

Abstract
Pseudomonas aeruginosa is a Gram-negative bacterium that does not contain large, nonspecific porins in its outer membrane. Consequently, the outer membrane is highly impermeable to polar solutes and serves as a barrier against the penetration of antimicrobial agents. This is one of the reasons why such bacteria are intrinsically resistant to antibiotics. Polar molecules that permeate across the outer membrane do so through substrate-specific channels proteins. To design antibiotics that target substrate-channel proteins, it is essential to first identify the permeation pathways of their natural substrates. In P. aeruginosa, the largest family of substrate-specific proteins is the OccD (previously reported under the name OprD) family. Here, we employ equilibrium and steered molecular-dynamics simulations to study OccD1/OprD, the archetypical member of the OccD family. We study the permeation of arginine, one of the natural substrates of OccD1, through the protein. The combination of simulation methods allows us to predict the pathway taken by the amino acid, which is enabled by conformational rearrangements of the extracellular loops of the protein. Furthermore, we show that arginine adopts a specific orientation to form the molecular interactions that facilitate its passage through part of the protein. We predict a three-stage permeation process for arginine.
Introduction
Nutrients in the form of small molecules enter Gram-negative bacteria through channel proteins that reside within their outer membranes. These channel proteins enable passive diffusion of solutes along a concentration gradient across the membrane. The general porins, such as OmpC and OmpF found in Escherichia coli, have large pores and are able to mediate the passage of a range of solutes, including drugs (1,2). The human pathogen Pseudomonas aeruginosa has a notoriously impermeable outer membrane. One of the reasons for this low permeability is that, in contrast to E. coli and some other Gram-negative bacteria, the outer membrane of P. aeruginosa does not contain general porins (3). As a consequence of the lack of general porins, the uptake of nutrients in P. aeruginosa occurs largely through substrate-specific channel proteins; for example, uptake of phosphate is achieved through the OprP channel (4). Substrate-specific channels have narrower pores compared with the general porins and they bind their substrates with higher affinities, which enables them to discriminate between molecular species (5–7). In P. aeruginosa, proteins belonging to the Occ family are responsible for uptake of the vast majority of the small, water-soluble nutrients needed by the bacterium. Nineteen proteins belonging to this family have been identified in P. aeruginosa. They are divided into two subfamilies, OccD and OccK, based upon phylogenetic analysis. Both subfamilies share a specificity for solutes containing the carboxylate group, whereas the OccD family of channels is specific toward basic amino acids and the OccK family of channels is less selective, with a preference for cyclic compounds (7).
In recent years, the x-ray structures of a number of the Occ family proteins have been determined (5,7,8). These channel proteins share a common architecture: 18 β-strands are connected by large extracellular loops and short turns on the periplasmic side to form barrels. Combined biophysical, structural, and computational studies have identified residues that are key for substrate specificity in some of these channels. Eren et al. (7) showed that central residues of the basic ladder, a row of arginine and lysine residues that leads toward and away from the binding site at the channel constriction, are key to the interaction with substrates, similar to what has been observed for the transient interaction of ampicillin with OmpF (9).
However, to our knowledge, neither the precise pathways of substrate translocation nor the orientation and conformation of substrates as they permeate through the channels are known for any of the OccD proteins. Computational approaches provide a route to establish the links between the structure and function of membrane proteins, and have previously been employed to study outer-membrane proteins, including those from P. aeruginosa (10–12). Here, we used a combination of docking, steered molecular-dynamics (SMD), and equilibrium MD simulations to study the permeation pathway of arginine through its substrate-specific channel, OccD1. Our results reveal that translocation through the protein is characterized by specific arginine binding along a portion of the lumen of the β-barrel, followed by random diffusion.
Materials and Methods
All MD simulations were performed with the GROMACS 4.5.5 package (13–15). We modeled 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipids using the GROMOS 53A6-Kukol force field (16), and protein and ions using the GROMOS 54A7 (17) force field. The SPC water model was used (18). The protein was embedded over a period of 10 ps using the g_membed implementation in GROMACS (19). The solvent, protein, and DMPC bilayer (the protein and DMPC lipids were treated as a single temperature-coupling group) were kept at 313 K using Nosé-Hoover temperature coupling (20,21) with a time constant of 0.5 ps. The Parrinello-Rahman barostat (22,23) was applied to maintain a constant pressure of 1 bar in a semi-isotropic fashion, i.e., the x and y axes were treated independently from the z axis. The pressure coupling scheme used a time constant of 0.5 ps. Electrostatic interactions were treated using the smooth particle mesh Ewald (SPME) algorithm (24) with a short-range cutoff of 0.9 nm. The van der Waals interactions used a cutoff of 1.4 nm with a long-range dispersion correction applied to the pressure and energy. The LINCS algorithm (25) was used to constrain all bonds and a time step of 2 fs was applied. SMD simulations were performed using a pull force of 25 kJ mol−1 nm−1.
