

Abstract
Lipid-linked oligosaccharides (LLOs) are the substrates of oligosaccharyltransferase (OST), the enzyme that catalyzes the en bloc transfer of the oligosaccharide onto the acceptor asparagine of nascent proteins during the process of N-glycosylation. To explore LLOs’ preferred location, orientation, structure, and dynamics in membrane bilayers of three different lipid types (dilauroylphosphatidylcholine, dimyristoylphosphatidylcholine, and dioleoylphosphatidylcholine), we have modeled and simulated both eukaryotic (Glc3-Man9-GlcNAc2-PP-Dolichol) and bacterial (Glc1-GalNAc5-Bac1-PP-Undecaprenol) LLOs, which are composed of an isoprenoid moiety and an oligosaccharide, linked by pyrophosphate. The simulations show no strong impact of different bilayer hydrophobic thicknesses on the overall orientation, structure, and dynamics of the isoprenoid moiety and the oligosaccharide. The pyrophosphate group stays in the bilayer head group region. The isoprenoid moiety shows high flexibility inside the bilayer hydrophobic core, suggesting its potential role as a tentacle to search for OST. The oligosaccharide conformation and dynamics are similar to those in solution, but there are preferred interactions between the oligosaccharide and the bilayer interface, which leads to LLO sugar orientations parallel to the bilayer surface. Molecular docking of the bacterial LLO to a bacterial OST suggests that such orientations can enhance binding of LLOs to OST.
Introduction
Lipid-linked oligosaccharides (LLOs) are the substrates of oligosaccharyltransferase (OST), which catalyzes the en bloc transfer of the oligosaccharide onto the acceptor asparagine of nascent proteins (1). This posttranslational modification is called N-glycosylation, which is important in many aspects of biology, such as protein folding and quality control in the endoplasmic reticulum, protein trafficking, and protein stability and function (2). Defects in N-glycosylation lead to congenital disorders of glycosylation, which are often fatal (3).
An LLO molecule is composed of an isoprenoid moiety and an oligosaccharide, which are linked by pyrophosphate (Fig. 1) (1). As the lipid component of LLOs, the isoprenoid moiety is dolichol in eukaryotic LLOs and undecaprenol in bacterial LLOs, and the number of isoprene units varies for different species (4). The oligosaccharide is mostly conserved in the eukaryotic LLO as Glc3-Man9-GlcNAc2 but becomes more diverse from bacteria to archaea (5). The lipid and oligosaccharide components of LLOs have been studied separately (6–16). However, to the best of our knowledge, there are no biochemical, biophysical, and computational studies of fully assembled LLOs in membrane bilayers, and thus the effects of membrane bilayers on the structure and dynamics of LLOs and their orientations with respect to the membrane remain elusive.
Figure 1.
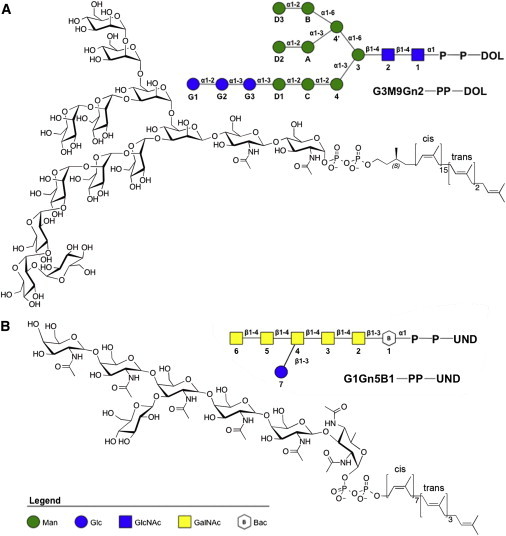
Chemical and schematic representations of the (A) eukaryotic and (B) bacterial LLOs used in this study. In the legend, Man: d-mannose; Glc: d-glucose; GlcNAc: N-acetyl-d-glucosamine; GalNAc: N-acetyl-d-galactosamine; Bac: d-bacillosamine. The oligosaccharide is covalently attached to a lipid chain by pyrophosphate. In (A), the oligosaccharide is composed of two GlcNAc residues joined to nine branching Man residues capped with three glucose residues. The lipid chain is composed of a saturated isoprenyl unit with an S chiral center, joined in series to 15 isoprenyl units with double bonds in cis configuration, followed by two isoprenyl units with trans double bonds, and a terminal isoprenyl unit. In (B), the oligosaccharide is composed of a Bac residue linked to five GalNAc residues, and a Glc residue attached to the third GalNAc. In the lipid chain, all isoprenyl units are unsaturated, the first seven units have cis double bonds and next three have trans with a terminal isoprenyl unit. Note that the anomeric center of the first sugar residue attached to pyrophosphate in both (A) eukaryotic and (B) bacterial LLOs is in the α configuration. To see this figure in color, go online.
Recently, the structure of PglB (PDB:3RCE, Fig. 2), a bacterial OST, was determined by x-ray crystallography (17). The structure reveals the LLO binding site positioned in a deep concave pocket, such that the oligosaccharide needs to be oriented toward the membrane surface to be bound to the OST. However, it is not yet known whether the LLO oligosaccharide samples this orientation with respect to the membrane surface before its binding to the OST or changes its orientation during its binding to the OST.
Figure 2.
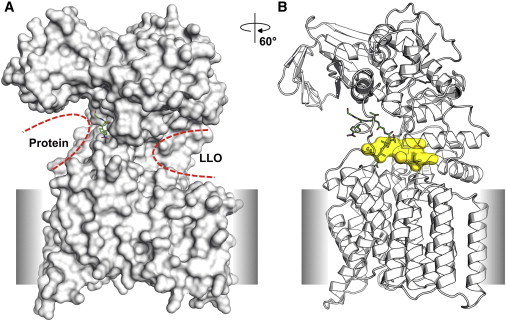
(A) Surface representation of PglB structure (PDB:3RCE) with protein and LLO binding sites marked by red dashed lines. The gray slab represents a tentative membrane region. (B) The inner shape of the LLO binding site. The inner shape is generated using a set of grid points located within 3 to 4.5 Å from the LLO binding site surface. Highly solvent exposed grids were discarded from the grid set by calculating enclosures. The enclosure is defined by the fraction of radial rays that strike the protein surface atoms among 146 evenly spaced 8 Å-long radial rays from a grid point. If the enclosure is less than 0.5, the grid is considered solvent exposed and thus removed. To see this figure in color, go online.
