

Abstract
The protein-protein docking problem is one of the focal points of activity in computational biophysics and structural biology. The three-dimensional structure of a protein-protein complex, generally, is more difficult to determine experimentally than the structure of an individual protein. Adequate computational techniques to model protein interactions are important because of the growing number of known protein structures, particularly in the context of structural genomics. Docking offers tools for fundamental studies of protein interactions and provides a structural basis for drug design. Protein-protein docking is the prediction of the structure of the complex, given the structures of the individual proteins. In the heart of the docking methodology is the notion of steric and physicochemical complementarity at the protein-protein interface. Originally, mostly high-resolution, experimentally determined (primarily by x-ray crystallography) protein structures were considered for docking. However, more recently, the focus has been shifting toward lower-resolution modeled structures. Docking approaches have to deal with the conformational changes between unbound and bound structures, as well as the inaccuracies of the interacting modeled structures, often in a high-throughput mode needed for modeling of large networks of protein interactions. The growing number of docking developers is engaged in the community-wide assessments of predictive methodologies. The development of more powerful and adequate docking approaches is facilitated by rapidly expanding information and data resources, growing computational capabilities, and a deeper understanding of the fundamental principles of protein interactions.
Introduction
Proteins recognize each other, typically in a crowded environment, and bind in a highly specific fashion. This process involves diffusion through a densely populated milieu of different proteins and other biomolecular structures, and binding (docking) to their designated protein partner in a structurally unique and precise way. Given the large size of these macromolecules, the great structural diversity, and the high density of the biomolecular environment, this constantly reoccurring process is truly remarkable.
Protein docking—prediction of the structure of a protein-protein complex from the structures of the individual proteins—has evolved significantly since its early days, by incorporating more adequate energy functions and powerful techniques to sample the energy landscapes, and by taking advantage of the rapidly growing body of knowledge on protein structures and interactions. Our current knowledge of protein interaction principles is far greater than before, helping design better docking approaches. The spectacular progress in computing hardware has obviously played a major role as well, opening new ways of thinking about modeling of protein interactions, and often allowing implementation of old but unfeasible at the time ideas. Still, some basic docking principles remain surprisingly unchanged, due to their true nature. Steric and physicochemical complementarity is still the foundation of most docking approaches, as it was in the beginning of the docking field.
Beginnings
The origins of the protein docking field can be traced to the earlier days of molecular modeling. Back than in the 70s, the force fields were simpler, the minds not clouded by the power of computers, and the goals clearer (e.g., to fold proteins from the sequence based on the physical forces alone).
The first docking approaches dealt not with protein-protein complexes per se, but rather with protein interactions with other ligands at predetermined binding sites (1–4). Despite the early times in molecular modeling, the approaches were remarkably sophisticated, implementing flexible docking, taking into account the internal coordinates of not only the ligand, but in some cases also the receptor—a challenging task largely avoided even in today’s community, with all its computing power and the history of methodology development. Protein-protein docking approaches followed shortly, implementing the global search for the docking pose in rigid-body approximation (5,6).
A significant uptake in the development of protein docking techniques (that continues to this day) occurred in the early 90s. Among most influential and consequential approaches put forward at that time were those based on efficient sampling techniques borrowed from computer science (7,8). The docking approach based on correlation by fast Fourier transform, commonly known as FFT docking (7), developed back then by an interdisciplinary group of biologists, chemists, physicists, and computer scientists, has become arguably the most popular protein docking algorithm, implemented over the years in many groups (9). The reason for its popularity is that, as opposed to employing a particular search strategy that may or may not lead to the global minimum (native complex), it allows computationally feasible exhaustive search of the full six-dimensional docking space. Although the space, for the purpose of the exhaustive sampling, has to be discretized, the atomic-size grid steps still provide the “comprehensive” solution to the rigid-body docking problem.
Docking Foundations
The protein-protein docking problem can be formulated as the prediction of the structure of the complex, given the structures of the individual proteins. In the general case, no information other than the structure of these individual proteins is available. Fig. 1 shows major steps in the proper development of a docking approach, involving scan (global search) of the docking space using simplified/coarse-grained protein representations, followed by a refinement to a higher resolution (local search), and systematic evaluation on comprehensive benchmark sets and blind community-wide assessments.
Figure 1.

The general scheme of protein docking methodology development. The scan (global search for complementarity) is performed on a simplified/coarse-grained representation of proteins (e.g., digitized on a grid, or discretized/approximated in other ways). The scan can be explicit (free) or based on similarity to known cocrystallized complexes (comparative). The refinement is supposed to bring back all or some structural resolution lost in the coarse-graining (e.g., by gradual transition from s smoothed intermolecular energy landscape to the one based on a physical force field, while tracking the position of the global minimum). The validity of the approach is determined by systematic benchmarking on representative sets of structures. To see this figure in color, go online.
In the heart of the docking methodology is the notion of steric complementarity at the protein-protein interface. These interfaces are indeed tightly packed, as observed in cocrystallized complexes in the Protein Data Bank (PDB). The steric complementarity has been the major driving force in the development of docking approaches, often with the addition of physicochemical complementarity—hydrophobicity, electrostatics, etc. (10,11), and statistics-based propensities (12,13). The structural complementarity has been observed at different resolutions, from the atomic to ultralow (14–18).
