclofenac residues in the tissues of recently dead cattle.
In the vulture, diclofenac is highly toxic with rapid mortality resulting from a single meal of 1 kg of meat rich in residue, with an estimated LD50 of 0.1–0.2 mg kg−1 [58]. Toxicity following exposure is also fairly predictable with birds showing signs of depression and head drooping as early as 24 h post exposure. Death is the typical endpoint with birds literally being described as falling dead from their perches. Based on the results from controlled toxicity studies, it has been shown that death after a single exposure consistently resulted within 48 h of exposure, with related massive increases in plasma uric acid and potassium concentrations and increased alkaline phosphatase activity. Necropsies are also very typical with signs of severe nephrosis, dehydration and accompanying diffuse visceral and articular gout. Histopathology indicated toxicity was characterized by necrosis of hepatocytes and the renal tubular epithelial cells (RTE) of the proximal convoluted tubules with associated uric acid tophi accumulation. While the mechanism of toxicity of diclofenac remains incompletely described, toxicity has been linked to RTE cell damage in a time-related manner, subsequent accumulation of uric acid, acidosis and terminal hyperkalaemia [59]. Results from various pharmacokinetics studies of diclofenac in different bird species, compared to the pharmacokinetic profiles of ketoprofen and meloxicam, which exhibit comparatively lower toxicity in the vulture, clearly indicate that toxicity is related to diclofenac's pharmacokinetics (figure 1).
Figure 1.
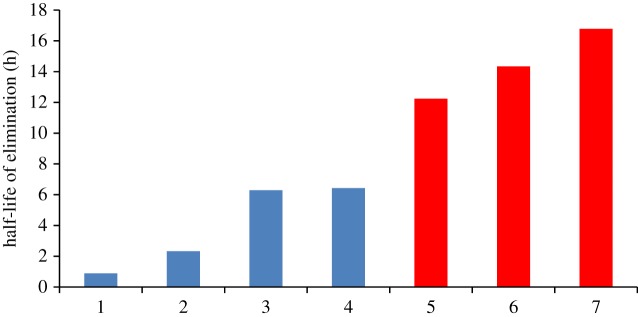
Estimated half-life of elimination for various avian species dosed with diclofenac in controlled toxicity studies. The half-lives have be ranked from fastest to slowest and represent: 1—G. domesticus (0.8 mg kg−1 oral); 2—C. albus (10 mg kg−1 oral); 3—C. aura (25 mg kg−1 oral); 4—C. aura (8 mg kg−1 oral); 5—G. coprotheres (0.8 mg kg−1 IV); 6—G. domesticus (5 mg kg−1 oral); 7—G. africanus (0.8 mg kg−1 oral). The red bars (5 to 7) indicate those doses associated with mortality. (Online version in colour.)
For the first of these studies, the pharmacokinetics of diclofenac was evaluated in the Cape Griffon vulture (Gyps coprotheres) [60]. While environmental toxicity has not been seen in this vulture, the species was specifically validated as a suitable model for further mechanistic studies on the toxicity of diclofenac and other NSAIDs. The choice of this species was twofold, firstly the easier availability to the study site, as well as being less endangered than the Indian vulture species. From this controlled acute toxicity study, the Cape Griffon vulture was shown to be equally susceptible to diclofenac as the oriental white-backed vulture at 0.8 mg kg−1 (intravenous (IV) dose) with exactly the same clinical signs, clinical pathological and histopathological changes. Non-compartmental analysis revealed a half-life of elimination (T1/2) of 12.24 ± 0.99 h, area under curve to the last quantifiable time point (AUClast) of 80.28 ± 51.26 µg ml h−1, and a mean residence time of 15.11 ± 4.13 h. To evaluate the importance of the pharmacokinetic profile obtained, Naidoo et al. [61] compared it to those published for other bird species (figure 2). This included the African white-backed vulture (Gyps africanus), the pied crow (Corvus albus), the turkey vulture (Cathartes aura) and the domestic chicken (Gallus domesticus). For these studies, no mortalities were reported for the pied crow (0.8 and 10 mg kg−1 oral), turkey vulture (8 and 25 mg kg−1 oral) and the domestic chicken (0.8 mg kg−1 oral), while toxicity was reported in the Cape Griffon (0.8 mg kg−1 IV), the African white-back (0.8 mg kg−1 oral) and one chicken at a higher dose (5 mg kg−1 oral). An important finding from these comparisons was a tentative link between the T1/2 and the occurrence of toxicity, with a T1/2 above 12 h being associated with death. Furthermore, zero-order metabolism was seen as a feature of toxicity as the T1/2 was increased in the one chicken that died, from 0.89 h at 0.8 mg kg−1 to 14.34 h at 5 mg kg−1 oral.
Figure 2.
