

Abstract
Amyloid fibrillation in water-organic mixtures has been widely studied to understand the effect of protein-solvent interactions on the fibrillation process. In this study, we monitored insulin fibrillation in formamide and its methyl derivatives (formamide, N-methyl formamide, N,N-dimethyl formamide) in the presence and absence of water. These model solvent systems mimic the cellular environment by providing denaturing conditions and a hydrophobic environment with limited water content. Thioflavin T (ThT) assay revealed that binary mixtures of water with formamide and its methyl derivatives enhanced fibrillation rates and β-sheet abundance, whereas organic solvents suppressed insulin fibrillation. We utilized solution small-angle x-ray scattering (SAXS) and differential scanning calorimetry (DSC) to investigate the correlation between protein-solvent interactions and insulin fibrillation. SAXS experiments combined with simulated annealing of the protein indicated that the degree of denaturation of the hydrophobic core region at residues B11–B17 determines the fibrillation rate. In addition, DSC experiments suggested a crucial role of hydrophobic interactions in the fibrillation process. These results imply that an environment with limited water, which imitates a lipid membrane system, accelerates protein denaturation and the formation of intermolecular hydrophobic interactions during amyloid fibrillation.
Introduction
Amyloidoses, including Alzheimer’s disease (1), Parkinson’s disease (2), type II diabetes (3), and spongiform encephalopathy (4), are associated with the formation of amyloid fibrils. The fibrillation process of amyloidogenic proteins is initiated by the conversion of innocuous proteins into oligomeric intermediates to form highly ordered, unbranched β-sheet structures (5). Due to the complexity of amyloidosis (6), amyloid fibrillation of proteins in vivo is not fully understood. However, a protein-misfolding event is highly correlated with amyloid fibrillation (7). Therefore, investigating the mechanistic details of the misfolding event is valuable for understanding amyloid fibrillation and developing a therapeutic strategy for amyloidosis.
Water-protein interactions are an important aspect of amyloid fibrillation. Water plays a pivotal role in the conformational search during protein folding (8). Hydrophilic residues are oriented toward the outer space of a protein, whereas hydrophobic residues are buried within the inner space due to intramolecular hydrophobic interactions (9,10). Thus, denaturation of a protein to expose its interior hydrophobic residues to external aqueous solution is not a spontaneous process (8,11). When the hydrophobic regions of a protein are exposed to water, self-assembled protein aggregation is initiated via intermolecular hydrophobic interactions to minimize contact with water molecules (6,12). Although the association rate is dependent on the protein sequence and the tertiary structure of a protein, hydrophobic interactions are considered a common driving force for protein aggregation (13). Various environmental factors for protein denaturation are present in cellular environments (14). For example, the presence of a lipid membrane restricts water contents near the water-lipid interface (15–17), resulting in the structural transformation of proteins (18–21). Thus, numerous studies have investigated the effect of environmental factors on amyloid fibrillation by mimicking the cellular environment using lipid vesicles (21–23), organic solvents (24–30), and artificial crowding agents (31–34). However, elucidating the role of denaturation and water in amyloid fibrillation remains challenging (8,35).
Insulin, one of the more than 30 amyloidogenic proteins that have been identified to date, causes injection-localized amyloidosis (Scheme 1) (5). Insulin fibrillation occurs in vitro as well and is a critical consideration in the commercial production of the protein for diabetes treatment (36). Numerous studies of the mechanism of insulin fibrillation have been performed at elevated temperature (37), low pH (38), and high ionic strength (39). Insulin is present as the associated oligomer in the native state through hydrophobic interactions of hydrophobic residues (A8–A18, B1–B3, B9–B18, and B25–28) (40,41). However, under the environmental stresses described above, the interface between the hydrophobic residues is destabilized and the associated insulin oligomer dissociates into monomers (37,42). The helix bundles of the insulin monomers then undergo partial denaturation, with the exception of residues B11–B17 of the B-chain (Scheme 1), which retains its α-helical structure before the formation of a nucleus for amyloid aggregates occurs (37). A pair of B11–B17 residues is eventually rearranged into a cross-β structure, which is assembled into the fibril spine of a mature insulin fibril (43). After protein denaturation, hydrophobic interactions are crucial for amyloid assembly of insulin. Residues B11–B17 and B24–B26 are related to the formation of dimeric building blocks, and the N-terminus of the B-chain governs the formation of oligomers during insulin fibrillation (44,45). Recently, our laboratory demonstrated effective inhibition of insulin fibrillation by capturing phenylalanine residues at B1 and B24 using cucurbit[7]uril (46), thereby inhibiting intermolecular hydrophobic interactions between the proteins. Thus, denaturation and hydrophobic interactions cooperate in each step of the protein fibrillation process.
Scheme 1.
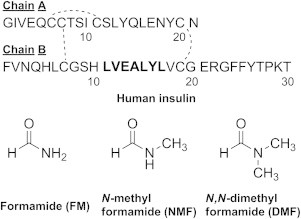
Structure of human insulin (top) and formamide derivatives (bottom). Residues B11–B17 are marked in bold.
The lipid membrane is closely associated with insulin fibrillation, as demonstrated by the formation of extracellular fibrils in response to frequent injection of insulin solution into the same general region near the subcutaneous fat layer in type I diabetes patients (47). Previous model studies have suggested that interactions between insulin and the headgroups of phospholipids cause protein denaturation in the lipid membrane (48–51). However, the effect of a low-water-density environment, which is a characteristic feature of the water-membrane interface, on the structural transition and related intermolecular interactions of insulin has not been investigated.
Water-organic cosolvent systems have been widely adopted as simplified model systems for membrane environments (28,53). The cosolvent system can modulate protein folding by changing the hydration shell (54), hydrophobic interactions (55), and intramolecular hydrogen bonds of the protein (18,56). This approach is also applicable for studying the fibrillation process of amyloidogenic proteins because protein fibrillation is a complex process that involves both intermolecular hydrophobic interactions and protein folding (9). The cosolvent system affects both the rearrangement of the protein structure in the initial state and protein aggregation in the denatured state (24,28,30). Thus, this system provides insight into the environmental factors that are closely related to amyloid fibrillation. Intermolecular interactions between water and formamide have been studied as a model system to mimic the hydration of a protein, because the functional group of formamide can represent the peptide backbone (57). When formamide is directly added to a protein solution, globular proteins are reported to unfold (58,59). Previous studies (60,61) have demonstrated that both the polar and nonpolar functional groups of formamide and its methyl derivatives (Scheme 1) contribute to the denaturation of globular proteins. The polar functional group (i.e., amide) solvates the peptide backbone, whereas the nonpolar functional group (i.e., the methyl group in the amide group) stabilizes the hydrophobic residues exposed to the exterior.
In this study, we investigated the role of denaturation and hydrophobic interactions on insulin fibrillation in formamide and its N-methyl derivatives (formamide (FM), N-methyl formamide (NMF), and N,N-dimethyl formamide (DMF)) as cosolvent systems (1:1 by volume). We then compared the insulin structures and fibrillations observed in the solutions of formamide and its methyl derivatives. We used the thioflavin T (ThT) assay to monitor the rate of fibrillation of insulin. Fibrillar morphology was confirmed by transmission electron microscopy (TEM). We further investigated insulin-solvent interactions and protein structures under these conditions by using solution small-angle x-ray scattering (SAXS) combined with theoretical structures obtained by simulated annealing and differential scanning calorimetry (DSC).