The conformations of the protein within the complex (bound structure) and the one outside the complex (unbound structure) are different. In some cases, this difference can be neglected or approximated (rigid body docking), or taken into account through conformational search (flexible docking). The rigid body docking involves six degrees of freedom of the two rigid bodies system (e.g., three translations and three rotations in the Cartesian coordinates). The flexible docking involves a much greater number of coordinates, given the conformational search in the internal coordinates of the proteins. However, this search typically does not involve solving the elusive ‘protein folding problem’, but rather can be restricted to a much more tractable unbound-to-bound conformational transition.
Originally, mostly the high-resolution, experimentally determined (primarily by x-ray crystallography) structures were considered. However, more recently, the focus has been shifting toward lower resolution modeled structures. The correct prediction of the complex does not mean the exact native (cocrystallized) complex per se, which is mathematically/computationally impossible, but rather a near-native approximation.
The general question is: what is the necessary level of structural accuracy for predicted protein complexes? In protein-protein interactions, many experimental and theoretical studies require simple knowledge of the residues at the interfaces (e.g., for further experimental analysis) and have no use for atomic resolution structural details of the complex (specific atom-atom, or even residue-residue contacts across the interface). For the interface (binding site) prediction, the high-resolution protein structures, generally, are not needed. That has been extensively shown by systematic studies over a number of years (15). Still, a high-resolution structure of the complex is required for a number of studies (e.g., for estimation of the binding affinity, certain approaches to inhibition of protein interactions, and such).
Bound and Unbound Docking
The bound docking problem, where the proteins within a cocrystallized complex are separated and redocked by a computational procedure, is a useful tool for the development of new docking approaches, but obviously has no practical value for biology. Docking becomes useful when it is able to predict complexes from the separately determined protein structures (unbound docking), thus becoming a tool for generating new knowledge.
Bound docking is the easiest docking case, because by definition it does not involve conformational change. Thus, the structures match ideally at the interface and the rigid body approach is the only tool required to deliver the correct solution. The bound docking problem has been considered solved for a number of years, in the sense that the existing docking approaches reliably and routinely deliver the near-native structures of the complex among the top predictions.
The approaches to the unbound docking problem have to deal with the conformational difference between the unbound and the bound structures. The change from the unbound to the bound conformation is the basis of the protein’s function in its interactions with other proteins. The intermolecular energy landscapes are characterized by conformational properties of the interacting proteins (19–21). One basic direction in the docking methodology involves coarse-graining (22,23). At lower levels of structural resolution, the difference between unbound and bound conformations is less significant (24,25), and ultimately disappears at ultralow (but still structurally meaningful) resolution (15,24,26). Such approaches allow prediction of the gross features of the complex, due to the large structural recognition factors, and the related funnel in the intermolecular energy landscape (27–29). However, prediction of the higher resolution structural details of interface requires modeling of the structural flexibility, at least at the interface regions.
Still, the majority of protein complexes in the nonredundant benchmark sets have small Cα root mean-square deviation (RMSD) between bound and unbound structures. Indeed, 71% of the Dockground set (30,31) has RMSD between superimposed unbound and bound proteins <2 Å for 71% of the complexes (31). The benchmark set from Weng’s group (32) has unbound/bound interface Cα RMSD (between Cα atoms of the interface residues only) <2.2 Å for 86% of complexes. In a number of cases, when the RMSD is large, the conformational change upon binding is a domain shift. The domains themselves do not undergo a significant conformational change. Thus, this docking still can be addressed by a rigid body approach (33).
Because most docking cases can be resolved by accounting for the flexibility of the surface side chains, the statistics of side-chain conformational changes is important. The results of a systematic large-scale study indicate that short and long side chains have different propensities for the conformational changes (34). Long side chains with three or more dihedral angles are often subject to large conformational transition. Shorter residues with one or two dihedral angles typically undergo local conformational changes not leading to a conformational transition. Most side chains undergo larger changes in the dihedral angle most distant from the backbone. The binding increases both polar and nonpolar interface areas. However, the increase of the nonpolar area is larger, suggesting that the protein association perturbs the unbound interfaces to increase the hydrophobic contribution to the binding free energy (34). Analysis of ensembles of bound and unbound conformations points to conformational selection as the binding mechanism for proteins. The bound and the unbound spectra of conformers also significantly overlap (35). An elastic network model, accounting for the mass distribution, was used to compare the binding site residues fluctuations with other surface residues, showing that, on average, the interface is more rigid (36).
Discretization of the conformational space into rotameric states is useful for the sampling of the conformational space in flexible docking (37,38). Such rotameric libraries for the surface side chains in bound and unbound proteins were generated and used to calculate the probabilities of the rotamer transitions upon binding (38). The stability of amino acids was quantified based on the transition maps. Most side chains changed conformation within the same rotamer or moved to an adjacent rotamer. The highest percentage of the transitions was observed primarily between the two most occupied rotamers (38).
Docking of Models
The docking problem is further complicated if the interacting proteins are models rather than the experimentally determined structures. The errors in such ‘double modeling’ (first of the individual proteins, then of the complex) accumulate, which presents a greater challenge, especially in higher resolution docking (Fig. 2). Thus, the use of approaches to dock these structures should be assessed by thorough benchmarking, specifically designed for protein models (39). To be credible, such benchmarking has to be based on carefully curated sets of structures with levels of distortion typical for modeled proteins. A suite of models was generated for the benchmark set of the x-ray structures from the Dockground resource (http://dockground.bioinformatics.ku.edu) by a combination of homology modeling and the nudged elastic band method (40). For each monomer, six models were generated with predefined Cα RMSD from the native structure (1, 2, …, 6 Å). The sets and the accompanying data provide a comprehensive resource for the development of docking methodology for modeled proteins (41). A new approach, implementing only actual modeling of new protein structures as in the real case scenario, was used by the same group of authors to generate a larger set of models (165 complexes, with full arrays of models for each).