The coordinates for a hexagonal DMPC bilayer were as described in a previous simulation study of an outer-membrane protein from Haemphilus influenza (26). The protein model for the OccD1 structure was obtained from the Protein Data Bank (PDB, accession code 3SY7). The wild-type (WT) protein and mutant (Y176R, Y282R, and D307H) models were completed as described in our previous study of OccD1 (27). The typical simulation system contained 35,000–40,000 atoms and was comprised of one protein, ∼170 DMPC lipid molecules, ∼14,000 water molecules, one arginine substrate, and the appropriate number of Cl− or Na+ ions to neutralize the system.
We embedded the OccD1 protein into the membrane using the GROMACS g_membed function. We then relaxed the WT and mutant simulation systems by performing simulations of 100 ns duration, with positional restraints using a force constant of 1000 kJ mol−1 applied to the backbone of the β-sheet regions of the proteins. Taking equilibrated system structures as the starting points for further simulations, we performed multiple independent simulations with (Sim_WT_B and Sim_Mut_B) and without (Sim_Mut_A and Sim_WT_A) the presence of the arginine substrate. Additional simulations were performed with 1 M NaCl (Sim_WT_C).
An initial set of six independent SMD simulations (SMD_WT) was performed. A pull force of 25 kJ mol−1 nm−1 acting on the arginine substrate perpendicular to the bilayer normal in the z dimension was used. Additional SMD simulations (SMD_WT_a–h) were performed with 25 kJ mol−1 nm−1 acting on a vector following the z dimension, and an additional weaker pull in the x and/or y axes, resulting in eight extra simulations (see Table 1). In all holo-SMD simulations, the arginine substrate was released ∼0.3 nm above center of the protein pore from the extracellular side. SMD simulations of the mutants were also performed. All of the simulation systems are summarized in Table 1.
Table 1.
Summary of the system details for each simulation
| Code | System composition | Substrate | SMD pull vectors axis |
Simulation length (ns) | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Sim_WT_A | WT-counterions | Apo- | n/a | n/a | n/a | 250 |
| Sim_Mut_A | Y176R, Y282R, D307H-counterions | Apo- | n/a | n/a | n/a | 250 |
| Sim_WT_B | WT-counterions | Holo- Arg | n/a | n/a | n/a | 250 |
| Sim_Mut_B | Y176R, Y282R, D307H-counterions | Holo- Arg | n/a | n/a | n/a | 250 |
| Sim_WT_C | WT-1M NaCl | Holo- Arg | n/a | n/a | n/a | 250 |
| SMD_WT | WT-counterions | Holo- Arg | 0 | 0 | 1 | 75 |
| SMD_WT_a | WT-counterions | Holo- Arg | 0.1 | 0 | 1 | 75 |
| SMD_WT_b | WT-counterions | Holo- Arg | 0.1 | 0.1 | 1 | 75 |
| SMD_WT_c | WT-counterions | Holo- Arg | 0 | 0.1 | 1 | 75 |
| SMD_WT_d | WT-counterions | Holo- Arg | −0.1 | 0.1 | 1 | 75 |
| SMD_WT_e | WT-counterions | Holo- Arg | −0.1 | 0 | 1 | 75 |
| SMD_WT_f | WT-counterions | Holo- Arg | −0.1 | −0.1 | 1 | 75 |
| SMD_WT_g | WT-counterions | Holo- Arg | 0 | −0.1 | 1 | 75 |
| SMD_WT_h | WT-counterions | Holo- Arg | 0.1 | −0.1 | 1 | 75 |
| SMD_MUT | Y176R, Y282R, D307H-counterions | Holo- Arg | 0 | 0 | 1 | 75 |
A pull force with a vector of 0, 0, 1, such as SMD_WT, has a pull force acting perpendicular to the bilayer normal.
Analyses were performed with standard GROMACS utilities and locally written scripts. Membrane thickness was calculated using the GridMat-MD code (28), by measuring the average distance between the nitrogen atoms in the choline groups on opposite sides of the DMPC bilayer. Molecular visualization was performed with the VMD package (29).
Results
Protein conformational drift and flexibility
To monitor the conformational drift and structural flexibility, we calculated the root mean-square deviations (RMSD) and fluctuations (RMSF) of the OccD1 protein in the apo-, holo-, and holo-SMD systems. RMSD calculations give an indication of conformational drift by comparing the movement of atoms within a system with the initial system coordinates at t = 0, whereas RMSF calculations are time-averaged analyses that enable identification of the residue-by-residue fluctuations of the protein.