Materials and Methods
Sample preparation
Human recombinant insulin (zinc free) and ThT were purchased from Sigma-Aldrich (St. Louis, MO). FM and NMF were purchased from Tokyo Chemical Industry (Tokyo, Japan), and DMF was purchased from Sigma-Aldrich. The stock solution of insulin was adjusted to 2 mg/mL, and 500 μL of the solution was transferred to a borosilicate glass vial. The final pH of the solution was adjusted to 7.0 using 0.5 M imidazole-HCl buffer. All insulin solutions were incubated at 60°C.
ThT binding assay
The ThT stock solution was freshly prepared at a concentration of 0.1 mg/mL by diluting 10 μL of the stock solution with 380 μL of buffer. A 10 μL aliquot of the incubated insulin solution was transferred to the diluted ThT solution. All fluorescence experiments were performed on a Quantmaster-3 spectrofluorometer (PTI, Birmingham, NJ) at an excitation wavelength of 452 nm and an emission wavelength of 475 nm. The fibrillation rate of amyloidogenic proteins is promoted by ThT in the in situ ThT assay (62,63). Thus, all insulin samples were incubated without ThT and fluorescence measurements were performed within 1 min. The intensities of all fluorescence measurements were normalized to the highest intensity in each plot except when the fibrillation process was not initiated during the incubation. The kinetics of fibril formation was analyzed using Origin 9.0 software and the t50 values were extracted from the sigmoidal curve (64). The t50 value was defined as the half-maximal fluorescence time point.
TEM
For TEM, 400-mesh formvar/carbon-coated copper grids were purchased from Electron Microscopy Science (Hatfield, PA). Aliquots of 5 μL of the incubated insulin solution were spotted on the grid for 0.5–5 min and then the grid was washed three times with 10 μL of water. Finally, 5 μL of 1 wt% uranyl acetate solution dissolved in water was spotted for 1 min to negatively stain the fibril species. The treated grids were analyzed using a JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan) at the Biotech Center of the Pohang University of Science and Technology (POSTECH).
Solution SAXS
All SAXS measurements were performed at the 4C SAXS II beamline of the Pohang Accelerator Laboratory, POSTECH. The concentration of insulin was adjusted to 2 mg/mL and the sample-to-detector distance was set to either 1 or 3 m. The magnitude of the scattering vectors (q = (4π/λ)sinθ) ranged from 0.01 to 0.76 Å−1. All scattering experiments were performed at 20°C. Each measurement was performed at least three times using fresh samples. The scattering patterns were recorded for 30 s. The procedures used to obtain the radius of gyration (Rg), zero-angle scattering intensity (I(0)), and p(r) function were as described in the literature (65).
3D modeling combined with theoretical structures of insulin
Simulated annealing was performed using Gromacs 4.5.5 (66) to generate a large pool of insulin conformation samples. The insulin structure (PDB code: 3E7Y) was annealed with a generalized Born implicit solvent model (67) with a stochastic dynamics integrator to perform an efficient search on a wide conformational space in the condensed phase. The initial structure was minimized in the implicit solvent using the steepest-descent method and subsequently subjected to 400 ps annealing cycles. In each annealing cycle, the temperature was maintained at 300 K for 100 ps for equilibration and then increased to 350, 400, 450, 500, 600, 700, 800, 900, and 1000 K for 100 ps. The high temperature was maintained for another 100 ps and the structure was then cooled to 300 K for 100 ps. The Amber99sb force field (68) was used and a total of 60,000 conformations were generated. The theoretical scattering profiles of the simulated structures were compared with the experimental scattering profiles of insulin for each solvent system using the CRYSOL program (69). The validity of the simulated structures was evaluated by comparing the χ2 values and Rg within the pool of insulin conformations. χ2-Rg diagrams from the pool of insulin conformation are presented in Fig. S1 in the Supporting Material. A relatively small χ2 value might indicate an increased probability of the simulated structure representing the actual conformation of the protein from SAXS analysis. The detailed equation for χ2 has been described previously (70). We also compared the p(r) function of the candidate structures with the experimental p(r) function to confirm the structural information of our theoretical insulin model in real space. We measured the solvent-accessible surface area (SASA) of the B11–B17 residues within the simulated structures by using the double cubic lattice method (71).
DSC
All DSC experiments were performed on a CSC 6100 Nano-Differential Scanning Calorimeter II (Calorimetry Sciences, Lindon, UT). The concentration of the insulin solution was adjusted to 2 mg/mL. The temperature range was set at 5–95°C with a scan rate of 1°C/min. A buffer-buffer reference scan was subtracted from each sample scan. The gains in enthalpy (ΔHu) and entropy (ΔSu) seen during thermal denaturation were calculated using Eqs. 1 and 2. The reference temperature (T0) of overall thermal denaturation was defined as the point at which the Gibbs free energy reached zero (Eq. 3) by enthalpy-entropy compensation. Thermal denaturation of the proteins in each solvent condition was thermodynamically favored above T0. Because this definition was derived from a general equation, it was applied to a non-two-state model (72,73).
| (1) |
| (2) |
| (3) |
Matrix-assisted laser desorption/ ionization mass spectrometry
Matrix-assisted laser desorption/ionization (MALDI) spectra of insulin within each solvent system were obtained using a Waters Synapt G2 HDMS MALDI spectrometer equipped with a 355 nm Nd:YAG laser (Waters, Manchester, UK) in the positive ion mode. The final concentrations of CHCA and insulin were reduced to 5 and 0.06 mg/mL, respectively, using the buffer solution. As shown in Fig. S2, all insulin samples obtained from the solvents investigated in this study exhibited a singly charged peak at m/z 5806.8, confirming that no chemical modifications were caused by the solvent.
Results and Discussion
Effect of formamide and its methyl derivatives on insulin fibrillation
The ThT binding assay was used to monitor fibrillation rates of insulin in various organic solvent systems composed of either formamide or its methyl derivatives. ThT emits fluorescence at ∼475 nm as it binds to β-sheets (74). As shown in Fig. 1 a, the ThT fluorescence in water (with 0.5 M imidazole-HCl buffer, pH 7.0) increased after a lag phase period of ∼13 h and the fluorescence reached a plateau at ∼26 h. The t50 value of the ThT fluorescence was measured as 18.7 ± 1.0 h. Insulin fibrillation became faster in binary mixtures of formamide and its methyl derivatives (1:1 water-organic solvent mixture with 0.5 M imidazole-HCl buffer, pH 7.0). The fibrillation process was further accelerated in binary mixtures because the number of hydrogen-bondable protons increases in organic solvent. The lag phase period decreased to ∼6 h, ∼5 h, and ∼3 h in 1:1 water cosolvents with DMF (DMF50), NMF (NMF50), and FM (FM50), respectively. Insulin then underwent fibrillation until it reached a plateau at 12, 10, and 8 h in DMF50, NMF50, and FM50, respectively. The t50 values of fibrillation kinetics were 7.9 ± 0.3, 7.0 ± 0.2, and 5.9 ± 0.1 h in DMF50, NMF50, and FM50, respectively. In contrast to the experiments in binary mixtures, the fibrillation rate of insulin was slower in organic solvents (0.5 M imidazole-HCl buffer, pH 7.0; Fig. 1 b). The ThT fluorescence intensity increased after a lag phase period of ∼35 h in FM (FM100) and reached a plateau at ∼90 h (t50 = 53.9 ± 7.7 h). No significant change in the ThT fluorescence was observed in NMF (NMF100) and DMF (DMF100) after incubation for 130 h.