Figure 2.
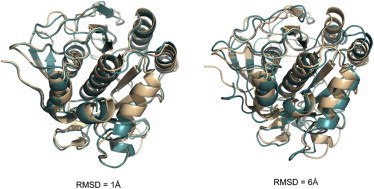
Structures with the increasing level of inaccuracy. The model structures (cyan) are overlapped with the x-ray structure (light brown). To see this figure in color, go online.
Physics Versus Knowledge-Based Docking
Solving the equations of motion for two proteins in arbitrary relative orientation, using atomic resolution force fields, does not dock them in the correct configuration. The reason is the extreme complexity of the energy landscape of the system—its span in multidimensional space of its coordinates and the multiplicity of the energy minima (24,42)—all compounded by the approximate nature of the landscape, with error bars that are often larger than the relative depth of the energy basins.
Still, if one is interested just in the location of the global minimum of the free energy, corresponding to the native structure of the complex, with no regard to the binding pathways, there are nonphysical sampling protocols that efficiently search the landscape and deliver the solution. These protocols treat the problem as global optimization, and find the global minimum through various nonlinear programming techniques, including the most trivial (and effective) one—systematics search (7). The main reason for their success, given the complexity of the landscape, is that the global minimum is significantly different from the local minima. It is not just deeper, as it is supposed to be by definition, but deeper by a significant margin (43), and has a number of other distinguishing characteristics, such as size and ruggedness (28,42). Thus, the unavoidable common approximations of the energy landscapes, although distorting the local minima hierarchy, are not approximate enough to eliminate the difference between the global and the local minima (or at least to remove the actual global minimum from the top candidates). Although such minimization protocols are nonphysical, because the landscape represents physics-based energy (even in its simplest form of steric complementarity, which is none other than the minimum of van der Waals energy) such approaches still pass as physical (often among nonphysicists, and physicists who know biology). Still, in many such approaches, the only physical concept is the trivial steric complementarity, and the rest are techniques borrowed from computer science and other engineering disciplines (pattern recognition, optimization, machine learning, etc.). Such approaches have been dominant in the protein docking field since its inception.
The whole notion of physics though goes out the window altogether with the recent docking approaches based solely on similarity to the existing experimentally determined complexes/templates. If two similar pairs of proteins generally bind in a similar way, and one of them is cocrystallized, for the other pair there may be no need to sample the extremely complex intermolecular landscape—one can simply get straight to the presumed global minimum (the correct structure of the complex) by assuming similarity to the experimentally determined complex.
Structural modeling by similarity (comparative modeling) of individual proteins has been around for a long time, since the establishment of the correlation between sequence and structure similarity in the 80s (44). Such similarity suggested that if the sequence of protein A, the structure of which is to be modeled, is similar to the sequence of protein A′, the structure of which is known, one can put protein A in the same fold as protein A′. That provided a dramatic improvement in terms of prediction reliability over the proverbial ‘protein folding problem’ where the protein structure is supposed to be modeled based on the amino acid sequence alone. The atomic resolution prediction of the protein structure was reduced to the repacking of the side chains, and tweaking of the backbone (often involving flexible loops)—a difficult, but quite tractable task, incomparable with the ultimate complexity of the structure prediction from the sequence alone. The critical aspect of such approach is the availability of the experimentally determined (largely by x-ray crystallography) templates. One can date the emergence of the comparative modeling to the expansion of the PDB that at the time had become large enough to provide a meaningful pool of templates. Currently, with the rapid growth of PDB, the template-based modeling of individual proteins is a dominant approach to the prediction of protein structures (45). The modeling of the folding-related physical processes is still a big challenge, and may well remain so in the future. However, the on-going expansion of PDB will arguably keep further simplifying the nonphysical prediction shortcut to the equilibrium structure, given the limited structural scope of the protein universe (46), which causes the reduction of the pool of proteins with the new fold (45).
In protein-protein docking, the similarity between proteins in complexes can be assessed through comparison/alignment of sequences (47–49), sequences and structures (threading) (50–52), or just the structures (52–58) because the structures of the protein to be docked are assumed to be known by the very definition of docking. However, the protein docking field, as opposed to the prediction of individual proteins, largely has not been taking advantage of the template-based modeling. One reason is that protein-protein docking is younger and thus less advanced (the protein docking community is also significantly smaller than the one in modeling of individual proteins, based on the number of participants in the community-wide prediction assessments (∼200 in CASP vs. ∼40 in CAPRI) and prediction targets (45,59).
Another reason has been the relative success of the traditional template free (ab initio) docking, as opposed to the ab initio modeling of the individual proteins. The rigid-body docking (six degrees of freedom) is a meaningful, working approximation for many complexes, whereas any practical approximation in protein folding involves the conformational search space of far greater dimensionality.