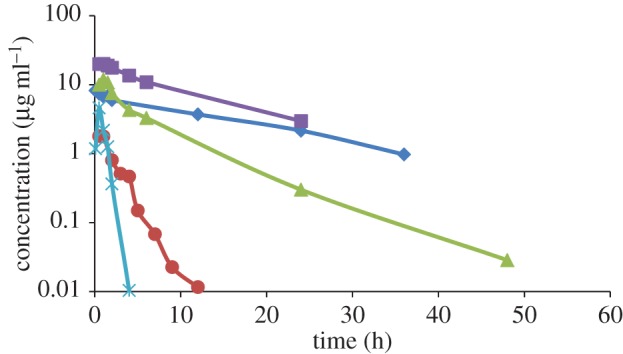
Plasma versus time profiles for diclofenac at 0.8 mg kg−1 IV in G. coprotheres (rhomboid); ketoprofen at 5 mg kg−1 oral for G. coprotheres that died (square); ketoprofen at 5 mg kg−1 oral in G. coprotheres that survived (triangle); diclofenac in chickens at 0.8 mg kg−1 oral (circle) and meloxicam in G. coprotheres at 2 mg kg−1 oral/intramuscular (cross). (Online version in colour.)
While diclofenac has received wide attention in published literature as a result of its environmental toxic effect, it is not, however, the only NSAID evaluated in vultures in terms of safety and pharmacokinetics. In an attempt to have diclofenac removed from the Indian veterinary market, a replacement for the drug needed to be found for use in cattle, as diclofenac was of valuable cultural benefit to the sick cattle being treated. Following an international survey, meloxicam and ketoprofen were identified as potential replacements: they were effective in cattle with some evidence of safety in captive vulture species [62,63]. Subsequently, both these drugs were evaluated in extensive safety studies including full characterization of their pharmacokinetics, once again in Cape Griffon as the model, with vastly contrasting results.
In the first ketoprofen study, Cape Griffon vultures treated at 1 mg kg−1 oral showed no indications of toxicity on both clinical and clinical pathological evaluations [64]. However, when a second group of vultures were treated at the increased dose of 5 mg kg−1 oral, the study resulted in mortalities in seven of the 11 birds treated, with the characteristic signs of toxicity seen in the diclofenac-treated birds. The most interesting finding for this study was a difference in the T1/2 between these two dose levels but also between the birds that died or survived at the 5 mg kg−1 dose. At 1 mg kg−1, the half-life was 2.66 ± 0.46 h. In the four birds that survived at 5 mg kg−1, the half-life was marginally higher at 3.24 ± 1.59 h. For the birds that died at the 5 mg kg−1 dose, the half-life had increased to 7.38 ± 1.72 h. With regards to AUClast, the four birds that survived had an AUClast fivefold higher, as expected for the fivefold increase in dose (9.79 ± 3.23 μg ml h−1 versus 50.31 ± 17.71 μg ml h−1, respectively). However, the birds that died at 5 mg kg−1 had an increased AUClast of 156.51 ± 33.14 μg ml h−1 and Cmax of 21.0 ± 1.88 μg ml−1 in comparison with 10.77 ± 3.26 μg ml−1 for the birds that survived. This once again supported previous findings that toxicity is related to zero-order metabolism. In addition, the increase in the AUClast and Cmax also indicated that toxicity resulted in saturation of presystemic elimination pathways [64].
In the last of the described pharmacokinetic studies, meloxicam was administered to Cape Griffon vultures in a two-way cross-over study at a dose of 2 mg kg−1 by either oral or intramuscular route, without any signs of toxicity or changes in the monitored clinical pathology parameters [62]. Meloxicam was characterized by a short half-life of elimination of 0.33 ± 0.167 h and 0.42 ± 0.11 h for the oral and intramuscular routes, respectively. This study further attempted to characterize the metabolites produced via liquid chromatography-tandem mass spectrometry (LC-MSMS) analysis. Two CYP metabolites, hydroxymethyl meloxicam (87%) and an unknown hydroxylated metabolite (7%), and one glucuronide (0.56%) metabolite were identified (figure 3). Based on literature for laboratory animals, it was suspected that the CYP most likely involved in metabolism was predominantly CYP2C9.
Figure 3.
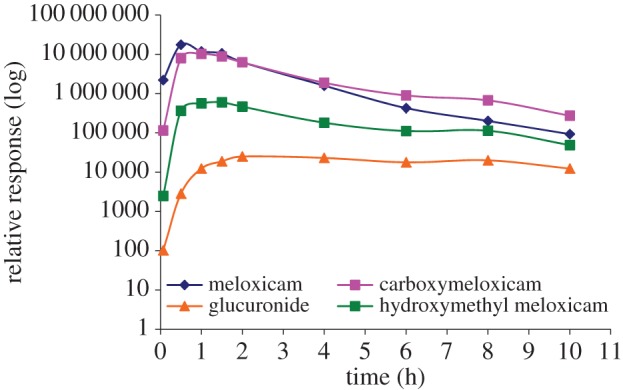
Relative response (AUC per peak on LC–MSMS chromatograms) versus time profiles for parent meloxicam and its three metabolites: hydroxymethyl meloxicam, an unidentified hydroxymethyl metabolite carboxymeloxicam and the glucuronide metabolite, following treatment at 2 mg kg−1 oral/intramuscular in G. coprotheres. (Online version in colour.)