Figure 1.
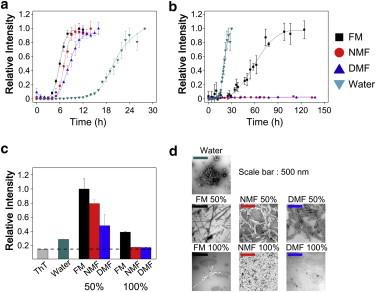
(a and b) Fibrillation kinetics of insulin in (a) binary mixtures and (b) organic solvents. The plot of water is inserted in each figure as the control experiment. All fluorescence plots in (a) and (b) are normalized to the highest fluorescence intensity in each plot, except for NMF100 and DMF100. (c) The ThT fluorescence of all incubated samples at the plateau was also measured to correct for the variable fluorescence of ThT in different solvents. All incubated solutions were diluted 80-fold with water. (d) TEM images of all incubated samples. To see this figure in color, go online.
The fluorescence intensity of ThT is sensitive to the solvent system as well as to the binding event with amyloid fibrils (75). To compare the intensity at the plateau without interference from solvent molecules, we also measured the fluorescence by diluting all incubated samples at the plateau 80-fold with water. We then compared the fluorescence intensity of the diluted sample with the reference, which was composed only of ThT dye in water. As shown in Fig. 1 c, all samples from the binary mixtures exhibited higher ThT intensities, suggesting the formation of more mature β-fibrils compared with those in the sample incubated in water. However, no significant increase in ThT fluorescence was observed with the samples incubated in organic solvents. Only the insulin sample from FM100 exhibited slightly increased ThT fluorescence. The relative ThT intensities at the plateau correlated with the fibrillation rates of insulin in different solvent systems. The morphology of the insulin fibrils was investigated using TEM. As shown in Fig. 1 d, the TEM image of the insulin fibrils supports the observed relative ThT fluorescence intensities. Insulin incubated in water formed short fibrils with amorphous aggregates. However, many elongated fibrils were formed in binary mixtures. By contrast, no significant protein fibril formation was observed for the insulin incubated in NMF100 and DMF100. Only sparse fibrils were observed for the insulin incubated in FM100. These results suggest that FM and its methyl derivatives promote the fibrillation of insulin in the presence of water. In addition, insulin fibrillation was enhanced as the number of hydrogen-bondable protons in the amide groups of organic solvent increased.
Structural analysis of insulin using Guinier and Kratky plots from SAXS profiles
A correlation between the initial structure of an amyloidogenic protein and its fibrillation rate has been proposed (76). The structural rigidity of an amyloidogenic protein produces constraints that prevent the intrinsic secondary structure of the core sequence from converting to the cross-β structure (7). The unfolded state of the amyloidogenic protein lowers the activation energy for the conformational rearrangement of the core sequence (76). Therefore, the formation of an intermediate in the unfolded state is expected to accelerate the fibrillation rate. To understand the relationship between the observed difference in fibrillation rates and the initial structures of the protein, we used solution SAXS to investigate the structures of insulin in organic solvents with and without water as a cosolvent. The scattering profiles of insulin are shown in Fig. 2 a and the Guinier plots (65) are shown in Fig. 2 b. Linearity in the small q range, which indicates a homogeneous size of insulin in solution, was observed in the Guinier plots of all solvent systems, with the exception of DMF100. The absence of linearity in DMF100 indicates that the protein is present as unknown aggregates (77). The Rg of insulin (Table 1) indicates that insulin is present as a tetramer in water (Rg = 21.2 Å), whereas the protein is present as a monomer in organic solvents, except DMF100 (Rg = 12.6 ∼13.9 Å) (41). The Rg value of insulin decreases as the number of hydrogen-bondable protons within the organic solvent decreases. The theoretical Rg calculated from the crystal structure of monomeric insulin (PDB code: 3E7Y) is ∼11.6 Å. Compared with the crystal structure, Rg values increased by +17%, +10%, and +8% in FM50, NMF50, and DMF50, respectively (Table S1). In FM100 and NMF100, Rg values increased by +19% and +10%, respectively.
Figure 2.
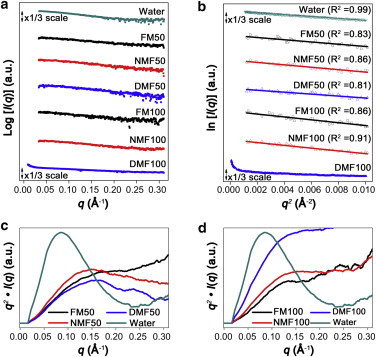
(a) Experimental SAXS scattering profiles of 2 mg/mL insulin. (b) Guinier plot from the small q range of the scattering profiles. (c and d) Kratky plots of insulin in (c) binary mixtures and (d) organic solvents. The y axes of the Kratky plots were normalized with the highest q2∙I(q) value of water.
Table 1.
Structural parameters of insulin from Guinier plots and pair distance distribution function p(r) in Figs. 3–5
| Rg,Guinier (Å) | Rg, p(r) (Å) | I(0) (a.u.) | Dmax (Å) | Association state | |
|---|---|---|---|---|---|
| Water | 21.2 ± 0.1 | 21.6 ± 0.1 | 37.4 ± 0.3 | 70 | tetramer |
| FM50 | 13.6 ± 0.5 | 13.4 ± 0.1 | 10.2 ± 0.1 | 45 | monomer |
| NMF50 | 12.8 ± 0.4 | 13.0 ± 0.2 | 11.6 ± 0.1 | 47 | monomer |
| DMF50 | 12.6 ± 0.4 | 12.8 ± 0.1 | 8.4 ± 0.1 | 43 | monomer |
| FM100 | 13.9 ± 1.5 | 13.6 ± 0.2 | 9.4 ± 0.2 | 44 | monomer |
| NMF100 | 12.8 ± 0.3 | 13.0 ± 0.1 | 11.4 ± 0.1 | 43 | monomer |
| DMF100 | not available | ||||
Kratky plots, which provide information about the folding state of polypeptide chains, are shown in Fig. 2, c and d (65). The Kratky plot of insulin in water exhibits a bell-shaped Gaussian curve, indicating a compact protein structure. By contrast, the Kratky plot of insulin within a binary mixture is consistent with the pattern of an unfolded protein (Fig. 2 c). As the number of hydrogen-bondable protons within the organic solvent increases, the compactness of insulin in solution increases. The Kratky plots of insulin in organic solvents indicate that insulin is present as loosely packed structures in FM100 and NMF100 (Fig. 2 d). Insulin in DMF100 exhibits features of an unfolded structure. However, the heterogeneity of DMF100 prevents a conclusive structural characterization. Both the Guinier and Kratky plots imply that organic solvents denature the native structure of insulin. Although the Guinier plots exhibit little difference in Rg values, the Kratky plots further support our contention that the degree of denaturation increases as the number of hydrogen-bondable protons within the organic solvent increases.