RMSD calculations of the Cα atoms of the protein in all WT simulations (MD and SMD) revealed plateau values of ∼0.2–0.3 nm (Fig. S1 in the Supporting Material). There was less structural drift in the β-sheet Cα atoms, which in general reached a plateau RMSD value of ∼0.1 nm. These values are comparable to those reported in previous simulation studies of other bacterial outer-membrane proteins, such as the values obtained for OmpT (∼0.1 nm for β-sheet Cα atoms, and ∼0.1–0.2 nm for whole-protein Cα atoms (30)), OmpA (∼0.1–0.2 nm for β-sheet Cα atoms (31)), and FecA (∼0.23–0.28 nm for β-barrel atoms). However, it should be noted that the OccD1 RMSD data presented here were gathered over a timescale 4, 10, and 20 times longer than those used for FecA, OmpA, and OmpT, respectively. This implies that the 2.15 Å structure is reliable and stable. In further agreement with the previous studies, the OccD1 simulations revealed that the greatest structural drift originates in the loop regions of the protein (Fig. S1). Loops L2, L7, L8, and T5 have RMSD values that plateau with an average of ∼0.5–0.6 nm, with spikes due to loop interactions.
The RMSF of the apo-OccD1 showed the greatest fluctuations in the loop regions L1, L2, L7, T7, and L8 (Fig. S2). In the holo- simulations, the dominant fluctuations occurred in the loop regions, specifically L1, L2, T4, T5, L7, T7, and L8 (Fig. 1). The flexibility of loops L2 and L7, as indicated by the fluctuations in the RMSF data, was unsurprising given the lack of structural data for these regions. L2 is comprised of 15 residues and alternates between lying on the membrane, interacting with the phospholipids, or being located away from the membrane and closer to the rest of the protein, and thus is able to adopt a range of conformations. A comparison of the apo- and holo- RMSFs indicates that the presence of the arginine substrate, ArgS, in the protein has a structurally stabilizing effect on L7. Conversely, it appears to increase fluctuations in L8. In agreement with Eren et al. (27), L7 fluctuations were largely within residues 285–294. Experimental studies indicated that the flexibility of L3 has an impact on the size of the protein pore and permeation (7). Our simulations revealed relatively low RMSF data for both apo- and holo- simulations, indicating a loop structure with lower flexibility than might be expected. This lower RMSD does not indicate a lack of importance and influence; rather, it shows that loop L3 is less flexible than other loops. Simulations of the mutant revealed behavior similar to that observed for the WT protein, with one notable difference: a decrease in the RMSF of the L2 and L7 loop regions (Fig. S2).
Figure 1.
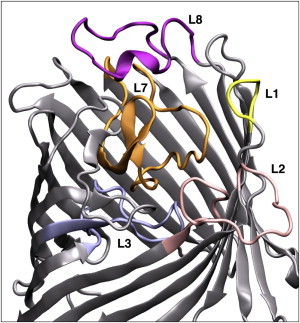
The modeled OccD1 protein, with loops of interest colored for clarity. The protein is shown in gray, with loops L1, L2, L3, L7, and L8 colored in yellow, pink, blue, orange, and purple, respectively. To see this figure in color, go online.
Influence of OccD1 on the local membrane environment
The three-dimensional thickness plot in Fig. S4 shows an average membrane thickness of ∼3.6 nm, which is measured as the distance between the nitrogen atoms in the choline headgroups of the DMPC. We note that Kucerka et al. (32) reported the thickness of a DMPC bilayer to be ∼4.4 nm as determined by small-angle neutron scattering. The 0.8 nm difference between the two values in part reflects the fact that Kucerka et al. obtained their measurements at a temperature of 303 K, whereas we performed our simulations at 313 K. The 10 K higher temperature in the simulations is likely to make the membrane more fluid and consequently slightly thinner. This difference in thickness is also due in part to the inability of the force field to reproduce the experimentally observed bilayer thickness (33). However, we observed an interesting influence on the bilayer thickness due to the presence of the protein, i.e., a depression in the inner- and outer-leaflet DMPC headgroups near strands S5–S8 of the barrel (Fig. S4). Residues H156 and D186 were located closer to the membrane core than the other charged residues at the extracellular end of the barrel. Similarly, residue E146 was also located closer to the membrane core than typical charged residues at the intracellular end of the barrel. These residues interacted with the zwitterionic headgroups of nearby DMPC lipids (Fig. 2). Close to strands S5 and S6, we observed three notable molecular interactions: 1), the phosphate group of a DMPC molecule formed a long-lived hydrogen-bond interaction with S168 backbone NH; 2), the choline headgroup formed a stable electrostatic interaction with the carboxylate side chain of E159; and 3), one of the carbonyl groups on a fatty tail formed a hydrogen-bond interaction with the K167 side-chain terminal amine. These lipid–protein interactions resulted in lipid headgroups being pulled toward the protein and closer to the membrane core, thereby thinning the bilayer such that the width became <3 nm.
Figure 2.
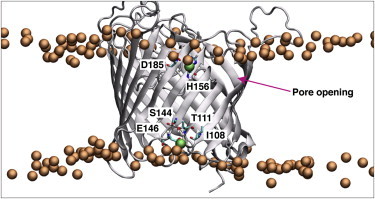
Snapshot of the DMPC membrane containing OccD1, showing membrane pinching around the region of the protein near strands S5–S8. The phosphate headgroups of the DMPC are shown in brown, with three phosphate headgroups of interest shown in green. To see this figure in color, go online.