Still, the main reason for the almost complete dominance of the ab initio docking arguably has been the presumed lack of protein-protein templates. Protein-protein complexes are generally more difficult to crystallize than single proteins, limiting the number of templates. Moreover, proteins potentially participate in multiple protein-protein interactions, making the number of protein-protein prediction targets larger than that of the individual proteins. The large-scale efforts to determine the structures of proteins, like the Protein Structure Initiative (60), which established a high-throughput structure determination pipeline, provided a substantial amount of structural information (and a significant portion of prediction targets for the community assessment of structure prediction CASP (45)). However, it has been much less instrumental in supplying the structures of protein-protein complexes (including the lack of targets to the community assessment of predicted interactions CAPRI (59)), presumably because of the relative difficulty of crystallizing protein complexes.
Thus, the general notion in the protein docking field has been that although the comparative docking may be more reliable and accurate than the traditional free docking, similar to the comparative modeling for individual protein modeling, the lack of the templates relegates the practical use of this approach to some future time. That was until the presumption of the lack of templates was actually checked on the existing PDB. The systematic study (61) showed that, surprisingly, docking templates are readily available for complexes representing almost all known protein-protein interactions, provided the components themselves have a known structure or can be homology built.
The study is based on 126,897 protein interactions involving pairs of proteins in 771 species. The structure alignment-based models of complexes were generated by TM-align (62). The structural similarity of two complexes was evaluated by the min TM-scores (the smallest of the receptor and the ligand TM-scores (62)). Fig. 3 shows how the interaction RMSD (47), a measure of the binding mode similarity, correlates with the min TM-score, a measure of the structural similarity between the complexes, in an all-to-all pairwise comparison of 989 cocrystallized complexes. The phase transition occurs near min TM-score = 0.4, with binding modes mostly similar above, and mostly different below.
Figure 3.
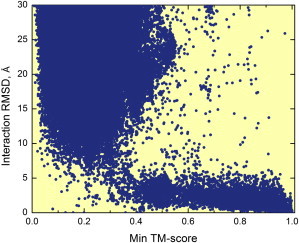
Structural similarity of the binding modes versus structural similarity of the interacting proteins. The structural similarity of the binding modes is described by interaction RMSD (47). The structural similarity of the interacting proteins is described by min TM-score—the lowest of the two components protein TM-scores (62). The sharp transition to small interaction RMSD occurs at the 0.4 value of structural similarity. To see this figure in color, go online.
To assess its predictive value, in the way the prediction would happen in a real case scenario, the approach was tested on newer protein-protein structures in PDB released in 2009–2011, using older template structures released before 2009. Fig. 4 shows the distribution of the interface RMSD (calculated between the ligand native interface in the x-ray and the predicted positions after superimposing receptors), with 36% of the modeled complexes close to the native structure of the complex (interface RMSD <5 Å).
Figure 4.

Benchmarking of template-based docking. The distribution of predicted complexes accuracies is shown relative to the cocrystallized structure. The benchmarking of PDB complexes released in 2009–2011 was based on template structures from 2008 and earlier. In 36% of the complexes, the predicted structure is close to the native (interface RMSD <5 Å). To see this figure in color, go online.
To determine the extent to which template-based docking can be used to model known protein interactions in whole genomes, homology models of individual proteins were built, and templates of their complexes searched for in PDB. Remarkably, structural templates were found for nearly all (99%) the complexes in which the structure of both components is known experimentally or could be built by homology (Fig. 5). As shown in Fig. 4 more than one-third of them are estimated to be correct. Thus, when no template was found for the complex, there were no templates to build models for one or both of its components. The coverage should therefore improve as more individual proteins have their structure determined.
Figure 5.
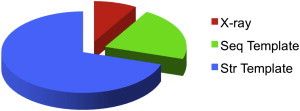
Structural coverage of protein interactions for five genomes with the largest number of known interactions. Complexes with the x-ray structure are in red, complexes with a sequence template are in green, and complexes for which the structure of the monomers is known are in blue—structural templates are found for ∼100% of such complexes. >1/3 of these templates are estimated to be correct (Fig. 4). To see this figure in color, go online.
Community-Wide Evaluation
When a research group develops a new docking approach and shows that it successfully predicts the structure of a protein-protein complex, the true value of this contribution to biological science is still unknown. To be properly evaluated, the approach has to be a), successful enough for all protein-protein complexes with known structure (or for a certain class of protein complexes, if it is specific to, e.g., antigen-antibody or enzyme-inhibitor complexes); and b), objectively compared to other existing approaches. It may not be always realistic to have both (a) and (b) evaluations at the same time, but they are the essential parts of an objective assessment. For practical reasons, and to avoid overrepresentation of certain complexes in PDB, which may not reflect their frequency in vivo, the (a) part most often is performed on a statistically significant nonredundant, representative subset of complexes (docking benchmark sets). The (b) part is performed by comparison of the performance of different approaches on these benchmark sets and in blind community-wide assessments.
The protein-docking community began to organize and actively develop such community-wide activities at the First Conference on Modeling of Protein Interactions (MPI) at Charleston, SC, 2001 (63). These activities were further developed at the subsequent MPI (http://www.bioinformatics.ku.edu/mpi-conference) and CAPRI (http://capri.ebi.ac.uk) conferences and other meetings.
The widely used benchmark sets of protein-protein complexes were developed in Weng’s group (32) and Vakser’s group (31). Both contain more than a hundred complexes of cocrystallized proteins and their separately crystallized components (unbound structures). The idea behind the benchmark is to provide pairs of unbound structures with the correct match known (cocrystallized complex) for the development of unbound docking algorithms.