Structure analysis of insulin in water based on SAXS
To obtain detailed information about the conformations of insulin in the solvent systems, we calculated the p(r) function from the SAXS data (65). The p(r) function for each solvent system is shown in Figs. 3–5 with the corresponding experimental SAXS profile. The values of Rg and I(0) obtained from the p(r) functions are summarized in Table 1. The Rg values calculated from the p(r) functions agree well with those obtained from the Guinier approximations. The I(0) value demonstrates that insulin in water prefers to exist as a tetramer at a concentration of 2 mg/mL (Table 1), in agreement with previous reports that the native oligomeric state (i.e., the association state) of insulin is a tetramer at neutral pH in the absence of divalent zinc ions (37,47).
Figure 3.
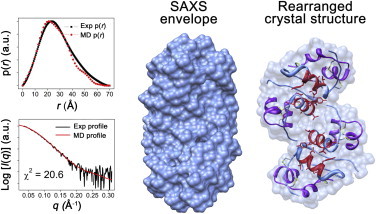
Reconstructed SAXS envelope in water and the rearranged crystal structure (PDB code: 3E7Y). Insulin chain A, chain B, and residues B11–B17 in the rearranged crystal structure are marked in purple, cornflower blue, and red, respectively. The scattering profiles and p(r) functions of the SAXS envelope and rearranged crystal structure are shown for comparison. To see this figure in color, go online.
Figure 4.
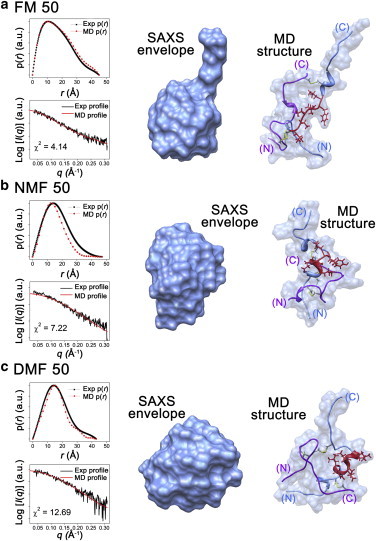
(a–c) Reconstructed SAXS envelopes and representative candidate structures from simulated annealing in (a) FM50, (b) NMF50, and (c) DMF50. Insulin chain A, chain B, and residues B11–B17 in the simulated structures are marked in purple, cornflower blue, and red, respectively. The scattering profiles and p(r) functions of the SAXS envelope and candidate structure are shown for comparison. The minor differences between the SAXS envelopes and representative structures were attributed to residues B24–B30. To see this figure in color, go online.
Figure 5.
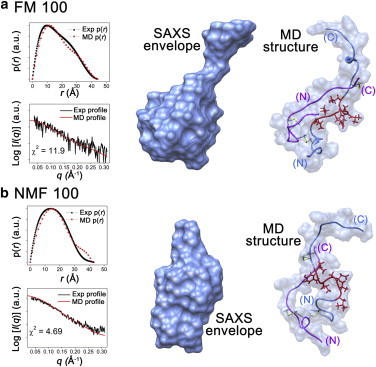
(a and b) Reconstructed SAXS envelopes and representative candidate structures from simulated annealing in (a) FM100 and (b) NMF100. Insulin chain A, chain B, and residues B11–B17 in the simulated structures are marked in purple, cornflower blue, and red, respectively. The profiles and p(r) functions of the SAXS envelope and candidate structure are shown for comparison. The minor differences between the SAXS envelopes and representative structures were attributed to residues B24–B30. To see this figure in color, go online.
Structural models (i.e., the SAXS envelope) of insulin were reconstructed from the p(r) function using the GASBOR program (78). Fig. 3 shows the SAXS envelope of insulin in water. To our knowledge, the crystal structure of the insulin tetramer has not been previously reported. Therefore, the conformation of a pair of insulin dimers was rearranged for comparison with the SAXS envelope. The theoretical scattering profile and p(r) function support a close match between the rearranged structure and the SAXS envelope (χ2 = 20.6). Disassembly of the associated insulin oligomer is one of the rate-determining steps in insulin fibrillation because fibrillation is initiated from the monomeric state of the protein (37). Therefore, the SAXS envelope of insulin suggests that the fibrillation rate in water is delayed by the disassembly of the tetramer into the monomer.
Structural conversion of B11–B17 core residues from α-helix to random coil in binary mixtures of water and organic solvents
Based on the I(0) values in Table 1, we conclude that insulin is present as a monomer in binary mixtures. The Rg values of monomeric insulin decrease as the number of hydrogen-bondable protons within the organic solvent decreases. The largest Rg value for insulin, 13.4 Å, was observed in FM50. In NMF50 and DMF50, the insulin structures become smaller, with Rg values of 13.0 and 12.8 Å, respectively. These results agree well with Rg values determined from the Guinier plots (Fig. 2 b). The difference in Rg values between the Guinier analysis and p(r) originates from the range of scattering profiles that are used in the analysis (65). However, trends of Rg values observed in both the Guinier plot and p(r) show good agreement.
Fig. 4 shows the SAXS envelopes of insulin within binary mixtures along with representative candidate structures obtained from sampling using simulated annealing (see Figs. S3–S5 for additional candidates). The candidate simulated structures in Fig. 4 were selected on the basis of good agreement between the theoretical and experimental scattering profiles (χ2 = 4.14, 7.22, and 12.69 for FM50, NMF50, and DMF50, respectively). The theoretical Rg values obtained from the simulated structures are also in good agreement with the experimental results. Minor differences between the SAXS envelopes and representative structures were observed, as were differences between the experimental and theoretical p(r) functions. The simulated structures demonstrate that the residues B24–B30 of the C-terminus do not match the SAXS envelopes. Because the structure of B24–B30 is flexible (79), the structure of this region is likely not detectable in scattering patterns (80).
We next investigated the secondary structure of the hydrophobic core region of the B-chain (residues B11–B17) from the simulated structures to characterize the role of FM and its methyl derivatives in insulin fibrillation. A gradual change from α-helical to random coil structure in residues B11–B17 was observed in the simulated structures as the number of hydrogen-bondable protons within the organic solvent increased (Figs. 4 and S3–S5). No trace of helical property was observed in insulin in FM50. Only three amino acid residues maintained α-helical structure in NMF50. By contrast, the entire B11–B17 region retained α-helical structure in DMF50. We also investigated the SASA to quantitatively analyze the degree of exposure of the B11–B17 region to solvents (Table S2). The SASA of residues B11–B17 was 6.8 nm2 in FM50 and decreased to 5.3 and 5.1 nm2 in NMF50 and DMF50, respectively. The lowest value of SASA for residues B11–B17, 4.9 nm2, was observed in the native state (PDB code: 3E7Y). These results demonstrate that residues B11–B17 in the simulated structures are gradually exposed to solvent during the structural transition from α-helix to random coil.
The SAXS results, combined with the theoretical structures obtained by simulated annealing, imply that organic solvents disassemble insulin from tetramer to monomer and denature the native structure in the initial state. The formation of multimeric insulin is sterically hindered by hydrogen-bond interactions between the protein and water molecules on multimer-forming surfaces (81). In the presence of organic solvents, the interaction between the protein and organic solvent is expected to be stronger than the interaction between the protein and water because amide-amide hydrogen bonds are more stable than hydroxyl-amide hydrogen bonds (82). Therefore, organic solvents may enhance the disassembly of tetrameric insulin into monomers, as compared with water. In addition, the interaction between the protein and organic solvents solvates peptide backbone, thereby denaturing the native structure of the protein (58). As the protons of the amide group are substituted for methyl groups, the hydrogen-bond interactions between the protein and organic solvent will most likely be reduced. Alternatively, the methyl groups could interact with the hydrophobic functional groups of the protein. Because multimer-forming surfaces include a large hydrophobic region (81), it is expected that methyl groups also enhance the disassembly of tetrameric insulin by solvation on multimer-forming surfaces. In contrast to the hydrogen-bond interaction between the protein and organic solvents, the hydrophobic environment of methyl groups increases α-helix propensity by enhancing hydrogen bonding of the protein and exposing hydrophobic side chains to the outside (83,84). Consequently, the α-helix propensity of residues B11–B17 of the protein is expected to increase as the number of substituted methyl groups in organic solvent increases.