A consistently thicker section of the membrane (∼3.7 × 3.7 nm; Fig. S4) involved another series of long-lived protein–lipid interactions on the inner leaflet of the membrane near strands S17 and S18 of the β-barrel. First, the choline headgroup of a DMPC lipid molecule formed an electrostatic interaction with the side-chain carboxylate of D385; second, a lipid phosphate group formed a hydrogen-bonding interaction with the side-chain terminal hydroxyl group of Y415; and finally, one of the carbonyl groups of one of the lipid tails formed a hydrogen-bonding interaction with the side-chain amine group of H421.
The interaction between loop L2 and the DMPC lipids, observed in simulations of the WT protein, occurred primarily due to an anchoring event involving K79. The side-chain of K79 was observed to penetrate in between the headgroups of the DMPC lipids, with the side-chain amine forming a hydrogen-bonding interaction with the fatty acid carbonyls of the DMPC molecules, as well as electrostatic interactions with the phosphate groups (Fig. 3). Similar local membrane perturbations in response to other outer-membrane proteins have previously been reported (34). Presumably, the specific membrane-protein interactions highlighted here facilitate and maintain an open orientation of the OccD1 within the membrane.
Figure 3.
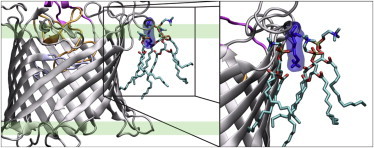
Snapshot showing the binding of the OccD1 loop L2 to the membrane. Residue K79 is highlighted in blue and the typical region occupied by the DMPC headgroups is shown by the green highlighted region. To see this figure in color, go online.
Equilibrium MD simulations
We performed a total of 10 independent equilibrium MD simulations of the holo- systems Sim_WT_B and Sim_Mut_B, in which ArgS was originally located in the extracellular loop region near the binding domain of Y176, Y282, and D307. In the WT simulations, the ArgS remained bound to the protein, with the guanidinium side chain of the substrate forming a number of hydrogen bonds and favorable electrostatic interactions in the protein-binding domain defined by residues Y176, Y282, and D307. The backbone of ArgS formed electrostatic interactions with the arginine ladder residues R410 and R391, in agreement with previously reported docking calculations (27). In all of the mutant simulations, ArgS was ejected from the binding domain of the protein within 20 ns toward the extracellular bulk water region. Subsequently, ArgS was unable to reenter the protein due to a conformational rearrangement of loop L7 that blocked access to the binding domain. Furthermore, movement of loop L2 also prevented access to the binding region. Specifically, movement of loop L2 initiated a series of long-lived interactions. Loops L7 and L9 were observed to engage in a number of different hydrogen bonding and electrostatic interactions with loop L2. For example, the amine group of the N84 side chain in L2 is engaged in hydrogen bonding with the carbonyl group of the E403 side chain in L9. Other hydrogen-bonding interactions included the carboxylate group of the D90 backbone in L2 with the guanidinium group of the R287 side chain in L7; the thioether M88 side chain in L2 with the hydroxyl side chain of S290 in L7. Electrostatic interactions between the carboxylate group of the D90 side chain in L2 with the guanidinium side chain of R287 in L7 were also observed. Other hydrogen bonding and electrostatic interactions were transiently observed between these loops. Closure of the loop L7 region and stabilization by the L2 loop region are reflected in the low RMSF values for loops L7 and L2. In combination, the conformational rearrangements of loops L7 and L2 provide a physical barrier against entry of arginine (Fig. 4).
Figure 4.
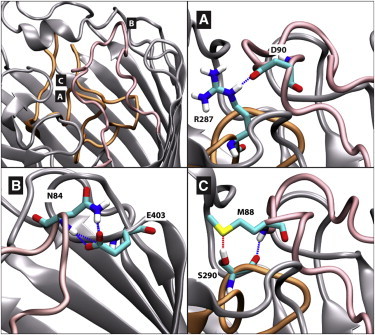
Top-left panel: an image showing loop L2 (pink) positioned on top of loop L7 (orange), with the locations of panels A, B, and C marked for clarity. Panels A, B, and C detail typical hydrogen-bonding interactions of loop L2 in mutant simulations, with residues colored by atom (carbon in cyan, sulfur in yellow, oxygen in red, nitrogen in blue, and hydrogen in white). To see this figure in color, go online.
Fluctuations of the loop T5 region (on the periplasmic side between strands S10 and S11) were observed in both the holo- and apo- simulations (Figs. S1 and S2). These fluctuations are a consequence of two modes of action: first, the Q227 residue changes orientation to form hydrogen-bonding interactions with residues in the neighboring strand (S12, Y260, and T261); second, residues P222 to Q227 change orientation to allow hydrogen-bonding interactions between the Q227 side-chain carboxyl or A224 backbone amine and the DMPC phosphatidyl groups and the fatty acid carbonyls (Fig. 5).
Figure 5.