While the metabolic pathway for diclofenac in the vulture is yet to be evaluated, the current pharmacokinetic information available allows for some conclusions to be drawn. The first of these is that toxicity is clearly linked to zero-order kinetics. For the NSAIDs, this deficiency could be at the level of the Phase I enzyme (CYP) system or Phase II glucuronidation, both of which have been previously described. Decreased CYP2C9 activity in people has been associated with resultant longer half-life of metabolized NSAIDs, whereas the absence of glucuronidation (UGT1A6) has been described as an important mechanism in the toxicity of paracetamol in the cat [19]. Limited glucuronide activity has also been described in people in association with aspirin toxicity. Based on the presence of a glucuronide metabolite for meloxicam, it is likely that toxicity in humans is not due to a complete absence of Phase II processes as in the cat. In addition, it is also doubtful that limited glucuronidation plays a role in human toxicity [65]. As a result, the rate-limiting step in avian metabolism is most likely at the level of the cytochrome P450 enzyme system. From medical literature, meloxicam is metabolized predominantly by CYP2C9 and, to a much lower extent, CYP3A4; diclofenac predominantly by CYP2C9, with some metabolism by CYP3A4 and CYP2C8 [66,67]; and ketoprofen by CYP2C9 [68]. When the half-life of elimination of diclofenac, ketoprofen and meloxicam in people is compared with the vulture, an important difference is noticed. In humans, the half-life of elimination of diclofenac, ketoprofen and meloxicam is typically 1–2, 2 and 15–20 h, respectively [69], while (as reported above) this is ±14, ±3 and 0.33 h, respectively for the vulture, with the metabolism of ketoprofen in vultures also being zero order. With CYP2C9 being the one common enzyme in metabolism, this is most probably the rate-limiting enzyme. With the rapid metabolism of meloxicam in vultures in contrast to humans, it may even be possible that the vulture is reliant on a Phase I system other than CYP2C9 for metabolism (in vultures, CYP3A4 seems a possibility). If this is the case, then the extreme sensitivity of the vulture to NSAID toxicity may be associated with the hepatotoxicity of diclofenac in humans, which is tentatively linked to CYP3A4 metabolism [70].
5. Conclusion
Pharmaceuticals provide many important health and economic benefits in the context of their capacity to generate desired and specific therapeutic effects in the target species (namely humans or in some cases, domestic animals and companion animals). In some cases, however, environmental exposures of wildlife to pharmaceutical residues can have dramatic consequences on non-mammalian species, as seen in the case of diclofenac and vultures [56–58] or fish populations in ecosystems exposed to synthetic oestrogens [71] (also see [72]). These notable examples, together with evidence of the widespread presence of pharmaceuticals in the environment, have been widely recognized to support the need for predictive environmental risk assessments [73–76] and consideration of the API residues in cattle and other livestock species [77].
A fundamental aspect of this challenge relates to the need to consider comparative metabolism for a range of non-mammalian species. Specifically, it is clear that there remain major knowledge gaps regarding the comparative metabolism of human and veterinary pharmaceuticals in non-mammalian species and this situation needs to be addressed in order to develop reliable environmental risk assessments for these important groups of medicines. It is proposed that this knowledge gap could be addressed in an efficient and ethical manner through the use of in vitro methods to define metabolism of reference APIs (selected from table 1) in hepatocytes from carnivorous birds compared with omnivorous bird species, for example, cormorants (Phalacrocorax auritus) and chickens (G. domesticus), respectively [78,79]. For fish, the same approach is feasible using in vitro hepatocyte assays for mainly carnivorous salmonid species such as rainbow trout (Oncorhynchus mykiss) versus the mainly herbivorous cyprinid species such as zebrafish (Danio rerio) or carp (Cyprinus carpio) [21,27]. For invertebrates, an in vivo approach would seem the best option and should be extended to both freshwater and marine species as part of an Adverse Outcome Pathways approach [41,80–82]. Subsequently, the in vitro avian and fish metabolic data and the in vivo invertebrate data for reference APIs could be used to develop and validate in silico tools to better predict which enzymes are responsible for API metabolism. If the measured or predicted metabolism of a human or veterinary drug in mammalian or non-mammalian wildlife species raised concerns, further work could be done to evaluate the in vitro metabolites data through computational toxicology or metabolic pathway analysis [52,83,84].
In the wider context, where predicted regional increases in drug use occur or measurements of APIs in the environment raise concerns, the availability of validated in silico and in vitro methods to predict comparative metabolism will be of immense use in conducting environmental risk assessments. Specifically, together with prioritization through the predicted exposure concentration approach, an understanding of ADMET can play an important role in defining and predicting no-observed effect concentrations for freshwater, terrestrial and other environmental compartments, including predators [74,75,85]. In addition to this predictive aspect of pharmaceutical risk assessment, an understanding of ADMET can provide an important role for targeted monitoring of wildlife species of concern (e.g. vultures and other ultra-carnivorous species [76,81]; also see [86]).