In this study, molecular modeling and simulations have been used to explore preferred orientation, structure, and dynamics of a eukaryotic LLO (Glc3-Man9-GlcNAc2-PP-Dolichol; G3M9Gn2-PP-Dol; Fig. 1 A) and a bacterial LLO (Glc1-GalNAc5-Bac1-PP-Undecaprenol; G1Gn5B1-PP-Und; Fig. 1 B) (18) in membrane bilayers with three different lipid types: dilauroylphosphatidylcholine (DLPC), dimyristoylphosphatidylcholine (DMPC), and dioleoylphosphatidylcholine (DOPC). The simulation results are discussed in terms of density distributions of each system component along the membrane normal, orientation and position of isoprenoid moieties in bilayers, oligosaccharide conformations and dynamics, oligosaccharide orientation and position with respect to the membrane, and interactions of oligosaccharides with the bilayers. Finally, the resulting position and orientation of the bacterial LLO are discussed in terms of its relevance as a substrate for the bacterial OST PglB based on results from LLO-OST molecular docking.
Methods
LLO force field parameters and initial structures
The CHARMM (19) inputs (llo_dol.inp and llo_und.inp in the Supporting Material) were used to generate the initial structures of G3M9Gn2-PP-Dol and G1Gn5B1-PP-Und, respectively. The CHARMM topology and parameter file, toppar_all36_lipid_llo.str (available at http://mackerell.umaryland.edu/charmm_ff.shtml), as part of the CHARMM additive force field (FF), contains additional residues and patches necessary for modeling and simulation of both LLO molecules. In the following paragraph, detailed explanations are given for the LLO FF parameters (toppar_all36_lipid_llo.str) and the LLO initial structures (llo_dol.inp and llo_und.inp).
To construct a CHARMM-compatible FF for LLO, we first modeled each LLO’s oligosaccharide portion in Fig. 1 with the CHARMM36 carbohydrate FF (20) that contains N-acetyl-d-glucosamine (GlcNAc) and N-acetyl-d-galactosamine (GalNAc) (21). The only exception was the unusual sugar bacillosamine (α-Bac) for G1Gn5B1-PP-Und in Fig. 1 B. Therefore, we constructed a CHARMM residue (RESID ABAC) in toppar_all36_lipid_llo.str based on α-GlcNAc and methylation patches in the CHARMM36 carbohydrate FF. Next, the diphosphate linkage and the isoprenoid chain were modeled based on the CHARMM General Force Field (CGenFF) version 2b7, exploiting the transferability between different parts of the CHARMM additive FF (22). To this end, using CGenFF atom types (23), the polyprenyl diphosphates were realized by constructing RESID DL19PP (with 19 isoprenyl subunits) for PP-Dol and UNDPP (with 11 subunits) for PP-Und in toppar_all36_lipid_llo.str. Because of the availability of directly relevant model compounds in CGenFF (i.e., methyldiphosphate and ethylphosphate for the pyrophosphate linkage, and retinol model compounds for the isoprenoid tail), it was deemed appropriate to manually assign the corresponding charges and bonded parameters by analogy, with guidance from the CGenFF program at https://cgenff.paramchem.org (24). Finally, using a similar methodology, a patch (PRES LLLO in toppar_all36_lipid_llo.str) was constructed to link any polyprenyl diphosphates to the C1 position of a given pyranose ring. The charges for this patch were assigned using the standard CHARMM FF charge assignment rules for combining fragments, and the new parameters that resulted from a covalent bond between a carbohydrate and a CGenFF group were assigned by analogy from the respective FF parameter files.
The CHARMM inputs (llo_dol.inp and llo_und.inp in the Supporting Material) were used to generate the oligosaccharides and the polyprenyl diphosphates, and to link them by the patch LLLO. The initial coordinates of G3M9Gn2-PP-Dol and G1Gn5B1-PP-Und were generated based on the internal coordinate (IC) information using the CHARMM IC functionality (19). Although most IC values in toppar_all36_lipid_llo.str were transferred from analogous residues, special attention was paid to the double-bond configurations (cis or trans) and chirality (Fig. 1), which were also maintained throughout the equilibration by applying corresponding dihedral restraints (see the next subsection). Because there is no oligosaccharide structure known for the bacterial LLO to the best of our knowledge, its initial structure was generated based on the IC glycosidic torsion angles given in the CHARMM36 carbohydrate topology file, which corresponds to the most favorable conformation of an arbitrary disaccharide. For the eukaryotic LLO, there are N-glycan crystal structures in the Protein Data Bank (PDB) (25), which partially cover its oligosaccharide structure. Therefore, we used the Glycan Fragment DB (http://www.glycanstructure.org) (26) to search for Man7-GlcNAc2 (i.e., up to D1, D2, D3 in each branch in Fig. 1 A) in the PDB glycan structures and to cluster the searched structures. The glycosidic torsion angles (given under IC EDIT in llo_dol.inp) in the top cluster were used to generate the initial oligosaccharide structure of G3M9Gn2-PP-Dol.
Molecular dynamics simulations
To simulate the LLO molecules in DLPC, DMPC, and DOPC membrane bilayers (with different hydrophobic thickness), we followed the general procedure of bilayer system building and equilibration in Membrane Builder (27,28) at the CHARMM-GUI website (http://www.charmm-gui.org/input/membrane) (29). First, the initial LLO structure was oriented along the Z axis and its diphosphate atoms (PP) were initially positioned around ZPP = 17 Å (in DLPC), 19 Å (DMPC), and 21 Å (in DOPC), so that the oligosaccharide portion and the isoprenoid chain were located above and below ZPP, respectively. In the present system setup, the bilayer center is located at Z = 0. Because the initial isoprenoid chain had a linear conformation (generated by the IC table), its last carbon atom was positioned at Z = −60 Å (for G3M9Gn2-PP-Dol) and −35 Å (G1Gn5B1-PP-Und), which were away from the bilayer hydrophobic core. To avoid unnecessary equilibration to bring this chain into the bilayer hydrophobic core and to reduce the system size, we performed a series of Langevin dynamics of a single LLO molecule in vacuum (at 310.15K) with a cylindrical restraint potential (with a force constant of 1.0 kcal/(mol⋅Å2)) to retain the LLO molecule within a radius of 20 Å in XY as well as with a positional restraint potential (with a force constant of 1.0 kcal/(mol⋅Å2)) to retain the PP atoms around ZPP. The Langevin dynamics simulation was stopped when the Z coordinate of the last carbon atom of the isoprenoid chain became less than −18 Å, which happened after 650 ps for Dol and 200 ps for Und. The rest of the system building followed the same steps in Membrane Builder (27,28), i.e., building a lipid bilayer (49 lipids in each leaflet) around the LLO molecule using the replacement method, a bulk water box, and K+ and Cl- ions (corresponding to 0.15 M KCl), and the assembly of each component (Fig. 3). These building steps were repeated three times with different random seed numbers to generate three independent systems for each LLO-bilayer type (yielding a total of 18 simulation systems) to improve sampling and to check simulation convergence. The system information including each system name is given in Table S1 in the Supporting Material.
