

Abstract
Protein kinase C (PKC) family members phosphorylate a wide variety of protein targets and are known to be involved in diverse cellular signaling pathways. However, the role of PKC in receptor activator of NF-κB ligand (RANKL) signaling has remained elusive. We now demonstrate that PKCβ acts as a positive regulator which inactivates glycogen synthase kinase-3β (GSK-3β) and promotes NFATc1 induction during RANKL-induced osteoclastogenesis. Among PKCs, PKCβ expression is increased by RANKL. Pharmacological inhibition of PKCβ decreased the formation of osteoclasts which was caused by the inhibition of NFATc1 induction. Importantly, the phosphorylation of GSK-3β was decreased by PKCβ inhibition. Likewise, down-regulation of PKCβ by RNA interference suppressed osteoclast differentiation, NFATc1 induction, and GSK-3β phosphorylation. The administration of PKC inhibitor to the RANKL-injected mouse calvaria efficiently protected RANKL-induced bone destruction. Thus, the PKCβ pathway, leading to GSK-3β inactivation and NFATc1 induction, has a key role in the differentiation of osteoclasts. Our results also provide a further rationale for PKCβ’s therapeutic targeting to treat inflammation-related bone diseases.
Keywords: glycogen synthase kinase-3β, osteoclast differentiation, protein kinase Cβ, receptor activator of NF-κB ligand
INTRODUCTION
Osteoclasts are the cells responsible for bone resorption during bone remodeling along with bone-forming osteoblasts and the fine balance between the activities of these two cells is essential for maintaining bone homeostasis (Boyle et al., 2003). Bone homeostasis is delicately regulated by various factors (Takayanagi, 2007), and an imbalance in bone remodeling can result in a variety of bone diseases in humans, such as osteoporosis and rheumatoid arthritis as well they play an essential role in the pathomechanism of multiple myeloma and bone metastasis of tumors.
Differentiation of osteoclasts is supported by two essential cytokines, macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL) (Anderson et al., 1997; Lacey et al., 1998; Suda et al., 1999; Wong et al., 1997; Yasuda et al., 1998). Binding of RANKL to its receptor RANK on the surface of precursor cells triggers formation of RANK-TRAF6 (TNF-receptor associated factor 6) complex, and this leads to activation of NF-κB and mitogen-activated protein kinases, including Jun N-terminal kinase and p38 (Kobayashi et al., 2001). The strong induction of nuclear factor of activated T cells, cytoplasmic 1 (NFATc1) which is a master transcription factor of osteoclast differentiation (Takayanagi et al., 2002) is required in osteoclastogenesis and this induction is dependent on both the TRAF6-NF-κB and the c-Fos pathways. Activated NFATc1 induces expression of many osteoclast-specific genes including TRAP, Cathepsin K, Oscar, and Atp6v0d2 (Crabtree and Olson, 2002; Hogan et al., 2003). Activity of NFAT members is regulated by its subcellular localization and nuclear export of NFAT members facilitated by phosphorylation. We have reported that glycogen synthase kinase-3β (GSK-3β) is inactivated by RANKL to induce NFATc1 expression and activation (Jang et al., 2011).
Protein kinase C (PKC) is expressed ubiquitously in many different cell types, where it regulates a variety of cellular processes that impact cell growth and differentiation, cytoskeletal remodeling, and gene expression in response to diverse stimuli. The PKC family comprises at least 10 mammalian isozymes of serine/threonine protein kinases (Saito et al., 2002) that can be grouped into three classes according to the presence or absence of motifs that dictate cofactor requirements for optimal catalytic activity (Steinberg, 2008). GSK-3β is intensively investigated serine/threonine kinase that phosphorylates and inactivates glycogen synthase in response to insulin stimulation (Lee and Kim, 2007). Extracellular stimuli promote GSK-3 inactivation by phosphorylating at the inhibitory serine residue (Ser9 for GSK-3β). A role for some PKC isoforms in GSK-3 inhibition has also been demonstrated (Fang et al., 2002), and Wang et al. showed downregulation of PKCβ1 by prolonged phorbolmyristic acid treatment in human colon cancer cells blocked neurotensin-mediated GSK-3β phosphorylation (Wang et al., 2006b). PKCβ was shown to be involved in regulation of osteoclast formation and function by participating in ERK signaling pathway of M-CSF and RANKL (Lee et al., 2003). However, whether PKCβ-GSK3β axis is involved in RANKL signaling is unknown.
In this study, we investigated whether PKCβ plays a regulatory role by inactivating GSK-3β during RANKL-induced osteoclastogenesis. Interestingly, PKCβ inhibition showed a strong inhibitory effect on osteoclast differentiation and GSK-3β phosphorylation in RANKL-stimulated osteoclast precursors. We also show that the inhibition of PKCβ may provide a molecular basis for a new therapeutic approach to bone disorders.