References
- 1.Yengi LG, Leung L, Kao J. 2007. The evolving role of drug metabolism in drug discovery and development. Pharm. Res. 24, 842–858. ( 10.1007/s11095-006-9217-9) [DOI] [PubMed] [Google Scholar]
- 2.LaLone CA, Berninger JP, Villeneuve DL, Ankley GT. 2014. Leveraging existing data for prioritization of the ecological risks of human and veterinary pharmaceuticals to aquatic organisms. Phil. Trans. R. Soc. B 369, 20140022 ( 10.1098/rstb.2014.0022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ping PH, Fouts JR. 1979. Drug metabolism in birds. Pharmacology 19, 289–293. ( 10.1159/000137327) [DOI] [PubMed] [Google Scholar]
- 4.Juskevich JC. 1987. Comparative metabolism in food producing animals: programs sponsored by the Center for Veterinary Medicines. Drug Metab. Rev. 18, 345–362. ( 10.3109/03602538708998312) [DOI] [PubMed] [Google Scholar]
- 5.Xu C, Li CY, Kong AN. 2005. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 28, 249–268. ( 10.1007/BF02977789) [DOI] [PubMed] [Google Scholar]
- 6.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, Savolainen J. 2008. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 7, 255–270. ( 10.1038/nrd2468) [DOI] [PubMed] [Google Scholar]
- 7.Lewis DFV. 1996. Cytochromes P450: structure, function and mechanism, pp. 348 London, UK: Taylor and Francis. [Google Scholar]
- 8.Nelson DR, Goldstone JV, Stegeman JJ. 2013. The cytochrome P450 genesis locus: the origin and evolution of animal cytochrome P450s. Phil. Trans. R. Soc. B 368, 20120474 ( 10.1098/rstb.2012.0474) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrlich PR, Raven PH. 1964. Butterflies and plants: a study in coevolution. Evolution 18, 586–608. ( 10.2307/2406212) [DOI] [Google Scholar]
- 10.Gonzalez FJ, Nebert DW. 1990. Evolution of the P450 gene superfamily: animal–plant ‘warfare’, molecular drive and human genetic differences in drug oxidation. Trends Genet. 6, 182–186. ( 10.1016/0168-9525(90)90174-5) [DOI] [PubMed] [Google Scholar]
- 11.Harborne JB. 1993. Introduction to ecological biochemistry, 4th edn London, UK: Taylor and Francis. [Google Scholar]
- 12.Nelson DR. 1998. Metazoan cytochrome P450 evolution. Comp. Biochem. Physiol. 121C, 15–22. [DOI] [PubMed] [Google Scholar]
- 13.Ronis MJJ, Walker CH. 1989. The microsomal monooxygenases of birds. Rev. Biochem. Toxicol. 10, 301–384. [Google Scholar]
- 14.Walker CH, Hopkin SP, Sibly RM, Peakall DB. 2012. Principles of ecotoxicology, 4th edn London, UK: Taylor and Francis. [Google Scholar]
- 15.Abass K, Reponen P, Mattila S, Rautio A, Pelkonen O. 2014. Comparative metabolism of benfuracarb in in vitro mammalian hepatic microsomes and its implications for chemical risk assessment. Toxicol. Lett. 224, 290–299. ( 10.1016/j.toxlet.2013.08.009) [DOI] [PubMed] [Google Scholar]
- 16.Walker CH. 1978. Species differences in microsomal monooxygenase activity and their relationship to biological half-lives. Drug Metab. Rev. 7, 295–323. ( 10.3109/03602537808993770) [DOI] [PubMed] [Google Scholar]
- 17.Walker CH. 1980. Species differences in some hepatic microsomal enzymes that metabolise xenobiotics. Prog. Drug Metab. 5, 118–164. [Google Scholar]
- 18.Walker CH. 1981. The correlation between in vivo metabolism and in vitro metabolism in vertebrates. In Progress in pesticide biochemistry, vol. 1 (eds Hutson DH, Roberts TR.), pp. 287–333. New York, NY: John Wiley and Sons. [Google Scholar]
- 19.Shrestha B, et al. 2011. Evolution of a major drug metabolizing enzyme defect in the domestic cat and other Felidae: phylogenetic timing and the role of hypercarnivory. PLoS ONE 6, e18046 ( 10.1371/journal.pone.0018046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Court MH. 2013. Feline drug metabolism and disposition: pharmacokinetic evidence for species differences and molecular mechanisms. Vet. Clin. N. Am. Small Anim. Pract. 43, 1039–1054. ( 10.1016/j.cvsm.2013.05.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiegand C, Pflugmacher S, Oberemm A, Steinberg C. 2000. Activity development of selected detoxication enzymes during the ontogenesis of the zebrafish (Danio rerio). Int. Rev. Hydrobiol. 85, 413–422. () [DOI] [Google Scholar]