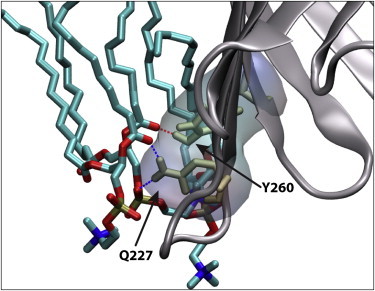
OccD1 lipid interactions near loop T5. Residues Q227 and Y260 are shown forming hydrogen-bonding and electrostatic interactions with DMPC phosphates and carbonyls. Carbon is shown in cyan, oxygen is in red, nitrogen is in blue, and phosphorus is in tan. To see this figure in color, go online.
Interestingly, a potential spontaneous gating motion was observed involving loop L8 residue Y359 and residues R287, N309, and D307 in the L7 loop region. This Y359 movement was present in all simulations, including those lacking ArgS permeation. The motion shows Y359 forming hydrogen-bonding interactions with residues N309 and D307 in one orientation and with R287 in another (Fig. 6). The interaction of loop L8 with R287 was previously documented by Eren et al. (27); here, however, we present a new rationale for the observed movement, using 2.9 μs simulation data from all of the WT, apo-, holo-, and SMD simulations. To monitor the motion of Y359 in the presence of ArgS, we recorded the minimum distance between residues Y359 and R287, D307, and ArgS over the course of the simulations. An interaction was counted when the Y359–R287 or Y359–D307 distance was ≤0.35 nm. Interactions typically involved Y359 side-chain hydroxyl hydrogen bonding to R287 side-chain guanidinium NH, with a minimum distance of ∼0.18 nm, or the Y359 side-chain hydroxyl hydrogen bonding to the D307 side-chain carbonyl oxygen, with a minimum distance of ∼0.16 nm. Typical hydrogen bonding can be seen in Fig. S5.
Figure 6.
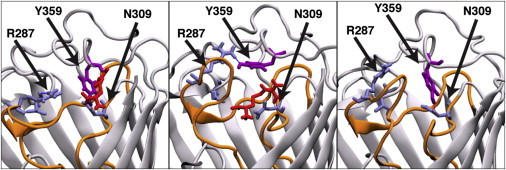
Gating event observed in SIM_REF. Y359 (violet) shows two distinct interactions: one with N309 (in lilac) in a downward-facing position and the other with R287 (lilac) in a more upward-facing position. It can be seen that the Y359 interaction fluctuated during the transport of ArgS (red). Loop L7 is shown in orange. Water and membrane have been omitted for clarity. To see this figure in color, go online.
Although ArgS is present in the extracellular binding region of OccD1 (for a total of 1.7 μs), determined as being within 1.2 nm of residue Y359, Y359 shows a preference to form an interaction with R287 over D307, with Y359–R287 interactions occurring 34% of the total time ArgS is present and Y359–D307 interactions occurring 20% of the total time. For the remaining 46% of the total time in which ArgS is present, Y359 does not interact with either D307 or R287. Although ArgS is absent from the protein-binding region (for a total of 1.2 μs), i.e., at a distance >1.2 nm from residue Y359, Y359 preferentially interacts with R287, with Y359–R287 interactions occurring 57% of the time, and does not bind favorably to either D307 or R287 for the remaining 46% of the time. Thus, the presence of ArgS in the binding region of the protein increases the frequency of the Y359–D307 interaction and decreases the frequency of the Y359–R287 interaction. These results complement those of Eren et al. (27), indicating that the frequency of interactions between the residues of loop L8 and residue R287 of loop L7 are influenced by the presence of ArgS.
SMD simulations
Investigations of the ArgS permeation pathway through OccD1 were primarily performed with SMD. ArgS was pulled through OccD1 along the principal axis of the protein (z dimension) using a constant force of 25 kJ mol−1. We monitored the orientation of the ArgS substrate with respect to the protein by calculating the relative COM z coordinate of the protein backbone compared with that of the ArgS molecule (Figs. 7 and S3). The graph shows a clear region at z = ∼4 nm with an orientation-based requirement for ArgS passage. This region contains the residues D307, S302, R410, R391, R389, and R131. Residues D307 and S302 interact with the guanidinium side chain of ArgS through electrostatic interactions and hydrogen bonding, respectively. The arginine ladder residues R410, R391, R389, and R131 engage in electrostatic interactions with the negative charge of the ArgS backbone carboxylate group. This interaction is typical of all permeation events observed in our simulations. A distinct pathway of permeation is visible when ArgS from five simulation replicas are overlaid on the protein (Fig. 8). This pathway reveals that the carboxylate group of the ArgS follows the arginine ladder substrates R30, R131, R410, R391, and R389 up until passage through the eyelet region (the narrowest section of the pore, near R410, S130, G294, V30, and Q304) of the protein.
Figure 7.