In water, residues B11–B17 are buried within the protein as an α-helix (Fig. 3). These residues retain helical structure in the partially folded intermediate (37) but eventually are converted to a cross-β structure during the fibrillation process (43). The activation energy for the structural conversion of residues B11–B17 from α-helix to cross-β is expected to lengthen the lag phase of insulin fibrillation in water (9). However, in binary mixtures, the overall structure of insulin is denatured, and residues B11–B17 change to random coil with increasing SAS values (Table S2). Therefore, the activation energy for rearrangement of residues B11–B17 is decreased compared with the protein in water. Within binary mixtures, the fibrillation rate of the protein is correlated with the degree of denaturation of residues B11–B17. The Rg values and Kratky plots also indicate that the degree of denaturation is proportional to the fibrillation rate. The randomness and SAS values of residues B11–B17 in the simulated structures also increase as the number of hydrogen-bondable protons within the organic solvent increases (Table S2). Therefore, a longer lag period is required to destabilize the helical structure of residues B11–B17 as the number of substituted methyl groups within the organic solvent increases.
Denatured structure of B11–B17 core residues in organic solvents
Insulin is also present as a monomer in FM100 and NMF100, as indicated by the values of I(0) (Table 1). The Rg values were 13.6 and 13.0 Å in FM100 and NMF100, respectively. Fig. 5 shows the SAXS envelopes of insulin in FM100 and NMF100 along with the simulated structures (see Figs. S6 and S7 for additional candidates), which were selected on the basis of Rg and χ2 values (11.9 for FM100 and 3.7 for NMF100). Residues B11–B17 of the simulated structures show only random coil (Fig. 5). The SAS values of the B11–B17 region from the simulated structures within FM100 and NMF100 are 7.1 and 8.8 nm2, respectively (Table S2). This result suggests that denaturation of residues B11–B17 is greater in organic solvents than in binary mixtures.
Insulin is present in a denatured state in FM100 and NMF100, but the fibrillation rates in these solvent mixtures are slower than in water and binary mixtures (Fig. 1). The simulated structure also suggests that the secondary structure of residues B11–B17 in FM100 and NMF100 is random coil, and the SAS values are larger than those of the binary mixtures. These results imply that the presence of water plays a crucial role in insulin fibrillation.
Thermal denaturation of insulin monitored by DSC
DSC was utilized to elucidate the role of protein-solvent interactions during insulin fibrillation based on the thermal stability of the protein in each solvent system (Fig. S8) (85,86). The reference temperature (T0), transition enthalpy (ΔHu), and entropy (ΔSu) upon protein unfolding in water were 67.1°C, 103.3 kJ/mol, and 304.1 J/K⋅mol, respectively (Table 2). ΔHu of insulin was larger in water than in binary mixtures or organic solvents, and this result was attributed to the native folding structure of the protein and its associated tetrameric state in water (Fig. 3). Insulin also exhibited a high T0 in water, indicating that the intramolecular hydrophobic interactions of the protein are stabilized to avoid the formation of a water-hydrophobic region interface. These values imply that a large amount of energy at high T0 is required to form a partially folded insulin intermediate, which then forms the dimeric building blocks of the nucleus of mature fibrils (44). As a result, insulin exhibits slower fibrillation and low β-sheet abundance in water compared with the protein in binary mixtures due to its high stability in water.
Table 2.
Thermal transition midpoint (T0), enthalpy (ΔHu), and entropy (ΔSu) during insulin unfolding based on the DSC thermograms shown in Fig. S8
| T0 (°C) | ΔHu (kJ/mol) | ΔSu (J/K⋅mol) | |
|---|---|---|---|
| Water | 67.1 ± 0.3 | 103.3 ± 0.2 | 304.1 ± 0.3 |
| FM50 | 31.9 ± 0.4 | 34.8 ± 0.1 | 113.9 ± 0.2 |
| NMF50 | 34.4 ± 0.1 | 52.8 ± 0.2 | 171.5 ± 0.1 |
| DMF50 | 29.5 ± 0.2 | 95.7 ± 0.3 | 315.7 ± 0.5 |
| FM100 | 48.0 ± 0.3 | 5.6 ± 0.1 | 17.3 ± 0.1 |
| NMF100 | 52.9 ± 0.7 | 17.2 ± 0.1 | 52.5 ± 0.1 |
| DMF100 | 68.2 ± 1.7 | 66.3 ± 0.4 | 193.5 ± 1.1 |
For insulin in binary mixtures, both the enthalpy and entropy changes during the thermal transition increase in the following order: DMF50 > NMF50 > FM50. The initial structure of insulin within each solvent system contributes to the trend of ΔHu because less thermal energy is required for the transition when the initial structure is more denatured. As discussed above, the degree of denaturation of insulin increases as the number of hydrogen-bondable protons in the organic solvent molecule increases (Fig. 2). In addition to the initial structure, the degree of interaction between the protein and organic solvent contributes to the degree of thermal transition of the protein from its initial state. Protein denaturation requires high energetic melting of cooperative intramolecular interactions in the protein (87), a process that is considered partially compensated for by formation of hydrogen-bonding interactions with organic solvent. Thus, the relatively lower increase in ΔHu in FM compared with other methyl derivatives can be explained by its maximum number of hydrogen-bondable protons. Consequently, the increase in ΔHu was correlated with the degree of N-methylation of the organic solvents. During protein denaturation, ΔHu is nearly compensated for by a corresponding change in ΔSu due to an increase in the degrees of freedom of the denatured protein (88). In addition, the entropic gain is attributed to rearrangement of the organic solvent molecules via hydrophobic interactions between the protein and the methyl groups of the organic solvent molecules.
The increase in the entropic gain is correlated with the degree of N-methylation of the organic solvent molecules. Residues B11–B17 are believed to form the intermolecular interface via hydrophobic interactions (37,43–45), and thus the increased hydrophobicity of the solvent system would suppress the association of residues B11–B17. Consequently, the β-sheet abundance would increase as the number of substituted methyl groups in the organic solvent decreases (Fig. 1 c).
Binary mixtures exhibit low T0 values (∼30°C) despite the presence of water, indicating that hydrophobic residues of insulin can be readily exposed to the outside. In other words, the observed structures in Fig. 4 are closer to an intermediate state than to the native state and can eventually form the nuclei of mature fibrils. When the structure of insulin is fully solvated after thermal denaturation, the water molecules in the binary mixtures will remain unstable on a hydrophobic interface exposed to the solvent. The enhancement of insulin fibrillation in binary mixtures therefore minimizes the water-hydrophobic region interface (Fig. 1 a).