- 22.Du B, et al. 2014. Bioaccumulation and trophic dilution of human pharmaceuticals across trophic positions of an effluent-dependent wadeable stream. Phil. Trans. R. Soc. B 369, 20140058 ( 10.1098/rstb.2014.0058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Randall DJ, Connell DW, Yang R, Wu SS. 1998. Concentrations of persistent lipophilic compounds in fish are determined by exchange across the gills, not through the food chain. Chemosphere 37, 1263–1270. ( 10.1016/S0045-6535(98)00124-6) [DOI] [PubMed] [Google Scholar]
- 24.Livingstone DR, Kirchin MA, Wiseman A. 1989. Cytochrome P450 and oxidative metabolism in molluscs. Xenobiotica 19, 1041–1062. ( 10.3109/00498258909043161) [DOI] [PubMed] [Google Scholar]
- 25.Livingstone DR. 1998. The fate of organic xenobiotics in aquatic ecosystems: quantitative and qualitative differences in biotransformation by invertebrates and fish. Comp. Biochem. Physiol. A 120, 43–49. ( 10.1016/S1095-6433(98)10008-9) [DOI] [PubMed] [Google Scholar]
- 26.Snyder MJ. 2000. Cytochrome P450 enzymes in aquatic invertebrates: recent advances and future directions. Aquat. Toxicol. 48, 529–547. ( 10.1016/S0166-445X(00)00085-0) [DOI] [PubMed] [Google Scholar]
- 27.Buhler DR, Wang-Buhler JL. 1998. Rainbow trout cytochrome P450s: purification, molecular aspects, metabolic activity, induction and role in environmental monitoring. Comp. Biochem. Physiol. 121, 107–137. [DOI] [PubMed] [Google Scholar]
- 28.Celander M, Leaver MJ, George SG, Forlin L. 1993. Induction of cytochrome P450 1A1 and conjugating enzymes in rainbow trout (Oncorhynchus mykiss) liver: a time course study. Comp. Biochem. Physiol. A 106, 343–349. ( 10.1016/0300-9629(93)90523-7) [DOI] [Google Scholar]
- 29.Solé M, Rodríguez S, Papiol V, Maynou F, Cartes JE. 2009. Xenobiotic metabolism markers in marine fish with different trophic strategies and their relationship to ecological variables. Comp. Biochem. Physiol. C 149, 83–89. ( 10.1016/j.cbpc.2008.07.008) [DOI] [PubMed] [Google Scholar]
- 30.Arnot JA, Mackay D, Parkerton T, Bonnell M. 2008. A database of fish biotransformation rates for organic chemicals. Environ. Toxicol. Chem. 27, 2263–2270. ( 10.1897/08-058.1) [DOI] [PubMed] [Google Scholar]
- 31.Gomez CF, Constantine L, Huggett DB. 2010. The influence of gill and liver metabolism on the predicted bioconcentration of three pharmaceuticals in fish. Chemosphere 81, 1189–1195. ( 10.1016/j.chemosphere.2010.09.043) [DOI] [PubMed] [Google Scholar]
- 32.Hasselberg L, Grøsvik BE, Goksøyr A, Celander MC. 2005. Interactions between xenoestrogens and ketoconazole on hepatic CYP1A and CYP3A, in juvenile Atlantic cod (Gadus morhua). Comp. Hepatol. 4, 2–8. ( 10.1186/1476-5926-4-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasselberg L, Westerberg S, Wassmur L, Celander MC. 2008. Ketoconazole, an antifungal imidazole, increases the sensitivity of rainbow trout to 17α-ethynylestradiol exposure. Aquat. Toxicol. 86, 256–264. ( 10.1016/j.aquatox.2007.11.006) [DOI] [PubMed] [Google Scholar]
- 34.Hegelund T, Ottosson K, Rådinger M, Tomberg P, Celander MC. 2004. Effects of the antifungal imidazole ketoconazole on CYP1A and CYP3A in rainbow trout and killifish. Environ. Toxicol. Chem. 23, 1326–1334. ( 10.1897/03-155) [DOI] [PubMed] [Google Scholar]
- 35.Jones HS, Trollope HT, Hutchinson TH, Panter GH, Chipman JK. 2009. Assessment of ibuprofen metabolism by zebrafish larvae, using liquid chromatography–mass spectrometry (LC–MS). Toxicology 262, 14–16. ( 10.1016/j.tox.2009.04.014) [DOI] [Google Scholar]
- 36.Owen SF, Huggett DB, Hutchinson TH, Hetheridge MJ, Kinter LB, Ericson JF, Sumpter JP. 2009. Uptake of propranolol, a cardiovascular pharmaceutical, from water into fish plasma and its effects on growth and organ biometry. Aquat. Toxicol. 93, 217–224. ( 10.1016/j.aquatox.2009.05.009) [DOI] [PubMed] [Google Scholar]
- 37.Smith EM, Chu S, Paterson G, Metcalfe CD, Wilson JY. 2010. Cross-species comparison of fluoxetine metabolism with fish liver microsomes. Chemosphere 79, 26–32. ( 10.1016/j.chemosphere.2010.01.058) [DOI] [PubMed] [Google Scholar]