Figure 3.
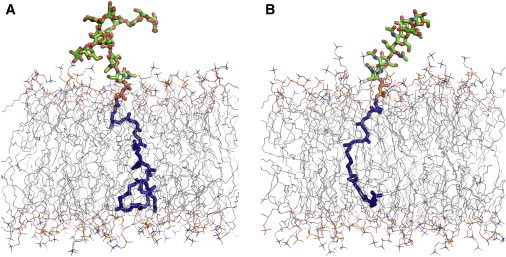
All-atom LLO simulation systems in DOPC bilayers: (A) Dol_DOPC for eukaryotic LLO (G3M9Gn2-PP-Dol) and (B) Und_DOPC for bacterial LLO (G1Gn5B1-PP-Und). The LLO molecules are displayed in stick representation. The figures were prepared using the last snapshot from the equilibration of each system. Water molecules and ions are not shown for clarity. To see this figure in color, go online.
The Membrane Builder’s standard six-step 375-ps equilibration simulations (27,28) were performed for each simulation system using CHARMM (19) with the C36 lipid (30) and carbohydrate FF (20,31,32), CGenFF (22), and a TIP3P water model (33). To ensure gradual equilibration of the assembled system, various restraints, including the restraints for the aforementioned LLO’s specific cis and trans double bonds and chirality (with an initial force constant of 250 kcal/(mol⋅rad2)), were applied to the LLO, lipid, and water molecules, and the restraint forces were gradually reduced during the equilibration. Additional dihedral angle restraints were applied to restrain all the sugar rings to the pertinent chair conformation. 50-ps NVT (constant particle number, volume, and temperature) dynamics was performed first, followed by 325-ps NPT (constant particle number, pressure, and temperature) dynamics for the equilibration.
After equilibration, a 350 ns NPT production run was performed with NAMD (34) for each system without any (dihedral) restraints. All simulations were performed under the following protocol. A time-step of 2 fs was used with the SHAKE algorithm (35). The van der Waals interactions were smoothly switched off over 10 to 12 Å by a force-switching function (36) and the long-range electrostatic interactions were calculated using the particle-mesh Ewald method (37) with a mesh size of ∼ 1 Å for fast Fourier transformation and a sixth-order B-spline interpolation. The temperature and pressure were held at 310.15 K and 1 bar, respectively. In the CHARMM simulations, Langevin temperature control was used for NVT dynamics. Temperature and pressure controls were achieved with a Hoover thermostat (38) and Langevin-piston for NPT dynamics (39,40). For the NAMD NPT simulations, Langevin dynamics was used to maintain constant temperature with a Langevin coupling coefficient set to 1 ps−1, and a Nosé-Hoover Langevin-piston (41,42) was used to maintain constant pressure with a piston period of 50 fs and a piston decay of 25 fs. Based on the X/Y system size time-series in Fig. S1 in the Supporting Material, the simulation results are presented for the last 330 ns of the trajectory of each system. Most results are presented by the averages of three independent runs and the standard errors.
Results and Discussion
In this section, the simulation results are first presented and discussed for the orientation, structure, and dynamics of the eukaryotic LLO, G3M9Gn2-PP-Dol, in the DOPC bilayers, and then those in the DLPC and DMPC bilayers. Subsequently, the orientation, structure, and dynamics of the bacterial LLO, G1Gn5B1-PP-Und, are compared with G3M9Gn2-PP-Dol. Finally, the docking results of G1Gn5B1-PP-Und to the bacterial OST PglB are presented and discussed.
Locations of eukaryotic LLO and structure and dynamics of isoprenoid chain in DOPC bilayers
Fig. 4 shows the density profiles of various components of G3M9Gn2-PP-Dol along the membrane normal (Z axis) in the DOPC bilayers. Clearly, most sugar residues (G3M9Gn2) stay above the lipid head group. However, as shown by the overlap with the membrane head group, a certain portion of the oligosaccharide is in frequent contact with the head group region and sometimes even buried in this region. This oligosaccharide-lipid interaction will be elaborated in detail in the next subsection. The pyrophosphate (PP) linkage stays close to, but slightly above the head group region on average. Note that there are sugar densities even below the average Z-position of the PP linkage. The isoprenoid (Dol) chain stays in the membrane hydrophobic region. Its distribution is asymmetric with more density found in the upper leaflet (63% at Z > 0 and 37% at Z < 0). The last carbon atom spends most of its time in the bilayer center but reaches into both leaflets and even stretches all the way to the head group of either leaflet with a slightly higher density in the lower leaflet.
Figure 4.
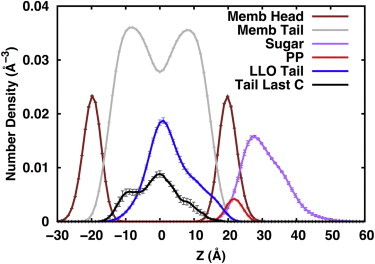
Density profiles of key components of the eukaryotic LLO, G3M9Gn2-PP-Dol, along the membrane normal (Z axis) in the DOPC bilayers: membrane head groups (brown), lipid acyl chains (gray), isoprenoid dolichol chain (blue), oligosaccharide (purple), pyrophosphate linkage (red), and the last carbon atom of dolichol chain (black). To aid viewing, the oligosaccharide distribution has been scaled by a factor of five and the pyrophosphate linkage, dolichol chain, and dolichol’s last carbon distributions by ten. The profiles are the averages of the three independent replicates, and the standard errors are also shown as the error bars. To see this figure in color, go online.
Fig. 5 A shows the distance distribution between Dol’s first carbon (C1A) and the middle carbon (C9E) and that between C1A and the last carbon (C19E). The C1A-C9E distribution indicates that C9E occupies a restricted region between 10 and 30 Å from C1A. The last carbon C19E can be close to C1A by 5 Å and stretched to 50 Å from C1A, illustrating the Dol tail’s flexibility. Fig. 5 B shows the relative orientation between the Dol tail and the oligosaccharide. The flexibility of the Dol tail and the extent of its motion are clearly visible, which arises from the dihedral angles (i.e., skew conformations around ±120°) before and after the double bonds in isoprenoids (Fig. S2). There is no strong preference of the Dol tail’s position with respect to the oligosaccharide on the XY plane (i.e., the membrane surface). The highly flexible nature of the Dol tail suggests a potential role of the Dol chain as a tentacle to search for certain binding sites in the transmembrane regions of the OST receptor or other glycosyltransferases. Some specific favorable interactions in membranes may cause the LLO tail to adhere to OST (or other glycosyltransferases), greatly increasing the likelihood of the LLO oligosaccharide interacting with the OST binding site. Indeed, previous NMR experiments showed specific binding between Dol and a polyisoprenol recognition sequence peptide from a protein in the multienzyme polysialyltransferase complex (7,8). Because of its flexibility within the membrane, the Dol chain appears to increase the flexibility of the DOPC acyl chains near the Dol chain, as the order parameters of the DOPC acyl chains near the Dol chain are slightly lower than those of the pure DOPC bilayers (Fig. S3). This is consistent with earlier experiments (9), and a similar observation was made in the simulation studies of lipid II, a precursor of the bacterial cell walls, in membrane bilayers (43,44); lipid II has an undecaprenyl lipid chain like the bacterial LLO.