The order of ΔHu and ΔSu in organic solvents is in agreement with the trend observed in binary mixtures, but ΔHu and ΔSu are much smaller in organic solvents than in binary mixtures (Table 2). The smaller changes in ΔHu and ΔSu indicate that the protein-solvent interaction does not significantly change during thermal denaturation. Therefore, the results indicate that organic solvents denature the entire structure of insulin more effectively than binary mixtures, thereby rendering the structure unsusceptible to the thermal heating process. The thermodynamic values of insulin in DMF100 (Table 2) may be attributed to the dissociation of unknown aggregates. Organic solvents exhibit higher T0 values than binary mixtures, indicating that ΔSu is decreased more than ΔHu in organic solvents compared with binary mixtures. This result implies that solvent molecules stabilize the hydrophobic region of insulin in an unfolded structure in the initial state. Thus, insulin fibrillation in organic solvents is highly suppressed due to the absence of water, which is the driving force for hydrophobic interactions (Fig. 1 b).
T0 values increased in organic solvents by the number of substituted methyl groups, but were unchanged in binary mixtures. This result may correlate with the presence of water in binary mixtures. Although methyl derivatives of formamide are fully miscible with water, it is expected that methyl groups within organic solvents do not prefer contact with water molecules (89). Therefore, when hydrophobic regions of the protein are exposed to the outside, methyl groups within organic solvents tend to interact with hydrophobic regions rather than with water. As a result, entropy gain is enhanced and the T0 values of NMF and DMF appear to shift in binary mixtures.
Importance of denaturation and hydrophobic interactions during insulin fibrillation
Our structural analysis via SAXS and simulated annealing indicated that FM and its methyl derivatives dissociate an insulin tetramer into monomers and concomitantly denature the protein into pseudo-intermediate monomers for protein fibrillation in the initial state (Fig. 6 a). Although all organic solvent systems investigated in this study yielded denatured structures of insulin in which residues B11–B17 were exposed (with the exception of DMF100), large variations in the fibrillation kinetics, fibril abundances, and morphologies were observed. The enhanced fibrillation of insulin in binary mixtures indicates that the cooperation between protein denaturation and hydrophobic interaction promotes insulin fibrillation (Fig. 6 b). In water, insulin prefers intramolecular hydrophobic interactions to avoid contact with water molecules. Consequently, a slow fibrillation rate is observed in water due to the high activation energy for the exposure of residues B11–B17. In binary mixtures, however, organic solvents denature the protein, and residues B11–B17 are exposed to solvent molecules (Table S2). As a result, the interface between water and the exposed B11–B17 region induces an intermolecular interaction among residues B11–B17 by a hydrophobic interaction. Organic solvents also induce denaturation of the protein and exposure of residues B11–B17 to the outside, but the driving force for the intermolecular hydrophobic interaction of the residues is diminished in the absence of water. Therefore, in organic solvents, insulin is stabilized as the denatured structure, which is nonamyloidogenic.
Figure 6.
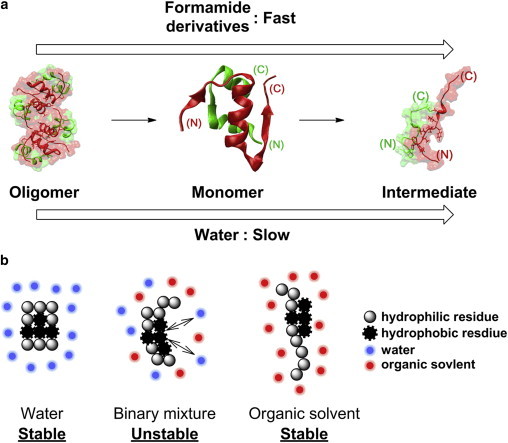
(a and b) Schematic overview of (a) the formation of the pseudo-intermediate structure for fibrillation and (b) the protein-solvent interactions in each solvent system. To see this figure in color, go online.
Conclusions
We have investigated the correlation between insulin fibrillation and the environmental factors that denature the native structure of the protein. The fibrillation process is accelerated in binary mixtures compared with water, whereas organic solvents delay fibrillation. The model system presented here may represent a heterogeneous biological system in which the water content is limited and the stability of the protein is reduced (e.g., the water-lipid interface in the cellular environment). In addition, this homogeneous system can provide insights into both the protein structure and the protein-medium interaction. Numerous studies have noted the importance of the denaturation of an amyloidogenic protein in fibrillation (76). This study also confirms that conformational change promotes fibrillation rates of insulin. However, the results obtained in organic solvents emphasize that the presence of water is essential for insulin fibrillation. Therefore, the ability to control these two important factors in abnormal protein-protein interactions may be a prerequisite for developing a therapeutic strategy for amyloidosis.
Acknowledgments
This work was supported by Basic Science Research (grant No. 2013R1A1A2008974) through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning, and by the Korea Health Technology R&D Project (grant No. HT13C-0011-040013) through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare. The synchrotron X-ray scattering measurements at the Pohang Accelerator Laboratory were supported by the Ministry of Education and Science Technology. Tae Su Choi acknowledge TJ Park Science Fellowship of POSCO TJ Park Foundation.
Supporting Material
Supporting Citations
Reference (90) appears in the Supporting Material.
References
- 1.Goedert M., Spillantini M.G. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.McLean P.J., Kawamata H., Hyman B.T. α-Synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience. 2001;104:901–912. doi: 10.1016/s0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]
- 3.Höppener J.W.M., Jacobs H.M., Ahrén B. Human islet amyloid polypeptide transgenic mice: in vivo and ex vivo models for the role of hIAPP in type 2 diabetes mellitus. Exp. Diabetes Res. 2008;2008:697035. doi: 10.1155/2008/697035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe D.J. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 7.Cohen F.E., Kelly J.W. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 8.Levy Y., Onuchic J.N. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 9.Chiti F., Dobson C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 10.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 11.England J.L., Haran G. Role of solvation effects in protein denaturation: from thermodynamics to single molecules and back. Annu. Rev. Phys. Chem. 2011;62:257–277. doi: 10.1146/annurev-physchem-032210-103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 13.Thirumalai D., Reddy G., Straub J.E. Role of water in protein aggregation and amyloid polymorphism. Acc. Chem. Res. 2012;45:83–92. doi: 10.1021/ar2000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldberg A.L. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 15.Kim H.I., Kim H., Beauchamp J.L. Interfacial reactions of ozone with surfactant protein B in a model lung surfactant system. J. Am. Chem. Soc. 2010;132:2254–2263. doi: 10.1021/ja908477w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H.I., Kim H., Beauchamp J.L. Time resolved studies of interfacial reactions of ozone with pulmonary phospholipid surfactants using field induced droplet ionization mass spectrometry. J. Phys. Chem. B. 2010;114:9496–9503. doi: 10.1021/jp102332g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko J.Y., Choi S.M., Kim H.I. Studying interfacial reactions of cholesterol sulfate in an unsaturated phosphatidylglycerol layer with ozone using field induced droplet ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2012;23:141–152. doi: 10.1007/s13361-011-0275-9. [DOI] [PubMed] [Google Scholar]