- 38.Thibaut R, Porte C. 2008. Effects of fibrates, anti-inflammatory drugs and antidepressants in the fish hepatoma cell line PLHC-1: cytotoxicity and interactions with cytochrome P450 1A. Toxicol. In Vitro 22, 1128–1135. ( 10.1016/j.tiv.2008.02.020) [DOI] [PubMed] [Google Scholar]
- 39.Bartram AE, et al. 2012. In vivo and in vitro liver and gill EROD activity in rainbow trout exposed to the beta-blocker propranolol. Environ. Toxicol. Chem. 27, 573–582. ( 10.1002/tox.20684) [DOI] [PubMed] [Google Scholar]
- 40.Wassmur B, Gräns J, Norström E, Wallin M, Celander MC. 2013. Interactions of pharmaceuticals and other xenobiotics on key detoxification mechanisms and cytoskeleton in Poeciliopsis lucida hepatocellular carcinoma, PLHC-1 cell line. Toxicol. In Vitro 27, 111–120. ( 10.1016/j.tiv.2012.10.002) [DOI] [PubMed] [Google Scholar]
- 41.Jeon J, Kurth D, Hollender J. 2013. Biotransformation pathways of biocides and pharmaceuticals in freshwater crustaceans based on structure elucidation of metabolites using high resolution mass spectrometry. Chem. Res. Toxicol. 26, 313–324. ( 10.1021/tx300457f) [DOI] [PubMed] [Google Scholar]
- 42.Canga AG, Sahagún Prieto AM, Liébana MJD, Martínez NF, Vega MS, García Vieitez JI. 2009. The pharmacokinetics and metabolism of ivermectin in domestic animal species. Vet. J. 179, 25–37. ( 10.1016/j.tvjl.2007.07.011) [DOI] [PubMed] [Google Scholar]
- 43.Carlsson G, Patring J, Kreuger J, Norrgren L, Oskarsson A. 2013. Toxicity of 15 veterinary pharmaceuticals in zebrafish (Danio rerio) embryos. Aquat. Toxicol. 126, 30–41. ( 10.1016/j.aquatox.2012.10.008) [DOI] [PubMed] [Google Scholar]
- 44.Parkinson A. 1996. Biotransformation of xenobiotics. In Casarett and Doull's toxicology, the basic science of poisons, unit 2 (ed. Klaassen CD.), ch. 6, pp. 133–186. New York, NY: McGraw-Hill. [Google Scholar]
- 45.Ogu CC, Maxa JL. 2000. Drug interactions due to cytochrome P450. Baylor Univ. Med. Center Proc. 13, 421–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. 2006. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34, D668–D672. ( 10.1093/nar/gkj067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohyama K, Maki S, Sato K, Kato Y. 2004. In vitro metabolism of [14C]methoxychlor in rat, mouse, Japanese quail and rainbow trout in precision-cut liver slices. Xenobiotica 34, 741–754. ( 10.1080/00498250400003455) [DOI] [PubMed] [Google Scholar]
- 48.Huggett DB, Cook JC, Ericson JE, Williams RT. 2003. A theoretical model for utilizing mammalian pharmacology and safety data to prioritize potential impacts of human pharmaceuticals to fish. J. Hum. Ecol. Risk. Assess. 9, 1789–1799. ( 10.1080/714044797) [DOI] [Google Scholar]
- 49.Schreiber R, Gündel U, Franz S, Küster A, Rechenberg B, Altenburger R. 2011. Using the fish plasma model for comparative hazard identification for pharmaceuticals in the environment by extrapolation from human therapeutic data. Reg. Toxicol. Pharmacol. 61, 261–275. ( 10.1016/j.yrtph.2011.08.006) [DOI] [PubMed] [Google Scholar]
- 50.Huber C, Bartha B, Schröder P. 2012. Metabolism of diclofenac in plants—hydroxylation is followed by glucose conjugation. J. Hazard. Mater. 243, 250–256. ( 10.1016/j.jhazmat.2012.10.023) [DOI] [PubMed] [Google Scholar]
- 51.Kirchmair J, Williamson MJ, Tyzack JD, Tan L, Bond PJ, Bender A, Glen RC. 2012. Computational prediction of metabolism: sites, products, SAR, P450 enzyme dynamics, and mechanisms. J. Chem. Inf. Model. 52, 617–648. ( 10.1021/ci200542m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obach RS, Baxter JG, Liston TE, Silber M, Jones BC, MacIntyre F, Rance DJ, Wastall P. 1997. The prediction of human pharmacokinetics parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 283, 46–58. [PubMed] [Google Scholar]
- 53.Barton HA, et al. 2006. The acquisition and application of absorption, distribution, metabolism and excretion (ADME) data in agricultural chemical safety assessments. Crit. Rev. Toxicol. 36, 9–35. ( 10.1080/10408440500534362) [DOI] [PubMed] [Google Scholar]
- 54.Nichols JW, McKim JM, Andersen ME, Gargas ML, Clewell HJ, III, Erickson RJ. 1990. A physiologically based toxicokinetic model for the uptake and disposition of waterborne organic chemicals in fish. Toxicol. Appl. Pharmacol. 106, 433–447. ( 10.1016/0041-008X(90)90338-U) [DOI] [PubMed] [Google Scholar]