Figure 5.
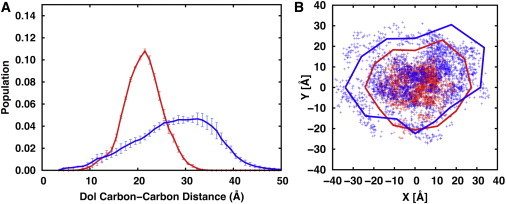
(A) Distributions of the distances of C1A-C9E atoms (red) and C1A-C19E atoms (blue) in the Dol tail in the DOPC bilayers; C1A for the first carbon of Dol, C9E the carbon at the end of 9th isoprenyl unit, and C19E the carbon at the end of 19th isoprenyl unit. The distributions are the averages of the three independent replicates, and the standard errors are also shown as the error bars. (B) Distributions of the XY locations of C9E (red) and C19E (blue). In this plot, C1A is always positioned at X = Y = 0, and the long-branch of the oligosaccharide (i.e., the 1C1-14C1 vector) is aligned along the positive X axis, where 1C1 is the C1 carbon of the first GlcNAc residue and 14C1 the C1 carbon of the last G1 Glc residue. The corresponding solid lines are polar histograms that express the angular preference, or lack thereof. If there is no angular preference, the lines should form a circle centered at the origin; the lines in (B) are close enough to be circular to claim no strong angular preference. Specifically, the plane was divided into twelve 30° sectors centered at the origin, and the solid lines connect the points located on the bisector of each sector at a distance equal to the cube root of the sum of the distances from the origin to each dot in that sector. The resulting polar graphs were scaled so that the shapes delineated by the solid lines fill the plot area but do not exceed it. This particular visualization was chosen because 1), a measure for the angular preference should not only take the number of points into account, but also their distance, as points close to the origin do not express a strong angular preference, and 2), for a sufficiently large number of bins and a sufficiently smooth distribution, the centroid of the shape delineated by the solid lines approaches the centroid of the cloud of dots, except for an arbitrary scaling factor. To see this figure in color, go online.
Orientation, structure, and dynamics of oligosaccharide of eukaryotic LLO in DOPC bilayers
To investigate the orientation of the eukaryotic LLO’s oligosaccharide (G3M9Gn2) in the DOPC bilayers, the distributions of tilt angles (from the membrane normal) of the long-branch (defined by a vector from the C1 carbon of residue 1 GlcNAc and the C4 carbon of residue 4 Man in Fig. 1 A) and the short-branch (defined by a vector from residue 1 C1 and residue 4' C4) are calculated and shown in Fig. S4. The average tilt angles are 66.6° ± 21.6° (long-branch) and 55.5° ± 15.1° (short-branch). On average, the long-branch has larger tilt angles, indicating that it stays closer to the membrane surface than the short-branch. Both branches have tilt angles that range from below 20° (< 2% population) to close to 90°, indicating the flexibility of the oligosaccharide orientation.
Fig. 6 A shows the average Z coordinate of the center of mass (ZCOM) of each sugar residue; see the residue names in Fig. 1 A. The standard errors (blue) from the three independent simulations are relatively small, indicating good convergence of this property. ZCOM shows each sugar’s preference for being close to the membrane. Notably, the standard deviation (red) of each sugar’s ZCOM within each simulation, i.e., the fluctuations of each sugar’s ZCOM, is large, so that even the G1 residue (with the average ZCOM of 34.5 Å) at the end of the long-branch can occasionally interact with the membrane head group. Fig. 6 B shows the fraction of the simulation time that each sugar interacts with the lipid head groups (orange) or tails (green). Clearly, except the first two GlcNAc residues, sugar residue 4 interacts the most with the membrane and is sometimes buried deep enough to interact with the lipid acyl chains. Interestingly, while residue 3 has almost the same ZCOM as residue 4, its interaction with the lipid head group is much smaller than residue 4. This difference arises from the oligosaccharide’s preferential orientation in the DOPC bilayers, i.e., the preference of the long-branch to be closer to the membrane surface (Fig. S4). Such preferential orientations of the long-branch and/or its interactions with the membrane head group also bring the A-D2 short chain close to the membrane surface. Fig. 7 shows a representative structure to illustrate the overall orientation of the oligosaccharide G3M9Gn2 of the eukaryotic LLO in a DOPC bilayer.
Figure 6.
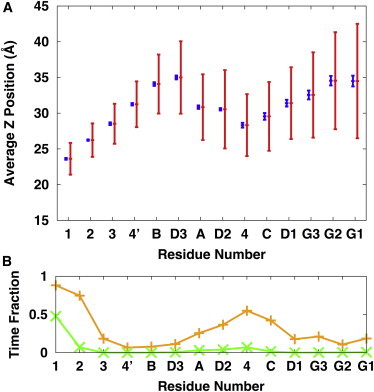
(A) Average Z distance of the center of mass of each sugar residue of the eukaryotic LLO from the bilayer center (i.e., Z = 0): the blue bars for the standard errors from the three independent simulations and the red bars for the standard deviations within each simulation. (B) Time fraction of each sugar residue in contact with lipid head groups (orange) or tails (green). The contact is counted when any heavy atom of each sugar residue is within 4.5 Å from any heavy atom from the head groups or the tails. The values are the averages of the three independent replicates, and the standard errors are smaller than the symbol sizes. To see this figure in color, go online.
Figure 7.
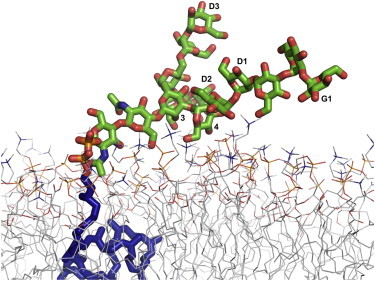
A representative snapshot to illustrate the orientation of the eukaryotic LLO’s oligosaccharide and its interactions with a DOPC bilayer. The snapshot was selected based on the most populated tilt angles in Fig. S4 and the average ZCOM of residues 4', D2, 4, and C in Fig. 6A. To see this figure in color, go online.