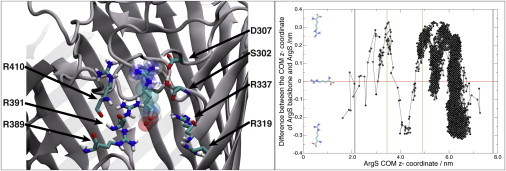
The ArgS orientation required for permeation is shown on the left, at the region z ∼ 4 nm. The carboxylate group of ArgS is interacting with a series of arginine ladder residues. The guanidinium group of the arginine is interacting with D307 and S302 of loop L7 in the protein. ArgS is shown in space-filling format for ease of visualization. Water molecules and the membrane have been omitted for clarity. On the right is a plot showing the orientation of ArgS through OccD1. The green lines indicate a region of specificity, requiring the substrate to be in a conformation such that the guanidinium side chain is pointing toward the periplasmic region, y < 0. The brown lines indicate the periplasmic and extracellular ends of the protein at ∼2.1 nm and ∼7.8 nm, respectively. To see this figure in color, go online.
Figure 8.
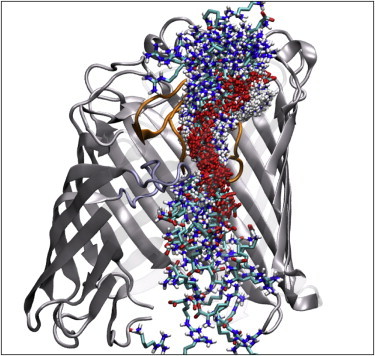
Overlap of the frames of all SMD transport simulations, showing a clear carboxylate pathway through the protein. The water and DMPC membrane, as well as the section of protein nearest to the camera, have been omitted for clarity. Loop L3 is shown in lilac and loop L7 is shown in orange. ArgS is colored by atom, with hydrogen in white, carbon in cyan, nitrogen in blue, and oxygen in red. To see this figure in color, go online.
Based on these simulations, we suggest that the permeation of ArgS occurs in three stages: 1), initial localization of the substrate to the pore opening; 2), reorientation at a checkpoint; and 3), eventual permeation and ejection. The first stage is where several simulations failed to show permeation, with the substrate either adopting an unfavorable conformation for passage through the protein or docking into nonpermeation poses with varying protein loops, and even membrane interaction. In three out of the five simulations in which permeation of Arg was observed, entry of ArgS into the extracellular opening of OccD1 involved binding near the region of loops L7, L8, and L9, incorporating hydrogen bonding between the ArgS guanidinium and Y359 side-chain hydroxyl group and between the ArgS backbone carboxylate and E403 backbone amine (Fig. 9 A). Following the initial binding pocket, the ArgS moved inward toward the eyelet pore of the protein. The freedom of orientation change for the ArgS on approach to the protein eyelet was much greater than that in the eyelet (Fig. 9), as can be seen by the spread of the guanidinium side chain shown in the extracellular region of the protein (Fig. 9, B and C). When it reached the binding pocket at the eyelet, a series of hydrogen bonds were formed, stabilizing the ArgS: the backbone carboxylate of ArgS formed hydrogen-bonding and electrostatic interactions with the S130 hydroxyl group and R410 guanidinium amines, and the side-chain guanidinium of ArgS formed electrostatic interactions with D307. The eyelet restriction around the ∼4 nm region acts as a checkpoint for the passage of ArgS, requiring the substrate to have the backbone pointing toward the periplasmic region.
Figure 9.
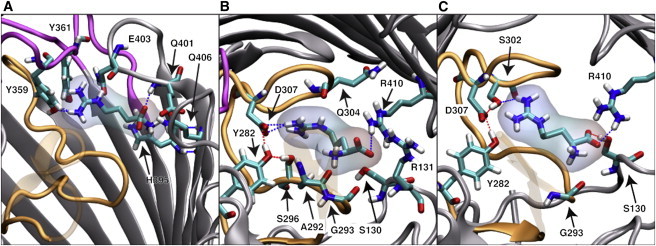
Snapshots from SMD simulations of OccD1, from left to right: (A) a binding site of the ArgS near the loop L9 region, (B) a binding conformation upon ArgS entrance to the eyelet region, and (C) a further binding pose within the eyelet toward ejection. To see this figure in color, go online.
The final stage again shows greater orientational freedom for ArgS (Fig. 8). After it passes through the eyelet of the protein, ArgS takes a seemingly random walk in the periplasmic region. The total time taken for ArgS to permeate through OccD1 ranged between ∼20 ns and ∼80 ns. The longer times are indicative of ArgS spending a greater length of time on approach to the eyelet region. The permeation of ArgS with the backbone carboxylate pointing toward the periplasmic region occurred fairly rapidly, taking only ∼20 ns (Fig. 10, A–C). After it reentered the protein through the periodic boundary in a different orientation, with the guanidinium side chain pointing toward the periplasmic region (Fig. 10, D–G), the ArgS entered into a bound conformation for >100 ns (not affording permeation).
Figure 10.