- 55.Schultz S, et al. 2004. Diclofenac poisoning is widespread in declining vulture populations across the Indian subcontinent. Proc. R. Soc. B 271, S458–S460. ( 10.1098/rsbl.2004.0223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cuthbert R, Green DR, Ranada S, Saravanan S, Pain D, Prakash V, Cunningham AA. 2006. Rapid population declines of Egyptian vulture (Neophron percnopterus) and red-headed vulture (Sarcogyps calvus) in India. Anim. Conserv. 9, 349–354. ( 10.1111/j.1469-1795.2006.00041.x) [DOI] [Google Scholar]
- 57.Cuthbert RJ, et al. 2014. Avian scavengers and the threat from veterinary pharmaceuticals. Phil. Trans. R. Soc. B 369, 20130574 ( 10.1098/rstb.2013.0574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swan GE, et al. 2006. Toxicity of diclofenac in Gyps vultures. Biol. Lett. 2, 1–4. ( 10.1098/rsbl.2005.0373) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naidoo V, Duncan N, Bekker L, Swan G. 2007. Validating the domestic fowl as a model to investigate the pathophysiology of diclofenac in Gyps vultures. Environ. Toxicol. Pharmacol. 24, 260–266. ( 10.1016/j.etap.2007.06.003) [DOI] [PubMed] [Google Scholar]
- 60.Naidoo V, Wolter K, Cuthbert R, Duncan N. 2009. Veterinary diclofenac threatens Africa's endangered vulture species. Reg. Toxicol. Pharmacol. 53, 205–208. ( 10.1016/j.yrtph.2009.01.010) [DOI] [PubMed] [Google Scholar]
- 61.Naidoo V, Mompati KF, Duncan N, Taggart MA. 2011. The pied crow (Corvus albus) is insensitive to diclofenac at concentrations present in carrion. J. Wildlife Dis. 47, 936–944. ( 10.7589/0090-3558-47.4.936) [DOI] [PubMed] [Google Scholar]
- 62.Naidoo V, Wolter K, Cromarty AD, Bartels P, Bekker L, McGaw L, Taggart MA, Cuthbert R, Swan GE. 2008. The pharmacokinetics of meloxicam in vultures. J. Vet. Pharmacol. Therap. 31, 128–134. ( 10.1111/j.1365-2885.2007.00923.x) [DOI] [PubMed] [Google Scholar]
- 63.Naidoo V, Wolter K, Cromarty D, Diekmann M, Duncan N, Meharg AA, Taggart MA, Venter L, Cuthbert R. 2010. Toxicity of non-steroidal anti-inflammatory drugs to Gyps vultures: a new threat from ketoprofen. Biol. Lett. 6, 339–341. ( 10.1098/rsbl.2009.0818) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naidoo V, Venter L, Wolter K, Taggart M, Cuthbert R. 2010. The toxicokinetics of ketoprofen in Gyps coprotheres: toxicity due to zero-order metabolism. Arch. Toxicol. 84, 761–766. ( 10.1007/s00204-010-0521-0) [DOI] [PubMed] [Google Scholar]
- 65.Walsh CT, Schwartz-Bloom RD. 2004. Levine's pharmacology: drug actions and reactions, 7th edn, pp. 561 Boca Raton, FL: CRC Press. [Google Scholar]
- 66.Yan Z, Li J, Huebert N, Caldwell GW, Du Y, Zhong H. 2005. Detection of a novel reactive metabolite of diclofenac: evidence for CYP2C9-mediated bioactivation via arene oxides. Drug Metab. Disp. 33, 706–713. ( 10.1124/dmd.104.003095) [DOI] [PubMed] [Google Scholar]
- 67.Dorado P, Cavaco I, Caceres MC, Piedade R, Ribeiro V, LLerena A. 2008. Relationship between CYP2C8 genotypes and diclofenac 5-hydroxylation in healthy Spanish volunteers. Eur. J. Clin. Pharmacol. 64, 967–970. ( 10.1007/s00228-008-0508-4) [DOI] [PubMed] [Google Scholar]
- 68.Zhou SF, Zhou ZW, Yang LP, Cai JP. 2009. Substrates, inducers, inhibitors and structure-activity relationships of human cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 16, 3480–3675. ( 10.2174/092986709789057635) [DOI] [PubMed] [Google Scholar]
- 69.Grosser T, Smyth E, FitzGerald GA. 2011. Anti-inflammatory, antipyretic and analgesic agents; pharmacotherapy of gout. In Goodman and Gilman's the pharmacological basis of therapeutics, 11th edn (eds Brunton L, Chabner B, Knollman B.), ch. 34, pp. 959–1004. New York, NY: McGraw-Hill. [Google Scholar]
- 70.Bort R, Macé K, Boobis A, Gómez-Lechón MJ, Pfeifer A, Castell J. 1999. Hepatic metabolism of diclofenac; role of human CYP in the minor oxidative pathways. Biochem. Pharmacol. 58, 787–796. ( 10.1016/S0006-2952(99)00167-7) [DOI] [PubMed] [Google Scholar]