- 18.Lee S.J.C., Lee J.W., Kim H.I. Probing conformational change of intrinsically disordered α-synuclein to helical structures by distinctive regional interactions with lipid membranes. Anal. Chem. 2014;86:1909–1916. doi: 10.1021/ac404132g. [DOI] [PubMed] [Google Scholar]
- 19.Hebda J.A., Miranker A.D. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: insights from type II diabetes. Annu. Rev. Biophys. 2009;38:125–152. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 20.Aisenbrey C., Borowik T., Gröbner G. How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J. 2008;37:247–255. doi: 10.1007/s00249-007-0237-0. [DOI] [PubMed] [Google Scholar]
- 21.Gorbenko G.P., Kinnunen P.K.J. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids. 2006;141:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Zhu M., Fink A.L. Lipid binding inhibits α-synuclein fibril formation. J. Biol. Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 23.Hellstrand E., Sparr E., Linse S. Retardation of Aβ fibril formation by phospholipid vesicles depends on membrane phase behavior. Biophys. J. 2010;98:2206–2214. doi: 10.1016/j.bpj.2010.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha A., Narayan S., Krishnamoorthy G. Solvent-induced tuning of internal structure in a protein amyloid protofibril. Biophys. J. 2012;103:797–806. doi: 10.1016/j.bpj.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fezoui Y., Teplow D.B. Kinetic studies of amyloid β-protein fibril assembly. Differential effects of α-helix stabilization. J. Biol. Chem. 2002;277:36948–36954. doi: 10.1074/jbc.M204168200. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi K., Naiki H., Goto Y. Mechanism by which the amyloid-like fibrils of a β2-microglobulin fragment are induced by fluorine-substituted alcohols. J. Mol. Biol. 2006;363:279–288. doi: 10.1016/j.jmb.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 27.Chatani E., Yagi H., Goto Y. Polymorphism of β2-microglobulin amyloid fibrils manifested by ultrasonication-enhanced fibril formation in trifluoroethanol. J. Biol. Chem. 2012;287:22827–22837. doi: 10.1074/jbc.M111.333310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munishkina L.A., Phelan C., Fink A.L. Conformational behavior and aggregation of α-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 2003;42:2720–2730. doi: 10.1021/bi027166s. [DOI] [PubMed] [Google Scholar]
- 29.Erlkamp M., Grobelny S., Winter R. Solvent effects on the dynamics of amyloidogenic insulin revealed by neutron spin echo spectroscopy. J. Phys. Chem. B. 2014;118:3310–3316. doi: 10.1021/jp500530h. [DOI] [PubMed] [Google Scholar]
- 30.Grudzielanek S., Jansen R., Winter R. Solvational tuning of the unfolding, aggregation and amyloidogenesis of insulin. J. Mol. Biol. 2005;351:879–894. doi: 10.1016/j.jmb.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 31.Munishkina L.A., Ahmad A., Uversky V.N. Guiding protein aggregation with macromolecular crowding. Biochemistry. 2008;47:8993–9006. doi: 10.1021/bi8008399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H.X., Rivas G., Minton A.P. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bokvist M., Gröbner G. Misfolding of amyloidogenic proteins at membrane surfaces: the impact of macromolecular crowding. J. Am. Chem. Soc. 2007;129:14848–14849. doi: 10.1021/ja076059o. [DOI] [PubMed] [Google Scholar]
- 34.Shtilerman M.D., Ding T.T., Lansbury P.T., Jr. Molecular crowding accelerates fibrillization of α-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- 35.Straub J.E., Thirumalai D. Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Annu. Rev. Phys. Chem. 2011;62:437–463. doi: 10.1146/annurev-physchem-032210-103526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brange J., Andersen L., Rasmussen E. Toward understanding insulin fibrillation. J. Pharm. Sci. 1997;86:517–525. doi: 10.1021/js960297s. [DOI] [PubMed] [Google Scholar]
- 37.Hua Q.X., Weiss M.A. Mechanism of insulin fibrillation: the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 2004;279:21449–21460. doi: 10.1074/jbc.M314141200. [DOI] [PubMed] [Google Scholar]
- 38.Whittingham J.L., Scott D.J., Guy Dodson G. Insulin at pH 2: structural analysis of the conditions promoting insulin fibre formation. J. Mol. Biol. 2002;318:479–490. doi: 10.1016/S0022-2836(02)00021-9. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen L., Khurana R., Fink A.L. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry. 2001;40:6036–6046. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 40.Pocker Y., Biswas S.B. Self-association of insulin and the role of hydrophobic bonding: a thermodynamic model of insulin dimerization. Biochemistry. 1981;20:4354–4361. doi: 10.1021/bi00518a019. [DOI] [PubMed] [Google Scholar]
- 41.Uversky V.N., Garriques L.N., Fink A.L. Prediction of the association state of insulin using spectral parameters. J. Pharm. Sci. 2003;92:847–858. doi: 10.1002/jps.10355. [DOI] [PubMed] [Google Scholar]
- 42.Bryant C., Spencer D.B., Brems D.N. Acid stabilization of insulin. Biochemistry. 1993;32:8075–8082. doi: 10.1021/bi00083a004. [DOI] [PubMed] [Google Scholar]
- 43.Ivanova M.I., Sievers S.A., Eisenberg D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA. 2009;106:18990–18995. doi: 10.1073/pnas.0910080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nayak A., Sorci M., Belfort G. A universal pathway for amyloid nucleus and precursor formation for insulin. Proteins. 2009;74:556–565. doi: 10.1002/prot.22169. [DOI] [PubMed] [Google Scholar]
- 45.Nielsen L., Frokjaer S., Fink A.L. Probing the mechanism of insulin fibril formation with insulin mutants. Biochemistry. 2001;40:8397–8409. doi: 10.1021/bi0105983. [DOI] [PubMed] [Google Scholar]
- 46.Lee H.H., Choi T.S., Kim H.I. Supramolecular inhibition of amyloid fibrillation by cucurbit[7]uril. Angew. Chem. Int. Ed. Engl. 2014;53:7461–7465. doi: 10.1002/anie.201402496. [DOI] [PubMed] [Google Scholar]
- 47.Dische F.E., Wernstedt C., Watkins P.J. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158–161. doi: 10.1007/BF00276849. [DOI] [PubMed] [Google Scholar]
- 48.Sharp J.S., Forrest J.A., Jones R.A.L. Surface denaturation and amyloid fibril formation of insulin at model lipid-water interfaces. Biochemistry. 2002;41:15810–15819. doi: 10.1021/bi020525z. [DOI] [PubMed] [Google Scholar]
- 49.Zhao H., Tuominen E.K.J., Kinnunen P.K.J. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 2004;43:10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- 50.Grudzielanek S., Smirnovas V., Winter R. The effects of various membrane physical-chemical properties on the aggregation kinetics of insulin. Chem. Phys. Lipids. 2007;149:28–39. doi: 10.1016/j.chemphyslip.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Fernández A., Berry R.S. Proteins with H-bond packing defects are highly interactive with lipid bilayers: Implications for amyloidogenesis. Proc. Natl. Acad. Sci. USA. 2003;100:2391–2396. doi: 10.1073/pnas.0335642100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reference deleted in proof.