To understand the energetics and origin of such flexibility and preferential orientations of eukaryotic LLO’s oligosaccharide, we estimated the potential of mean force (PMF) profile along the tilt angle (τ) of the long-branch, which was calculated directly from the tilt angle distribution (Fig. S4). As shown in Fig. 8 A, at small tilt angles (τ < 20°), the estimated PMF is dominated by the precession entropy (Jacobian) contribution, which is consistent with previous results from the PMF calculation of a transmembrane helix along the tilt angle (45,46). In the tilt angle range of 30° to 100°, the favorable interactions between sugar and its environment dominates the PMF, which is shown as a basin. This τ-range is thermally accessible (2kBT range from the PMF minimum, where kB is the Boltzmann constant and T is temperature) (47), and thus the long-branch of sugar can swing around the PMF-minimum τ without much energetic cost in this range. Although it is qualitative, the insight into the nature of the favorable interactions between the sugar and its surrounding environment can be obtained from the mean interaction energy calculation (48). As shown in Fig. 8 B, as τ increases, there arises an energetic penalty because of the loss of water solvation, but this energetic penalty is compensated by more favorable sugar-lipid interactions, which results in the overall favorable interactions. This result suggests that the sugar-lipid interactions are the driving force for the preferential orientation of sugar (parallel to membrane). To further identify the nature of the sugar-lipid interactions, the mean sugar-lipid interaction energy was decomposed into the contributions from van der Waals and electrostatic interactions. As shown in Fig. 8 C, both van der Waals and electrostatic interactions are favorable, but the latter contributes more in the case of eukaryotic LLO’s oligosaccharide, indicating that favorable electrostatic interactions between sugar and lipid head groups (presumably hydrogen bonds) can stabilize the preferential orientation of the oligosaccharide. Because of the limited sampling, we did not consider the energetics at large tilt angles (> 100°). The energetics at these tilt angles can be studied by calculating the PMF using the umbrella sampling method (46), which is beyond the scope of the current study.
Figure 8.
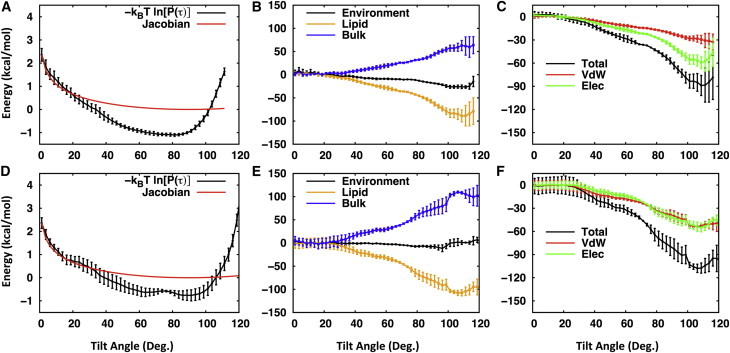
(A and D) The potential of mean force (PMF) along the tilt angle (τ) of the long-branch of (A) eukaryotic and (D) bacterial LLO’s oligosaccharide in DOPC lipid bilayers. The PMF was calculated from its distribution P(τ) (Fig. S4) by –kBT ln[P(τ)]. Shown together (in red) is the precession entropy contribution to the PMF, which given by –kBT ln[sin(τ)]. (B and E) The mean interaction energy between the oligosaccharide ((B) eukaryotic and (E) bacterial) and its environment (black), lipids (orange), and bulk (blue). (C) The mean interaction energy between the oligosaccharide ((C) eukaryotic and (F) bacterial) and lipids (black) and its decomposition into the van der Waals (red) and electrostatic (green) interaction energies. For all the panels, the error bars are the standard errors over the three independent replicates and those for the PMF are estimated from the standard errors for the corresponding tilt angle distributions. To see this figure in color, go online.
The root-mean squared deviation (RMSD) of M9Gn2 (i.e., up to D1, D2, D3 in each branch in Fig. 1 A) with respect to the initial structure obtained from Glycan Fragment DB (26) (see Methods) is ∼ 3 Å, suggesting that the M9Gn2 conformations in PDB glycoproteins are more or less maintained in the eukaryotic LLO in membranes on the time scale of the present simulations. As shown in Figs. 9 A and B, whereas the distance between the C1 atoms of residue 1 and residue G1 (d1-G1) has a narrow distribution around 28 ± 2 Å, dD3-G1 and dD2-G1 show slightly wider distributions around 18 ± 4 Å and 21 ± 3 Å, respectively. Fig. 9 C shows two different views of the representative oligosaccharide structures of the top five clusters (see Fig. 9 C legend), which cover ∼ 81% of the entire trajectories. Clearly, dD3-G1 is shorter than dD2-G1, although dD2-G1 seems to be longer than dD3-G1 in Fig. 1 A. With the first three residues 1, 2, and 3 used for alignment, the root-mean squared fluctuation (RMSF) of each sugar residue indicates that the D2 (5.2 ± 0.2 Å) and D3 (6.5 ± 0.3 Å) sugars are slightly more flexible than the D1 sugar (4.5 ± 0.2 Å), due to the flexibility of the 1-6 linkages to D2 (through 3-4') and D3 (through 4'-B) (26,49). The RMSF of the G1 sugar at the end of the long-branch show the greatest flexibility (10.2 ± 0.4 Å). It is such flexibility that not only makes dD3-G1 and dD2-G1 wider than d1-G1, but also allows G1 to interact with membranes occasionally, although its ZCOM is above the membrane (Fig. 6 A).
Figure 9.
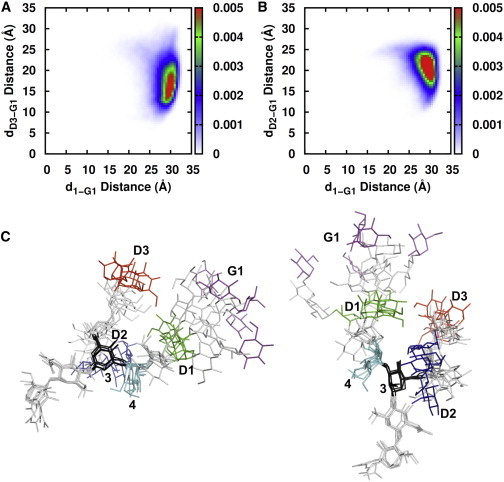
(A and B) Two-dimensional populations of (A) d1-G1 and dD3-G1 and (B) d1-G1 and dD2-G1 where d1-G1 is the distance between 1 C1 and G1 C1 atoms, dD3-G1 the distance between D3 C1 and G1 C1 atoms, and dD2-G1 the distance between D2 C1 and G1 C1 atoms. The bin size is 0.5 Å in X and Y. (C) Two different views of the overlay of representative G3M9Gn2 conformations from the Dol_DOPC simulations. The representative snapshots correspond to the centroid structures of the top five clusters of G3M9Gn2 conformations, which cover ∼ 81% of the conformational space accessed during the simulation. For clustering, a total of 898 snapshots was collected from the simulation trajectories using a simulation time interval of 1.2 ns and then clustered using a RMSD cutoff of 2.5 Å. The clustering analysis was performed by in-house software using the average linkage clustering method. The RMSD between the different snapshots was calculated using M9Gn2. To prepare the figures, the five G3M9Gn2 conformations were superposed again only using the first five residues (1, 2, 3, 4, and 4'). To see this figure in color, go online.