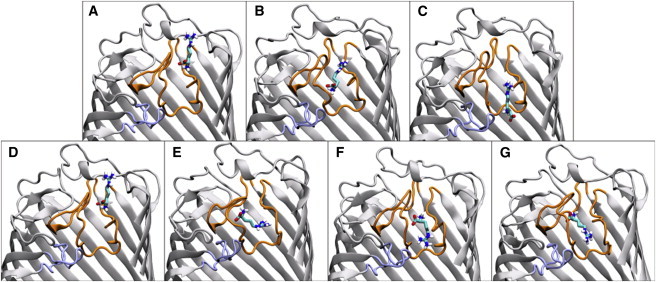
(A–C) Transport of the ArgS through OccD1, with the carboxylate in a downward position. The total time required for transport of the substrate was ∼10 ns. (D–G) Reentry of ArgS into OccD1 through the periodic boundary, with the guanidinium group pointing downward. The total time ArgS remained bound was >100 ns. To see this figure in color, go online.
Water permeation through OccD1
The main pore of OccD1 remained open throughout the simulations, with the flow of water being obstructed only by the presence of ArgS in the pore in the relevant simulations. Water was observed to flow in both directions through the protein pore. Over the course of 200 ns simulations, only one or two Na+ ions were observed to permeate through the pore of the protein.
β-Sheet strands S5 and S6 are several residues shorter than the neighboring strands S4 and S7, and this reduced β-sheet length provides a possible channel-like lateral opening into the protein. Residues E117, A100, R99, and L161 formed a channel directly adjacent to the extracellular end of strands S5 and S6, through which water was observed to permeate from within the protein to the bulk water region, albeit at a very low rate of ∼1–2 molecules over the course of 200 ns (Fig. 2). The side chain of K167 formed a seal between the channel and the extracellular water. If the alternative channel does act as a pathway for molecules through the OccD1 protein, the method of permeation either incorporates the movement of K167 or permeation directly into the membrane.
One of the limitations of this study is that we used a simplified, symmetrical model of the membrane. Although this reflects the setup used in the majority of experimental studies of OccD1 and outer-membrane proteins in general, a more complex model would be more representative of the in vivo environment. Recently, lipopolysaccharide (LPS)-containing, asymmetrical models of the E. coli and P. aeruginosa outer membrane have been reported (35–37). However, we note that due to the much slower diffusion time of LPS compared with phospholipids (35), we would have struggled to observe rearrangements of loops in atomistic simulations of even hundreds of nanoseconds.
Conclusions
Arginine permeation through OccD1 occurs via a series of binding sites, which are present in the majority of the permeation simulations. The initial binding site incorporates electrostatic and hydrogen-bonding interactions with loops L7, L8, and L9. The permeation of ArgS through the extracellular loop region is not a well-defined motion, with the guanidinium side chain fluctuating until it reaches the eyelet region of the protein. On the other hand, the backbone motion is conserved, with all repeat simulations depicting a clear pathway for the carboxylate group all the way to the eyelet region. The motion of ArgS through OccD1 involves electrostatic interactions between the ArgS backbone and the arginine ladder, as predicted in previous studies (5,7,27).
Acknowledgments
The authors thank Dr. Thomas Piggot for providing the membrane model and for his advice throughout this study. We also acknowledge use of the University of Southampton HPC resources Iridis3 and Iridis4.
Supporting Material
References
- 1.Jaffe A., Chabbert Y.A., Semonin O. Role of porin proteins OmpF and OmpC in the permeation of β-lactams. Antimicrob. Agents Chemother. 1982;22:942–948. doi: 10.1128/aac.22.6.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahendran K.R., Kreir M., Winterhalter M. Permeation of antibiotics through Escherichia coli OmpF and OmpC porins: screening for influx on a single-molecule level. J. Biomol. Screen. 2010;15:302–307. doi: 10.1177/1087057109357791. [DOI] [PubMed] [Google Scholar]
- 3.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moraes T.F., Bains M., Strynadka N.C.J. An arginine ladder in OprP mediates phosphate-specific transfer across the outer membrane. Nat. Struct. Mol. Biol. 2007;14:85–87. doi: 10.1038/nsmb1189. [DOI] [PubMed] [Google Scholar]
- 5.Biswas S., Mohammad M.M., van den Berg B. Structural insight into OprD substrate specificity. Nat. Struct. Mol. Biol. 2007;14:1108–1109. doi: 10.1038/nsmb1304. [DOI] [PubMed] [Google Scholar]
- 6.Nikaido H., Rosenberg E.Y. Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J. Bacteriol. 1983;153:241–252. doi: 10.1128/jb.153.1.241-252.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eren E., Vijayaraghavan J., van den Berg B. Substrate specificity within a family of outer membrane carboxylate channels. PLoS Biol. 2012;10:e1001242. doi: 10.1371/journal.pbio.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biswas S., Mohammad M.M., van den Berg B. Crystal structure of the outer membrane protein OpdK from Pseudomonas aeruginosa. Structure. 2008;16:1027–1035. doi: 10.1016/j.str.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Kumar A., Hajjar E., Ceccarelli M. Molecular simulations reveal the mechanism and the determinants for ampicillin translocation through OmpF. J. Phys. Chem. B. 2010;114:9608–9616. doi: 10.1021/jp9110579. [DOI] [PubMed] [Google Scholar]