Several groups put together decoy sets of protein-protein complexes. These sets contain false positive matches of proteins and are useful in the development of better potentials and scoring functions for the discrimination of false positive predictions (thus they are sometimes called scoring benchmarks). An early set of protein-protein docking decoys was compiled in Vakser’s group in the 90s. The current version is based on the Dockground resource (64). Other decoy sets are available from Weng’s group (http://zdock.umassmed.edu/software), and others. Fig. 6 shows an example of the docking decoys from the Dockground set for one complex.
Figure 6.
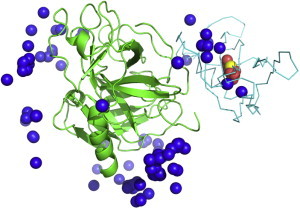
Example of docking decoys. Matches represented by the ligand’s center of mass are shown for 1bui enzyme-substrate complex. The receptor (in green) and the ligand (in cyan) are shown in cocrystallized configuration. The native match is in yellow, 10 near-native matches are in red, and 100 nonnative matches are in blue. To see this figure in color, go online.
The community-wide experiments on Critical Assessment of Structure Prediction (CASP; http://predictioncenter.org) and Critical Assessment of PRedicted Interactions (CAPRI; http://capri.ebi.ac.uk) allow a comparison of different computational methods on a set of prediction targets (experimentally determined structures unknown to the predictors). The protein-protein docking category was introduced at CASP2 (43,65) and has been successfully continued in CAPRI. CAPRI solicits yet unpublished structures of cocrystallized protein-protein complexes from experimentalists (primarily, x-ray crystallographers) and distributes the separately crystallized structures of the components, or their homologs, to the predictors’ community. The participants are groups of researchers (who often use available biological information on the targets to narrow down the docking search) and automated servers. A separate prediction category is scoring of the docked complexes. CAPRI is conducted on a continuing basis (currently in its 30th round), upon the availability of the targets. CAPRI generated great interest in the scientific community and has led to significant progress in the docking methodologies.
Large-Scale Docking
Most challenging docking tasks address large-scale (up to the level of the entire genome) modeling of protein interaction networks (52,66–69), where most proteins have to be models themselves, and the docking has to be performed by high-throughput (and thus less accurate) approaches (67). The approaches to genome-wide structural modeling of protein interactions are either traditional free docking (70,71) or the template-based docking (54,61,72). Modeling templates are available for a significant part of soluble proteins in genomes (73), including those in known protein interactions (61,74). For systematic evaluation of potential accuracy in high-throughput modeling of binding sites, a statistical analysis of target-template sequence alignments was performed for a representative set of protein complexes (75). For most of the complexes, alignments containing all residues of the interface were found. The full interface alignments were obtained even in the case of poor overall alignments where a relatively small part of the target sequence (as low as 40%) aligned to the template sequence, with a low overall alignment identity (<30%). Although such poor overall alignments might be considered inadequate for modeling of whole proteins, the alignment of the interfaces was strong enough for docking. Overall, about half of the complexes with the interfaces modeled by high-throughput techniques had accuracy suitable for meaningful docking. This percentage should grow with the increasing availability of cocrystallized protein-protein complexes.
Challenges
Determination of the static protein interactome, restricted to the most energetically stable (and potentially crystallizable) equilibrium configurations of the complexes, is already within reach, at least at the low-resolution, first-pass approximation. Protein complexes are more difficult to crystallize than the individual proteins, and thus are less represented in PDB. However, the structural diversity of the stable protein interactions is limited to such an extent, that it is already covered by the current PDB, for interacting proteins with known structures (61). High-quality (according to an unbiased a priori evaluation) structural templates are available for virtually all such complexes, and more than one-third of them reproduce the cocrystallized targets in comprehensive benchmarking. Leaving aside speculations that the remaining two-thirds, or some part of it, may correspond to the noncrystallizable (or not crystallized yet) interacting modes of the given proteins, one can safely assume that with further growth of PDB the current one-third portion will grow as well. Based on the current statistics, one can also extrapolate that for most of the newly determined (experimentally or by homology) protein structures, their interactions already will have had structural homologs in PDB. Thus, the remaining challenge for the determination of the static interactome is a), determination of the interactors (both the fact that protein A interacts with protein B, and their respective structures—currently 85% of all interacting proteins in Escherichia coli and 39% in yeast (61)), rather than the structure of the interaction; and b), the refinement of the interaction to the atomic resolution, when needed.
These challenges will have to be addressed in the coming years. However, conceptually, in terms of the broader picture, the focus is inevitably shifting toward more dynamic and realistic representation of the protein interactome in vivo, beyond the cocrystallizable subset. Experimental techniques other than x-ray crystallography, such as nuclear magnetic resonance spectroscopy, electron microscopy, and a number of low-resolution methodologies (76) will play an increasing role in providing directions and constraints for the modeling. The new frontier for structural modeling is the cell itself, including molecular association and dissociation rates and molecular crowding (77–79), and other aspects of biomolecular interactions in cellular environment, involving myriads of intermolecular encounters (80) and conformational changes associated with them.
Acknowledgments
The author is grateful to Petras Kundrotas and other co-workers and colleagues for discussions and results that provided the foundation for his current understanding of the protein docking field.
The support from National Institutes of Health (NIH) grant R01GM074255 and National Science Foundation (NSF) grant DBI1262621 is acknowledged.