Locations, orientation, structure, and dynamics of eukaryotic LLO in DLPC and DMPC bilayers
As shown in Fig. S5, the peaks of the PP linkage along the Z axis are shifted to larger values to match with the head group positions as the membrane thickness increases: 18.8 Å (DLPC), 19.8 Å (DMPC), and 21.8 Å (DOPC). Similar to the Dol_DOPC system, the Dol chain stays in the membrane hydrophobic core in the DLPC and DMPC bilayers, but its distribution becomes wider as the membrane thickness increases. The population of the Dol tail with Z > 0 is similar in all three bilayer types: 0.60 ± 0.03 (DLPC), 0.60 ± 0.02 (DMPC), and 0.63 ± 0.01 (DOPC). As shown in Fig. S6, the distributions of the C1A-C9E (red) and C1A-C19E (blue) distances as well as their average distances (18.6 ± 2.6 Å and 26.7 ± 4.0 Å in DLPC, 19.5 ± 2.7 Å and 26.9 ± 4.6 Å in DMPC, and 21.2 ± 2.3 Å and 27.7 ± 4.7 Å in DOPC) are all similar in different membrane bilayers. In addition, the XY distribution patterns of C9E and C19E appear to be all random and similar in different membranes (Fig. S7). Overall, the Z position of the PP linkage is dependent on the membrane thickness, whereas the Dol chain distribution and flexibility are not. Similar to the case in the DOPC bilayer, the flexibility of the Dol chain makes the DMPC and DLPC acyl chains more disordered than in pure DMPC and DLPC bilayers (Fig. S3).
Although the ZCOM of the eukaryotic LLO’s sugars are overall shifted to larger values because of the shift of the PP linkage as the membrane thickness increases (Fig. S8), the characteristics of the oligosaccharide orientations are similar in different membranes, i.e., the long-branch has larger tilt angles than the short-branch (Fig. S4). Because of such orientational similarity, the overall patterns of the sugar and lipid interactions are also similar; i.e., the sugar residue 4 (except the first two GlcNAc residues) interacts the most with membranes (Fig. S9). The characteristics of the sugar-lipid interaction energy are the same as in Fig. 8 C; i.e., the electrostatic interactions contribute more to the sugar-lipid interaction energy (data not shown). Overall, as shown in Fig. 7, it is a general orientational feature of the eukaryotic LLO’s oligosaccharide in membranes that the oligosaccharide prefers to be tilted toward the membranes with preference for the long-branch to be closer to the membrane surface.
Locations, orientation, structure, and dynamics of bacteria LLO in bilayers
As shown in Fig. S5, similar to the eukaryotic LLO in bilayers, the PP linkage of a bacterial LLO, G1Gn5B1-PP-Und, is within the lipid head group region and its Und chain is within the bilayer hydrophobic core with larger populations at Z > 0. The distributions of the last Und carbon in different membranes are similar, unlike the Dol systems where its distribution becomes wider as the membrane thickness increases. These observations are related to the shorter length of the Und chain. The effective (relaxed) length of the Und chain is optimal even in DLPC bilayers, whereas that of the Dol chain appears to easily cover the DOPC bilayer thickness. The average distances of C1A-C6E (Und middle carbon) and C1A-C11E (Und last carbon) and their distributions are similar (given their wide distributions) in different membranes (Fig. S6). Interestingly, the average C1A-C11E distance in Und (21.6 ± 3.45 Å in DLPC, 23.0 ± 3.6 Å in DMPC, and 24.9 ± 3.7 Å in DOPC) is comparable with that of C1A-C19E in Dol (26.7 ± 4.0 Å in DLPC, 26.9 ± 4.6 Å in DMPC, and 27.7 ± 4.7 Å in DOPC), although the maximum stretch of the Und chain is 10 Å shorter than that of Dol (Fig. S6). Clearly, the Und chain is more restricted in the XY projection than the Dol chain (Fig. S7), not simply because the Und chain length is shorter, but partially because the last carbon stays in the lower leaflet more frequently. This suggests that the Und chain may search for the OST in a shorter range than the Dol chain. If we consider that the OST-LLO complex may form when the LLO tail makes contact with the transmembrane region of OST, a longer reach gives more opportunity to make contact with OST. We hypothesize that contact of the hydrocarbon portion of LLO with the OST is a favorable interaction increasing the likelihood of LLO-OST complex formation. To a lesser extent than Dol, the Und chain makes the lipid acyl chains more disordered, although this is less evident in Und_DOPC (Fig. S3).
Similar to the eukaryotic LLO, the orientation of the bacterial LLO’s oligosaccharide is largely titled with respect to the Z axis, yet very dynamic (Fig. S4). Each sugar’s ZCOM of G1Gn5B1 shows more variations (in terms of the standard errors) in DMPC and DOPC than in DLPC and also than the eukaryotic LLO’s oligosaccharide in all membranes (Fig. S8). This could arise from limited sampling, particularly for the Und_DOPC system. For example, as shown in Fig. S10, the mode of the tilt angle made by oligosaccharide residue 1 and residue 5 in one independent Und_DOPC simulation is ∼ 40° less than the modes in the other two simulations. Such different modes are likely attributable to an intrinsic dynamic nature of the oligosaccharide as well as the context of sugar-lipid interactions, i.e., G1Gn5B1 has different extents of interactions with lipids. As shown in Fig. S9, there is an increasing tendency of G1Gn5B1 to interact with lipids as the membrane thickness increases, which eventually makes each sugar’s ZCOM lower or similar to that in DLPC. Such increasing interactions of G1Gn5B1 with DOPC make the tilt distribution of G1Gn5B1 shifted to larger tilt angles (Fig. S4). These observations are also consistent with the fact that more sugar density is found in head group or even in lipid tail regions in Und_DOPC (Fig. S7). Sugar residue 7 interacts slightly more with lipids than sugar residue 5, although they are in structurally similar positions (Fig. 1). Overall, the general orientational features of the bacterial LLO’s oligosaccharide in membranes are similar to that of the eukaryotic LLO’s oligosaccharide in that the oligosaccharide prefers to be tilted toward the membrane surface. The estimated PMF from the tilt angle distributions of the long-branch shows the similar behavior to that for the eukaryotic LLO’s oligosaccharide: At small tilt angles, the PMF is dominated by the precession entropy contribution and then followed by a basin (Fig. 8 D), whose overall favorable interactions are driven by sugar-lipid interactions (Fig. 8 E). However, the characteristics of sugar-lipid interactions are different from the eukaryotic LLO systems in that the contribution from the electrostatic interactions becomes comparable with that from the van der Waals interactions (Fig. 8 F).