- 10.Carpenter T., Khalid S., Sansom M.S.P. A multidomain outer membrane protein from Pasteurella multocida: modelling and simulation studies of PmOmpA. Biochim. Biophys. Acta. 2007;1768:2831–2840. doi: 10.1016/j.bbamem.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 11.Khalid S., Bond P.J., Sansom M.S.P. OmpA: gating and dynamics via molecular dynamics simulations. Biochim. Biophys. Acta. 2008;1778:1871–1880. doi: 10.1016/j.bbamem.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 12.Pongprayoon P., Beckstein O., Sansom M.S.P. Simulations of anion transport through OprP reveal the molecular basis for high affinity and selectivity for phosphate. Proc. Natl. Acad. Sci. USA. 2009;106:21614–21618. doi: 10.1073/pnas.0907315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berendsen H.J.C., Vanderspoel D., Vandrunen R. Gromacs—a message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 14.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 15.Van Der Spoel D., Lindahl E., Berendsen H.J.C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 16.Kukol A. Lipid models for united-atom molecular dynamics simulations of proteins. J. Chem. Theory Comput. 2009;5:615–626. doi: 10.1021/ct8003468. [DOI] [PubMed] [Google Scholar]
- 17.Schmid N., Eichenberger A.P., van Gunsteren W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 18.Berendsen H., Postma J., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. Reidel Publishing; Boston: 1981. pp. 331–342. [Google Scholar]
- 19.Wolf M.G., Hoefling M., Groenhof G. g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010;31:2169–2174. doi: 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- 20.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 21.Nose S. A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 1984;81:511–519. [Google Scholar]
- 22.Nose S., Klein M.L. Constant pressure molecular-dynamics for molecular-systems. Mol. Phys. 1983;50:1055–1076. [Google Scholar]
- 23.Parrinello M., Rahman A. Polymorphic transitions in single-crystals—a new molecular-dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 24.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 25.Hess B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 26.Holdbrook D.A., Piggot T.J., Khalid S. Stability and membrane interactions of an autotransport protein: MD simulations of the Hia translocator domain in a complex membrane environment. Biochim. Biophys. Acta. 2013;1828:715–723. doi: 10.1016/j.bbamem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Eren E., Parkin J., van den Berg B. Toward understanding the outer membrane uptake of small molecules by Pseudomonas aeruginosa. J. Biol. Chem. 2013;288:12042–12053. doi: 10.1074/jbc.M113.463570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen W.J., Lemkul J.A., Bevan D.R. GridMAT-MD: a grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009;30:1952–1958. doi: 10.1002/jcc.21172. [DOI] [PubMed] [Google Scholar]
- 29.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 30.Baaden M., Sansom M.S.P. OmpT: molecular dynamics simulations of an outer membrane enzyme. Biophys. J. 2005;87:2942–2953. doi: 10.1529/biophysj.104.046987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bond P.J., Faraldo-Gómez J.D., Sansom M.S. OmpA: a pore or not a pore? Simulation and modeling studies. Biophys. J. 2002;83:763–775. doi: 10.1016/S0006-3495(02)75207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kucerka N., Kiselev M.A., Balgavý P. Determination of bilayer thickness and lipid surface area in unilamellar dimyristoylphosphatidylcholine vesicles from small-angle neutron scattering curves: a comparison of evaluation methods. Eur. Biophys. J. 2004;33:328–334. doi: 10.1007/s00249-003-0349-0. [DOI] [PubMed] [Google Scholar]
- 33.Piggot T.J., Pineiro A., Khalid S. Molecular dynamics simulations of phosphatidylcholine membranes: a comparative force field study. J. Chem. Theory Comput. 2012;8:4593–4609. doi: 10.1021/ct3003157. [DOI] [PubMed] [Google Scholar]
- 34.Khalid S., Bond P.J., Sansom M.S.P. Modeling and simulations of a bacterial outer membrane protein: OprF from Pseudomonas aeruginosa. Proteins. 2006;63:6–15. doi: 10.1002/prot.20845. [DOI] [PubMed] [Google Scholar]
- 35.Piggot T.J., Holdbrook D.A., Khalid S. Electroporation of the E. coli and S. Aureus membranes: molecular dynamics simulations of complex bacterial membranes. J. Phys. Chem. B. 2011;115:13381–13388. doi: 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- 36.Wu E.L., Fleming P.J., Im W. E. coli outer membrane and interactions with OmpLA. Biophys. J. 2014;106:2493–2502. doi: 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nascimento A., Jr., Pontes F.J.S., Soares T.A. Hydration, ionic valence and cross-linking propensities of cations determine the stability of lipopolysaccharide (LPS) membranes. Chem. Commun. (Camb.) 2014;50:231–233. doi: 10.1039/c3cc46918b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.