References
- 1.Platzer K.E.B., Momany F.A., Scheraga H.A. Conformational energy calculations of enzyme-substrate interactions. II. Computation of the binding energy for substrates in the active site of alpha-chymotrypsin. Int. J. Pept. Protein Res. 1972;4:201–219. doi: 10.1111/j.1399-3011.1972.tb03420.x. [DOI] [PubMed] [Google Scholar]
- 2.Pincus M.R., Zimmerman S.S., Scheraga H.A. Prediction of three-dimensional structures of enzyme-substrate and enzyme-inhibitor complexes of lysozyme. Proc. Natl. Acad. Sci. USA. 1976;73:4261–4265. doi: 10.1073/pnas.73.12.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wodak S.Y., Liu M.Y., Wyckoff H.W. The structure of cytidilyl(2′,5′)adenosine when bound to pancreatic ribonuclease S. J. Mol. Biol. 1977;116:855–875. doi: 10.1016/0022-2836(77)90275-3. [DOI] [PubMed] [Google Scholar]
- 4.Pincus M.R., Scheraga H.A. Conformational energy calculations of enzyme-substrate and enzyme-inhibitor complexes of lysozyme. 2. Calculation of the structures of complexes with flexible enzyme. Macromolecules. 1979;12:633–644. [Google Scholar]
- 5.Wodak S.J., Janin J. Computer analysis of protein-protein interaction. J. Mol. Biol. 1978;124:323–342. doi: 10.1016/0022-2836(78)90302-9. [DOI] [PubMed] [Google Scholar]
- 6.Greer J., Bush B.L. Macromolecular shape and surface maps by solvent exclusion. Proc. Natl. Acad. Sci. USA. 1978;75:303–307. doi: 10.1073/pnas.75.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katchalski-Katzir E., Shariv I., Vakser I.A. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer D., Norel R., Nussinov R. Surface motifs by a computer vision technique: searches, detection, and implications for protein-ligand recognition. Proteins. 1993;16:278–292. doi: 10.1002/prot.340160306. [DOI] [PubMed] [Google Scholar]
- 9.Vajda S., Hall D.R., Kozakov D. Sampling and scoring: a marriage made in heaven. Proteins. 2013;81:1874–1884. doi: 10.1002/prot.24343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vakser I.A., Aflalo C. Hydrophobic docking: a proposed enhancement to molecular recognition techniques. Proteins. 1994;20:320–329. doi: 10.1002/prot.340200405. [DOI] [PubMed] [Google Scholar]
- 11.Mandell J.G., Roberts V.A., Ten Eyck L.F. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001;14:105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 12.Kozakov D., Brenke R., Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 13.Mintseris J., Pierce B., Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins. 2007;69:511–520. doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 14.Vakser I.A. Main-chain complementarity in protein-protein recognition. Protein Eng. 1996;9:741–744. doi: 10.1093/protein/9.9.741. [DOI] [PubMed] [Google Scholar]
- 15.Vakser I.A., Matar O.G., Lam C.F. A systematic study of low-resolution recognition in protein-protein complexes. Proc. Natl. Acad. Sci. USA. 1999;96:8477–8482. doi: 10.1073/pnas.96.15.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicola G., Vakser I.A. A simple shape characteristic of protein-protein recognition. Bioinformatics. 2007;23:789–792. doi: 10.1093/bioinformatics/btm018. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q., Sanner M., Olson A.J. Shape complementarity of protein-protein complexes at multiple resolutions. Proteins. 2009;75:453–467. doi: 10.1002/prot.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kundrotas P.J., Vakser I.A. Protein-protein alternative binding modes do not overlap. Protein Sci. 2013;22:1141–1145. doi: 10.1002/pro.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trizac E., Levy Y., Wolynes P.G. Capillarity theory for the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 2010;107:2746–2750. doi: 10.1073/pnas.0914727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J., Faeder J.R., Camacho C.J. Toward a quantitative theory of intrinsically disordered proteins and their function. Proc. Natl. Acad. Sci. USA. 2009;106:19819–19823. doi: 10.1073/pnas.0907710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu X., Gan L., Wang J. Quantifying the topography of the intrinsic energy landscape of flexible biomolecular recognition. Proc. Natl. Acad. Sci. USA. 2013;110:E2342–E2351. doi: 10.1073/pnas.1220699110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saunders M.G., Voth G.A. Coarse-graining of multiprotein assemblies. Curr. Opin. Struct. Biol. 2012;22:144–150. doi: 10.1016/j.sbi.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Baaden M., Marrink S.J. Coarse-grain modelling of protein-protein interactions. Curr. Opin. Struct. Biol. 2013;23:878–886. doi: 10.1016/j.sbi.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Vakser I.A. Long-distance potentials: an approach to the multiple-minima problem in ligand-receptor interaction. Protein Eng. 1996;9:37–41. doi: 10.1093/protein/9.1.37. [DOI] [PubMed] [Google Scholar]
- 25.Gray J.J., Moughon S., Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 26.Vakser I.A. Protein docking for low-resolution structures. Protein Eng. 1995;8:371–377. doi: 10.1093/protein/8.4.371. [DOI] [PubMed] [Google Scholar]
- 27.Tovchigrechko A., Vakser I.A. How common is the funnel-like energy landscape in protein-protein interactions? Protein Sci. 2001;10:1572–1583. doi: 10.1110/ps.8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Toole N., Vakser I.A. Large-scale characteristics of the energy landscape in protein-protein interactions. Proteins. 2008;71:144–152. doi: 10.1002/prot.21665. [DOI] [PubMed] [Google Scholar]