- 71.Kidd KA, Blanchfield PJ, Mills KH, Palace VP, Evans RE, Lazorchak JM, Flick RW. 2007. Collapse of a fish population after exposure to a synthetic estrogen. Proc. Natl Acad. Sci. USA 104, 8897–8901. ( 10.1073/pnas.0609568104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown AR, Gunnarsson L, Kristiansson E, Tyler CR. 2014. Assessing variation in the potential susceptibility of fish to pharmaceuticals, considering evolutionary differences in their physiology and ecology. Phil. Trans. R. Soc. B 369, 20130576 ( 10.1098/rstb.2013.0576) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daughton CG, Ternes TA. 1999. Pharmaceuticals and personal care products in the environment: agents of subtle change? Environ. Health Perspect. Suppl. 107(S6), 907–938. ( 10.1289/ehp.99107s6907) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmitt H, et al. 2010. Recommendations on the environmental risk assessment of pharmaceuticals: effect characterization. Int. Environ. Assess. Manag. 6, 588–602. ( 10.1897/IEAM_2009-053.1) [DOI] [PubMed] [Google Scholar]
- 75.Küster A, et al. 2010. Environmental risk assessment of human pharmaceuticals in the European Union: a case study with the beta-blocker atenolol. Int. Environ. Assess. Manag. 6, 514–523. ( 10.1897/IEAM_2009-050.1) [DOI] [PubMed] [Google Scholar]
- 76.Boxall A, et al. 2012. Pharmaceuticals and personal care products in the environment: what are the big questions? Environ. Health Perspect. 120, 1221–1229. ( 10.1289/ehp.1104477) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taggart MA, Senacha KR, Green RE, Jhala YV, Raghaven B, Rahmani AR, Cuthbert R, Pain DJ, Meharg AA. 2007. Diclofenac residues in carcasses of domestic ungulates available to vultures in India. Environ. Int. 33, 759–765. ( 10.1016/j.envint.2007.02.010) [DOI] [PubMed] [Google Scholar]
- 78.Verbrugge LA, Giesy JP, Verbrugge DA, Woodin BR, Stegeman JJ. 2001. Catalytic and immunochemical properties of hepatic cytochrome P450 1A in three avian species treated with beta-naphthoflavone or isosafrole. Comp. Biochem. Physiol. C 130, 67–83. ( 10.1016/S1095-6433(01)00365-8) [DOI] [PubMed] [Google Scholar]
- 79.Kubota A, Kim EY, Iwata H. 2009. Alkoxyresorufin (methoxy-, ethoxy-, pentoxy- and benzyloxyresorufin) O-dealkylase activities by in vitro-expressed cytochrome P450 1A4 and 1A5 from common cormorant (Phalacrocorax carbo). Comp. Biochem. Physiol. C 149, 544–551. ( 10.1016/j.cbpc.2008.12.004) [DOI] [PubMed] [Google Scholar]
- 80.David P, Dauphin-Villemant C, Mesneau A, Meyran C. 2003. Molecular approach to aquatic environmental bioreporting: differential response to environmental inducers of cytochrome P450 monooxygenase genes in the detritivorous subalpine planktonic Crustacea, Daphnia pulex. Mol. Ecol. 12, 2473–2481. ( 10.1046/j.1365-294X.2003.01913.x) [DOI] [PubMed] [Google Scholar]
- 81.Hutchinson TH, Lyons BP, Thain J, Law RJ. 2013. Evaluating legacy contaminants and emerging chemicals in marine environments using adverse outcome pathways and biological effects-directed analysis. Mar. Poll. Bull. 74, 517–525. ( 10.1016/j.marpolbul.2013.06.012) [DOI] [PubMed] [Google Scholar]
- 82.Celander M, Goldstone J, Denslow N, Iguchi T, Kille P, Meyerhoff R, Smith B, Hutchinson TH, Wheeler JR. 2011. Species extrapolation for the 21st century. Environ. Toxicol. Chem. 30, 52–63. ( 10.1002/etc.382) [DOI] [PubMed] [Google Scholar]
- 83.Kolanczyk RC, et al. 2012. MetaPath: an electronic knowledge base for collating, exchanging and analyzing case studies of xenobiotic metabolism. Regul. Toxicol. Pharmacol. 63, 84–96. ( 10.1016/j.yrtph.2012.02.013) [DOI] [PubMed] [Google Scholar]
- 84.Li S, Pozhitkov A, Ryan RA, Manning CS, Brown-Peterson N, Brouwer M. 2010. Constructing a fish metabolic network model. Genome Biol. 11, R115 ( 10.1186/gb-2010-11-11-r115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murray-Smith RJ, Coombe VT, Haag Grönlund M, Waern F, Baird JA. 2012. Managing emissions of active pharmaceutical ingredients from manufacturing facilities: an environmental quality standard approach. Integ. Environ. Assess. Manag. 8, 320–330. ( 10.1002/ieam.1268) [DOI] [PubMed] [Google Scholar]
- 86.Shore RF, Taggart MA, Smits J, Mateo R, Richards NL, Fryday S. 2014. Detection and drivers of exposure and effects of pharmaceuticals in higher vertebrates. Phil. Trans. R. Soc. B 369, 20130570 ( 10.1098/rstb.2013.0570) [DOI] [PMC free article] [PubMed] [Google Scholar]