Docking of bacteria LLO to bacteria OST PglB
An x-ray structure of a bacterial OST, the PglB protein from Campylobacter lari (17), has been recently reported (PDB:3RCE; Fig. 2 and Fig. 10 A). This structure is in complex with an acceptor peptide (-Asp-Gln-Asn-Ala-Thr-) and a divalent cation M2+, which is known to be critical in catalyzing N-glycosylation between asparagine in an acceptor protein and an oligosaccharide in a donor LLO. To investigate potential binding modes of a bacterial LLO, G1Gn5B1-PP-Und, to the OST PglB, we performed molecular docking using the conformations and orientations of the bacterial LLO sampled during the LLO/DOPC simulations (see Supporting Material for the docking procedure).
Figure 10.
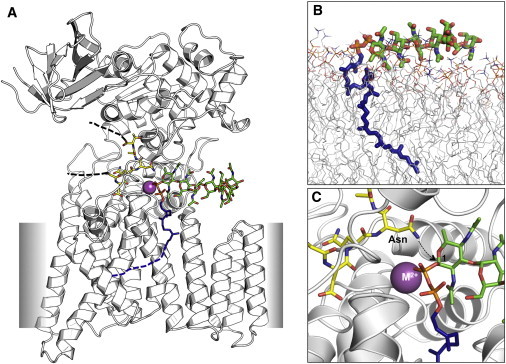
(A) A predicted binding mode of the bacterial LLO, G1Gn5B1-PP-Und, in the bacterial OST PglB from Campylobacter lari (PDB:3RCE). A divalent cation, which can be Mg2+ or Mn2+ according to reference (17), is represented by a purple sphere labeled M2+. (B) A representative snapshot from the MD simulations of G1Gn5B1-PP-Und in the DOPC bilayers. The snapshot was selected on the basis of the most populated tilt angles and average ZCOM of sugar residues 5, 6, and 7 (Fig. 1B). (C) An enlarged figure of the docking model, focusing on the active site in the OST. We used the docking procedure described in the Supporting Material. To see this figure in color, go online.
Fig. 10 A shows a final docking model of the bacterial LLO in PglB, which is a docking pose with the minimum number of bad contacts among all the docking poses (number of bad contacts = 4). The successful docking models clearly show that the orientation of the bacterial LLO is significantly tilted toward the membrane surface to form a favorable complex within the N-glycosylation active site of PglB. These docking poses also have similar orientation to a representative simulation snapshot selected on the basis of the most populated tilt angles and average ZCOM of sugar residues 5, 6, and 7 (Fig. 10 B). When we measured tilt angles of 1-5 and 1-7 sugar residue pairs for the docked bacterial LLOs, the average tilt angles and their distributions (85.8° ± 14.8° and 86.7° ± 18.8° in Fig. S11) are comparable with those from the full simulation trajectories (83.8° ± 18.3° and 88.1° ± 21.1° in Fig. S4 Und_DOPC). Our simulation results show that the eukaryotic LLO also has the orientations of both the short and long chains that are tilted toward the membrane (Figs. 7 and S4), suggesting that this preferential orientation is a conserved feature of LLOs across species. All these observations suggest that 1), the orientation of the oligosaccharide of LLOs in membrane bilayers before its interaction with OST corresponds to the orientation required for the OST binding, thereby facilitating binding, and 2), the eukaryotic LLO likely binds to its enzyme with a pose similar to that of the bacterial LLO proposed in this study.
Conclusions
This computational study of eukaryotic and bacterial LLOs, an N-glycan substrate of OST for N-glycosylation, uses molecular modeling, simulations, and docking. We describe in detail the molecular modeling procedure of each LLO, including the LLOs’ unique structural features such as sugar linkages, double-bond configuration, and chirality, as well as the assignment of FF parameters consistent with available C36 lipid and carbohydrate FF.
The molecular dynamics simulations of both eukaryotic and bacterial LLOs in membrane bilayers of three different lipids (DLPC, DMPC, and POPC) reveal: 1), a consistent location of the different LLO moieties (oligosaccharide, pyrophosphate linkage, and isoprenoid chain) in bilayers, where most LLO sugar residues stay above the lipid head group, the PP linkages are within the lipid head group, and the isoprenoid chains are within the bilayer hydrophobic core, 2), a potential role of the isoprenoid chain as a tentacle to search for (specific favorable) OST (binding sites) inside the bilayer due to its flexibility, 3), similar N-glycan conformations in both LLOs and glycoproteins, 4), the oligosaccharide’s preferential orientations parallel to the bilayer surface due to favorable interactions between the oligosaccharide and the bilayer head groups (or sometimes with the hydrophobic core), and 5), overall similar orientation, structure, and dynamics of eukaryotic and bacterial LLOs in bilayers with different hydrophobic thickness.
The molecular docking of a bacterial LLO to OST PglB from Campylobacter lari suggests that 1), the preferential orientations of LLO’s oligosaccharide parallel to the bilayer surface correspond to the orientation required in the LLO binding site positioned in a deep concave pocket of PglB and 2), similar binding modes exist in eukaryotic LLOs due to the overall similar orientation of eukaryotic and bacterial LLOs’ oligosaccharides in bilayers.
Acknowledgments
This work was supported in part by NSF MCB-1157677, NSF ABI-1145987, HFSP RGP0064/2011, XSEDE MCB070009 (to W. I.); K-INBRE undergraduate research scholarship (NIH P20 GM103418) and KU undergraduate research award (to N. R. K.); NSF DBI-1145652 (to J. B. K.); and NIH GM070855 and GM072558 (to A. D. M.).
Footnotes
Nathan R. Kern, Hui Sun Lee, and Emilia L. Wu contributed equally to this work.