- 29.Hunjan J., Tovchigrechko A., Vakser I.A. The size of the intermolecular energy funnel in protein-protein interactions. Proteins. 2008;72:344–352. doi: 10.1002/prot.21930. [DOI] [PubMed] [Google Scholar]
- 30.Douguet D., Chen H.C., Vakser I.A. DOCKGROUND resource for studying protein-protein interfaces. Bioinformatics. 2006;22:2612–2618. doi: 10.1093/bioinformatics/btl447. [DOI] [PubMed] [Google Scholar]
- 31.Gao Y., Douguet D., Vakser I.A. DOCKGROUND system of databases for protein recognition studies: unbound structures for docking. Proteins. 2007;69:845–851. doi: 10.1002/prot.21714. [DOI] [PubMed] [Google Scholar]
- 32.Hwang H., Vreven T., Weng Z. Protein-protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emekli U., Schneidman-Duhovny D., Haliloglu T. HingeProt: automated prediction of hinges in protein structures. Proteins. 2008;70:1219–1227. doi: 10.1002/prot.21613. [DOI] [PubMed] [Google Scholar]
- 34.Ruvinsky A.M., Kirys T., Vakser I.A. Side-chain conformational changes upon Protein-Protein Association. J. Mol. Biol. 2011;408:356–365. doi: 10.1016/j.jmb.2011.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruvinsky A.M., Kirys T., Vakser I.A. Ensemble-based characterization of unbound and bound states on protein energy landscape. Protein Sci. 2013;22:734–744. doi: 10.1002/pro.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruvinsky A.M., Vakser I.A. Sequence composition and environment effects on residue fluctuations in protein structures. J. Chem. Phys. 2010;133:155101. doi: 10.1063/1.3498743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beglov D., Hall D.R., Vajda S. Minimal ensembles of side chain conformers for modeling protein-protein interactions. Proteins. 2011;80:591–601. doi: 10.1002/prot.23222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirys T., Ruvinsky A.M., Vakser I.A. Rotamer libraries and probabilities of transition between rotamers for the side chains in protein-protein binding. Proteins. 2012;80:2089–2098. doi: 10.1002/prot.24103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tovchigrechko A., Wells C.A., Vakser I.A. Docking of protein models. Protein Sci. 2002;11:1888–1896. doi: 10.1110/ps.4730102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elber R., Karplus M. A method for determining reaction paths in large molecules - application to myoglobin. Chem. Phys. Lett. 1987;139:375–380. [Google Scholar]
- 41.Anishchenko I., Kundrotas P.J., Vakser I.A. Protein models: the Grand Challenge of protein docking. Proteins. 2014;82:278–287. doi: 10.1002/prot.24385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruvinsky A.M., Vakser I.A. Chasing funnels on protein-protein energy landscapes at different resolutions. Biophys. J. 2008;95:2150–2159. doi: 10.1529/biophysj.108.132977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vakser I.A. Evaluation of GRAMM low-resolution docking methodology on the hemagglutinin-antibody complex. Proteins. 1997;(Suppl 1):226–230. [PubMed] [Google Scholar]
- 44.Sánchez R., Sali A. Advances in comparative protein-structure modelling. Curr. Opin. Struct. Biol. 1997;7:206–214. doi: 10.1016/s0959-440x(97)80027-9. [DOI] [PubMed] [Google Scholar]
- 45.Moult J., Fidelis K., Tramontano A. Critical assessment of methods of protein structure prediction (CASP)–round x. Proteins. 2014;82(Suppl 2):1–6. doi: 10.1002/prot.24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y., Hubner I.A., Skolnick J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. USA. 2006;103:2605–2610. doi: 10.1073/pnas.0509379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aloy P., Ceulemans H., Russell R.B. The relationship between sequence and interaction divergence in proteins. J. Mol. Biol. 2003;332:989–998. doi: 10.1016/j.jmb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 48.Kundrotas P.J., Lensink M.F., Alexov E. Homology-based modeling of 3D structures of protein-protein complexes using alignments of modified sequence profiles. Int. J. Biol. Macromol. 2008;43:198–208. doi: 10.1016/j.ijbiomac.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Rodrigues J.P.G.L.M., Melquiond A.S.J., Bonvin A.M.J.J. Defining the limits of homology modeling in information-driven protein docking. Proteins. 2013;81:2119–2128. doi: 10.1002/prot.24382. [DOI] [PubMed] [Google Scholar]
- 50.Lu L., Lu H., Skolnick J. MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins. 2002;49:350–364. doi: 10.1002/prot.10222. [DOI] [PubMed] [Google Scholar]
- 51.Guerler A., Govindarajoo B., Zhang Y. Mapping monomeric threading to protein-protein structure prediction. J. Chem. Inf. Model. 2013;53:717–725. doi: 10.1021/ci300579r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szilagyi A., Zhang Y. Template-based structure modeling of protein-protein interactions. Curr. Opin. Struct. Biol. 2014;24:10–23. doi: 10.1016/j.sbi.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Günther S., May P., Preissner R. Docking without docking: ISEARCH–prediction of interactions using known interfaces. Proteins. 2007;69:839–844. doi: 10.1002/prot.21746. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q.C., Petrey D., Honig B. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghoorah A.W., Devignes M.D., Ritchie D.W. Spatial clustering of protein binding sites for template based protein docking. Bioinformatics. 2011;27:2820–2827. doi: 10.1093/bioinformatics/btr493. [DOI] [PubMed] [Google Scholar]