- 53.Anderson V.L., Ramlall T.F., Eliezer D. Identification of a helical intermediate in trifluoroethanol-induced α-synuclein aggregation. Proc. Natl. Acad. Sci. USA. 2010;107:18850–18855. doi: 10.1073/pnas.1012336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toba S., Hartsough D.S., Merz K.M. Solvation and dynamics of chymotrypsin in hexane. J. Am. Chem. Soc. 1996;118:6490–6498. [Google Scholar]
- 55.Oakenfull D., Fenwick D.E. Hydrophobic interaction in aqueous organic mixed solvents. J. Chem. Soc., Faraday Trans. 1. 1979;75:636–645. [Google Scholar]
- 56.Hirota N., Mizuno K., Goto Y. Group additive contributions to the alcohol-induced α-helix formation of melittin: implication for the mechanism of the alcohol effects on proteins. J. Mol. Biol. 1998;275:365–378. doi: 10.1006/jmbi.1997.1468. [DOI] [PubMed] [Google Scholar]
- 57.Blanco S., López J.C., Alonso J.L. Microsolvation of formamide: a rotational study. J. Am. Chem. Soc. 2006;128:12111–12121. doi: 10.1021/ja0618393. [DOI] [PubMed] [Google Scholar]
- 58.Herskovits T.T., Behrens C.F., Pandolfelli E.R. Solvent denaturation of globular proteins: unfolding by the monoalkyl- and dialkyl-substituted formamides and ureas. Biochim. Biophys. Acta. 1977;490:192–199. doi: 10.1016/0005-2795(77)90119-2. [DOI] [PubMed] [Google Scholar]
- 59.Klyosov A.A., Van Viet N., Berezin I.V. The reactions of α-chymotrypsin and related proteins with ester substrates in non-aqueous solvents. Eur. J. Biochem. 1975;59:3–7. doi: 10.1111/j.1432-1033.1975.tb02417.x. [DOI] [PubMed] [Google Scholar]
- 60.Knubovets T., Osterhout J.J., Klibanov A.M. Structure of lysozyme dissolved in neat organic solvents as assessed by NMR and CD spectroscopies. Biotechnol. Bioeng. 1999;63:242–248. [PubMed] [Google Scholar]
- 61.Bloemendal M., Somsen G. Properties of some protein denaturants in N,N-dimethylformamide. Enthalpic interaction coefficients of urea and substituted urea compounds. J. Am. Chem. Soc. 1985;107:3426–3431. [Google Scholar]
- 62.D’Amico M., Di Carlo M.G., Leone M. Thioflavin t promotes Aβ (1–40) amyloid fibrils formation. J. Phys. Chem. Lett. 2012;3:1596–1601. doi: 10.1021/jz300412v. [DOI] [PubMed] [Google Scholar]
- 63.Alí-Torres J., Rimola A., Sodupe M. Insights on the binding of Thioflavin derivative markers to amyloid-like fibril models from quantum chemical calculations. J. Phys. Chem. B. 2013;117:6674–6680. doi: 10.1021/jp402807g. [DOI] [PubMed] [Google Scholar]
- 64.Ruschak A.M., Miranker A.D. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. USA. 2007;104:12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Putnam C.D., Hammel M., Tainer J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 66.Hess B., Kutzner C., Lindahl E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 67.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 68.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Svergun D., Barberato C., Koch M.H.J. Crysol–a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 70.Bernadó P., Mylonas E., Svergun D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 71.Eisenhaber F., Lijnzaad P., Scharf M. The double cubic lattice method—efficient approaches to numerical-integration of surface-area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 1995;16:273–284. [Google Scholar]
- 72.Boudker O., Todd M.J., Freire E. The structural stability of the co-chaperonin GroES. J. Mol. Biol. 1997;272:770–779. doi: 10.1006/jmbi.1997.1263. [DOI] [PubMed] [Google Scholar]
- 73.Freire E., Murphy K.P., Privalov P.L. The molecular basis of cooperativity in protein folding. Thermodynamic dissection of interdomain interactions in phosphoglycerate kinase. Biochemistry. 1992;31:250–256. doi: 10.1021/bi00116a034. [DOI] [PubMed] [Google Scholar]
- 74.Groenning M., Norrman M., Frokjaer S. Binding mode of thioflavin T in insulin amyloid fibrils. J. Struct. Biol. 2007;159:483–497. doi: 10.1016/j.jsb.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 75.Maskevich A.A., Stsiapura V.I., Turoverov K.K. Spectral properties of thioflavin T in solvents with different dielectric properties and in a fibril-incorporated form. J. Proteome Res. 2007;6:1392–1401. doi: 10.1021/pr0605567. [DOI] [PubMed] [Google Scholar]
- 76.Uversky V.N., Fink A.L. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 77.Mertens H.D.T., Svergun D.I. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 78.Svergun D.I., Petoukhov M.V., Koch M.H.J. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hua Q.X., Gozani S.N., Weiss M.A. Structure of a protein in a kinetic trap. Nat. Struct. Biol. 1995;2:129–138. doi: 10.1038/nsb0295-129. [DOI] [PubMed] [Google Scholar]
- 80.Ciszak E., Beals J.M., Smith G.D. Role of C-terminal B-chain residues in insulin assembly: the structure of hexameric LysB28ProB29-human insulin. Structure. 1995;3:615–622. doi: 10.1016/s0969-2126(01)00195-2. [DOI] [PubMed] [Google Scholar]
- 81.Bagchi K., Roy S. Sensitivity of water dynamics to biologically significant surfaces of monomeric insulin: role of topology and electrostatic interactions. J. Phys. Chem. B. 2014;118:3805–3813. doi: 10.1021/jp411136w. [DOI] [PubMed] [Google Scholar]
- 82.Habermann S.M., Murphy K.P. Energetics of hydrogen bonding in proteins: a model compound study. Protein Sci. 1996;5:1229–1239. doi: 10.1002/pro.5560050702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cammers-Goodwin A., Allen T.J., Oslick S.L., McClure K.F., Lee J.H., Kemp D.S. Mechanism of stabilization of helical conformations of polypeptides by water containing trifluoroethanol. J. Am. Chem. Soc. 1996;118:3082–3090. [Google Scholar]
- 84.Duhamel J., Kanagalingam S., Ingratta M.W. Side-chain dynamics of an α-helical polypeptide monitored by fluorescence. J. Am. Chem. Soc. 2003;125:12810–12822. doi: 10.1021/ja035947q. [DOI] [PubMed] [Google Scholar]
- 85.Liu L., Yang C., Guo Q.X. A study on the enthalpy-entropy compensation in protein unfolding. Biophys. Chem. 2000;84:239–251. doi: 10.1016/s0301-4622(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 86.Batchelor J.D., Olteanu A., Pielak G.J. Impact of protein denaturants and stabilizers on water structure. J. Am. Chem. Soc. 2004;126:1958–1961. doi: 10.1021/ja039335h. [DOI] [PubMed] [Google Scholar]
- 87.Back J.F., Oakenfull D., Smith M.B. Increased thermal stability of proteins in the presence of sugars and polyols. Biochemistry. 1979;18:5191–5196. doi: 10.1021/bi00590a025. [DOI] [PubMed] [Google Scholar]
- 88.Jackson W.M., Brandts J.F. Thermodynamics of protein denaturation. A calorimetric study of the reversible denaturation of chymotrypsinogen and conclusions regarding the accuracy of the two-state approximation. Biochemistry. 1970;9:2294–2301. doi: 10.1021/bi00813a011. [DOI] [PubMed] [Google Scholar]
- 89.Lei Y., Li H.R., Han S.J. Structures and hydrogen bonding analysis of N,N-dimethylformamide and N,N-dimethylformamide-water mixtures by molecular dynamics simulations. J. Phys. Chem. A. 2003;107:1574–1583. [Google Scholar]
- 90.Bekard I.B., Dunstan D.E. Tyrosine autofluorescence as a measure of bovine insulin fibrillation. Biophys. J. 2009;97:2521–2531. doi: 10.1016/j.bpj.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.