Supporting Material
References
- 1.Weerapana E., Imperiali B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology. 2006;16:91R–101R. doi: 10.1093/glycob/cwj099. [DOI] [PubMed] [Google Scholar]
- 2.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaeken J. Congenital disorders of glycosylation. Ann. N. Y. Acad. Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- 4.Jones M.B., Rosenberg J.N., Krag S.S. Structure and synthesis of polyisoprenoids used in N-glycosylation across the three domains of life. Biochim. Biophys. Acta. 2009;1790:485–494. doi: 10.1016/j.bbagen.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz F., Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011;21:576–582. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 6.de Ropp J.S., Troy F.A. 2H NMR investigation of the organization and dynamics of polyisoprenols in membranes. J. Biol. Chem. 1985;260:15669–15674. [PubMed] [Google Scholar]
- 7.Zhou G.-P., Troy F.A., 2nd NMR study of the preferred membrane orientation of polyisoprenols (dolichol) and the impact of their complex with polyisoprenyl recognition sequence peptides on membrane structure. Glycobiology. 2005;15:347–359. doi: 10.1093/glycob/cwi016. [DOI] [PubMed] [Google Scholar]
- 8.Zhou G.P., Troy F.A., 2nd Characterization by NMR and molecular modeling of the binding of polyisoprenols and polyisoprenyl recognition sequence peptides: 3D structure of the complexes reveals sites of specific interactions. Glycobiology. 2003;13:51–71. doi: 10.1093/glycob/cwg008. [DOI] [PubMed] [Google Scholar]
- 9.Valtersson C., van Duÿn G., Dallner G. The influence of dolichol, dolichol esters, and dolichyl phosphate on phospholipid polymorphism and fluidity in model membranes. J. Biol. Chem. 1985;260:2742–2751. [PubMed] [Google Scholar]
- 10.Murgolo N.J., Patel A., Wong T.K. The conformation of dolichol. Biochemistry. 1989;28:253–260. doi: 10.1021/bi00427a035. [DOI] [PubMed] [Google Scholar]
- 11.Wilson I.B., Webberley M.C., Flitsch S.L. Dolichol is not a necessary moiety for lipid-linked oligosaccharide substrates of the mannosyltransferases involved in in vitro N-linked-oligosaccharide assembly. Biochem. J. 1995;310:909–916. doi: 10.1042/bj3100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartley M.D., Imperiali B. At the membrane frontier: a prospectus on the remarkable evolutionary conservation of polyprenols and polyprenyl-phosphates. Arch. Biochem. Biophys. 2012;517:83–97. doi: 10.1016/j.abb.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackeen M.M., Almond A., Wormald M.R. The conformational properties of the Glc3Man unit suggest conformational biasing within the chaperone-assisted glycoprotein folding pathway. J. Mol. Biol. 2009;387:335–347. doi: 10.1016/j.jmb.2009.01.043. [DOI] [PubMed] [Google Scholar]
- 14.Petrescu A.J., Butters T.D., Wormald M.R. The solution NMR structure of glucosylated N-glycans involved in the early stages of glycoprotein biosynthesis and folding. EMBO J. 1997;16:4302–4310. doi: 10.1093/emboj/16.14.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Höög C., Widmalm G. Molecular dynamics simulation and nuclear magnetic resonance studies of the terminal glucotriose unit found in the oligosaccharide of glycoprotein precursors. Arch. Biochem. Biophys. 2000;377:163–170. doi: 10.1006/abbi.2000.1752. [DOI] [PubMed] [Google Scholar]
- 16.Alvarado E., Nukada T., Ballou C.E. Conformation of the glucotriose unit in the lipid-linked oligosaccharide precursor for protein glycosylation. Biochemistry. 1991;30:881–886. doi: 10.1021/bi00218a001. [DOI] [PubMed] [Google Scholar]
- 17.Lizak C., Gerber S., Locher K.P. X-ray structure of a bacterial oligosaccharyltransferase. Nature. 2011;474:350–355. doi: 10.1038/nature10151. [DOI] [PubMed] [Google Scholar]
- 18.Weerapana E., Glover K.J., Imperiali B. Investigating bacterial N-linked glycosylation: synthesis and glycosyl acceptor activity of the undecaprenyl pyrophosphate-linked bacillosamine. J. Am. Chem. Soc. 2005;127:13766–13767. doi: 10.1021/ja054265v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guvench O., Greene S.N., Mackerell A.D., Jr. Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 2008;29:2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guvench O., Mallajosyula S.S., Mackerell A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011;7:3162–3180. doi: 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanommeslaeghe K., Hatcher E., Mackerell A.D., Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanommeslaeghe K., MacKerell A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012;52:3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanommeslaeghe K., Raman E.P., MacKerell A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012;52:3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman H.M., Westbrook J., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo S., Im W. Glycan fragment database: a database of PDB-based glycan 3D structures. Nucleic Acids Res. 2013;41:D470–D474. doi: 10.1093/nar/gks987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo S., Kim T., Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jo S., Lim J.B., Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jo S., Kim T., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 30.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatcher E., Guvench O., Mackerell A.D. CHARMM additive all-atom force field for aldopentofuranoses, methyl-aldopentofuranosides, and fructofuranose. J. Phys. Chem. B. 2009;113:12466–12476. doi: 10.1021/jp905496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guvench O., Hatcher E.R., Mackerell A.D. CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 34.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 36.Steinbach P.J., Brooks B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994;15:667–683. [Google Scholar]
- 37.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 38.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 39.Nose S., Klein M.L. A study of solid and liquid carbon tetrafluoride using the constant pressure molecular dynamics technique. J. Chem. Phys. 1983;78:6928–6939. [Google Scholar]
- 40.Andersen H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980;72:2384–2393. [Google Scholar]
- 41.Feller S.E., Zhang Y., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 42.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 43.Jia Z., O’Mara M.L., Mark A.E. The effect of environment on the recognition and binding of vancomycin to native and resistant forms of lipid II. Biophys. J. 2011;101:2684–2692. doi: 10.1016/j.bpj.2011.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chugunov A., Pyrkova D., Efremov R. Lipid-II forms potential “landing terrain” for lantibiotics in simulated bacterial membrane. Sci. Rep. 2013;3:1678. doi: 10.1038/srep01678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J., Im W. Restraint potential and free energy decomposition formalism for helical tilting. Chem. Phys. Lett. 2007;441:132–135. [Google Scholar]
- 46.Lee J., Im W. Transmembrane helix tilting: insights from calculating the potential of mean force. Phys. Rev. Lett. 2008;100:018103. doi: 10.1103/PhysRevLett.100.018103. [DOI] [PubMed] [Google Scholar]
- 47.Park S., Kim T., Im W. Transmembrane helix assembly by window exchange umbrella sampling. Phys. Rev. Lett. 2012;108:108102. doi: 10.1103/PhysRevLett.108.108102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song K.C., Livanec P.W., Im W. Orientation of fluorescent lipid analogue BODIPY-PC to probe lipid membrane properties: insights from molecular dynamics simulations. J. Phys. Chem. B. 2011;115:6157–6165. doi: 10.1021/jp109629v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Re S., Miyashita N., Sugita Y. Structural diversity and changes in conformational equilibria of biantennary complex-type N-glycans in water revealed by replica-exchange molecular dynamics simulation. Biophys. J. 2011;101:L44–L46. doi: 10.1016/j.bpj.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.