- 56.Tuncbag N., Keskin O., Gursoy A. Fast and accurate modeling of protein-protein interactions by combining template-interface-based docking with flexible refinement. Proteins. 2012;80:1239–1249. doi: 10.1002/prot.24022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinha R., Kundrotas P.J., Vakser I.A. Docking by structural similarity at protein-protein interfaces. Proteins. 2010;78:3235–3241. doi: 10.1002/prot.22812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kundrotas P.J., Vakser I.A. Global and local structural similarity in protein-protein complexes: implications for template-based docking. Proteins. 2013;81:2137–2142. doi: 10.1002/prot.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lensink M.F., Wodak S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins. 2013;81:2082–2095. doi: 10.1002/prot.24428. [DOI] [PubMed] [Google Scholar]
- 60.Khafizov K., Madrid-Aliste C., Fiser A. Trends in structural coverage of the protein universe and the impact of the Protein Structure Initiative. Proc. Natl. Acad. Sci. USA. 2014;111:3733–3738. doi: 10.1073/pnas.1321614111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kundrotas P.J., Zhu Z., Vakser I.A. Templates are available to model nearly all complexes of structurally characterized proteins. Proc. Natl. Acad. Sci. USA. 2012;109:9438–9441. doi: 10.1073/pnas.1200678109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vajda S., Vakser I.A., Janin J. Modeling of protein interactions in genomes. Proteins. 2002;47:444–446. doi: 10.1002/prot.10112. [DOI] [PubMed] [Google Scholar]
- 64.Liu S., Gao Y., Vakser I.A. DOCKGROUND protein-protein docking decoy set. Bioinformatics. 2008;24:2634–2635. doi: 10.1093/bioinformatics/btn497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dixon J.S. Evaluation of the CASP2 docking section. Proteins. 1997;(Suppl 1):198–204. doi: 10.1002/(sici)1097-0134(1997)1+<198::aid-prot26>3.3.co;2-d. [DOI] [PubMed] [Google Scholar]
- 66.Wass M.N., David A., Sternberg M.J.E. Challenges for the prediction of macromolecular interactions. Curr. Opin. Struct. Biol. 2011;21:382–390. doi: 10.1016/j.sbi.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 67.Vakser I.A. Low-resolution structural modeling of protein interactome. Curr. Opin. Struct. Biol. 2013;23:198–205. doi: 10.1016/j.sbi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mosca R., Pons T., Aloy P. Towards a detailed atlas of protein-protein interactions. Curr. Opin. Struct. Biol. 2013;23:929–940. doi: 10.1016/j.sbi.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 69.Wodak S.J., Vlasblom J., Pu S. Protein-protein interaction networks: the puzzling riches. Curr. Opin. Struct. Biol. 2013;23:941–953. doi: 10.1016/j.sbi.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 70.Zhu Z., Tovchigrechko A., Vakser I.A. Large-scale structural modeling of protein complexes at low resolution. J. Bioinform. Comput. Biol. 2008;6:789–810. doi: 10.1142/s0219720008003679. [DOI] [PubMed] [Google Scholar]
- 71.Mosca R., Pons C., Aloy P. Pushing structural information into the yeast interactome by high-throughput protein docking experiments. PLOS Comput. Biol. 2009;5:e1000490. doi: 10.1371/journal.pcbi.1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kar G., Keskin O., Gursoy A. Human proteome-scale structural modeling of E2-E3 interactions exploiting interface motifs. J. Proteome Res. 2012;11:1196–1207. doi: 10.1021/pr2009143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levitt M. Nature of the protein universe. Proc. Natl. Acad. Sci. USA. 2009;106:11079–11084. doi: 10.1073/pnas.0905029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kundrotas P.J., Vakser I.A., Janin J. Structural templates for modeling homodimers. Protein Sci. 2013;22:1655–1663. doi: 10.1002/pro.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kundrotas P.J., Vakser I.A. Accuracy of protein-protein binding sites in high-throughput template-based modeling. PLOS Comput. Biol. 2010;6:e1000727. doi: 10.1371/journal.pcbi.1000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Russell R.B., Alber F., Sali A. A structural perspective on protein-protein interactions. Curr. Opin. Struct. Biol. 2004;14:313–324. doi: 10.1016/j.sbi.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 77.Zhou H.X., Bates P.A. Modeling protein association mechanisms and kinetics. Curr. Opin. Struct. Biol. 2013;23:887–893. doi: 10.1016/j.sbi.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kastritis P.L., Bonvin A.M.J.J. Molecular origins of binding affinity: seeking the Archimedean point. Curr. Opin. Struct. Biol. 2013;23:868–877. doi: 10.1016/j.sbi.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 79.Feig M., Sugita Y. Variable interactions between protein crowders and biomolecular solutes are important in understanding cellular crowding. J. Phys. Chem. B. 2012;116:599–605. doi: 10.1021/jp209302e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kozakov D., Li K., Vajda S. Encounter complexes and dimensionality reduction in protein-protein association. eLife. 2014;3:e01370. doi: 10.7554/eLife.01370. [DOI] [PMC free article] [PubMed] [Google Scholar]