MATERIALS AND METHODS
Cells and reagents
Primary bone marrow macrophages (BMMs) were generated from murine bone marrow precursors of 4-6-week-old male C57BL/6 mice (The Jackson Laboratory, USA) and cultured in α-minimum essential medium (α-MEM; HyClone, USA) supplemented with 10% fetal bovine serum (FBS) and antibiotics. Retrovirus packaging cell line, PLAT-E cells (kindly gifted from T. Kitamura, University of Tokyo, Japan) and macrophage cell line, RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; HyClone) with 10% FBS and antibiotics. Gö6976 was purchased from Sigma (USA) and GF109203X and Rottlerin were obtained from Biomol (USA).
Protein analysis and fractionation
Equal amount of cell lysates harvested under the indicated conditions were subjected to Western blot analysis using specific antibodies against NFATc1, PKCα, PKCβ, tubulin, β-actin (Santa Cruz Biotechnology, USA), GSK-3β, phospho-GSK-3β, phospho-PKCβ (Cell Signaling Technology, USA), TATA-binding protein (TBP) (Abcam, UK), and GAPDH (Ab Frontier, Korea). Anti-Atp6v0d2 antibody was kindly provided by Y. Choi (University of Pennsylvania, USA). Harvested cells were fractionated using Nuclear and Cytoplasmic Extraction Reagent (Pierce, USA) according to the manufacturer’s protocol.
In vitro osteoclast differentiation
Osteoclasts were prepared from bone marrow cells using a standard method (Kim et al., 2009; Suda et al., 1997). In brief, bone marrow cells were cultured with 30 ng/ml M-CSF for 3 days to obtain osteoclast precursor cells of the monocyte/macrophage lineage. The precursors were cultured with 30 ng/ml M-CSF and 100 ng/ml RANKL for the indicated time periods. The PKC inhibitors including GF109203X, Gö6976, and Rottlerin were added at the time of RANKL addition. The osteoclast marker tartrate-resistant acid phosphatase-positive (TRAP+) cells with more than three nuclei or cells larger than 100 μm in diameter containing more than 20 nuclei were counted as TRAP+ multinucleated cells (TRAP+ MNCs).
Transfection and reporter assay
RAW264.7 cells were seeded at a density of 105 cells per well in a 12-well plate 1 day prior to transfection using LipofectamineTM2000 (Invitrogen, USA) according to the manufacturer’s protocol. RAW264.7 cells were cultured with or without RANKL (400 ng/ml) and Gö6976 for 2 days after transfection as indicated. Luciferase reporter constructs driven by the NFAT-responsive elements were described previously (Kim et al., 2010). Luciferase activity was measured by the dual luciferase reporter assay system (Promega, USA) according to the manufacturer’s protocol and divided by the Renilla luciferase activity (control reporter) to normalize for transfection efficiency. Data were obtained from three independent transfections and presented as fold induction in luciferase activity [mean ± standard deviation (SD)] relative to the control.
RNA interference and retroviral infection
Custom SMARTpool plus small interfering RNA (siRNA) to target mouse PKCα (catalog no. M-040348) was designed and synthesized by Dharmacon (Lafayette, USA). A pool of three target-specific siRNA for mouse PKCβ was purchased from Santa Cruz Biotechnology (sc-36255). siRNAs (20 nmol) were transfected into BMMs using LipofectamineTM2000 (Invitrogen) according to the manufacturer’s protocol. After transfection, BMMs were cultured with M-CSF and RANKL for 3–4 days and then harvested for protein analysis or differentiated into osteoclasts. RNA interference oligonucleotides targeting mouse PKCβ (5′-TCTGCTGCTTTGTTGTACA-3′) (Zhu et al., 2010) were synthesized by Bioneer (Korea) and cloned into the retroviral short-hairpin RNA (shRNA) vector pSuper-retro Puro (OligoEngine, USA) following the manufacturer’s protocol. The plasmids were transfected into PLAT-E cells using LipofectamineTM2000, and the supernatant was collected 24–36 h after transfection. BMMs were cultured with viral supernatant for 6 h, and then changed to media with M-CSF and puromycin (2 μg/ml) for 2 days. Puromycin-resistant BMMs were cultured with M-CSF and RANKL for 3–4 days to generate osteoclasts.
Real-time polymerase chain reaction (real time-PCR)
Total RNA was isolated from cells using TRIzol Reagent (Invitrogen), and cDNA was synthesized by reverse transcriptase (Invitrogen). To quantify gene expression real-time PCR was performed on an ABI Prism 7000 sequence detection system (Applied Biosystems, USA) using KAPA SYBR FAST ABI Prism qPCR kit (KapaBiosystems, USA) according to the manufacturer’s instructions. All reactions were run in triplicate and the mRNA copy number of a specific gene in total RNA was calculated and normalized to total RNA with GAPDH as the internal control. Primer sequences are listed in Supplementary Table S1.
RANKL-induced bone loss
PBS or RANKL (2 mg/kg) in a volume of 50 μl was injected subperiosteally in the midline calvaria of 6-week-old male mice at 2-day intervals for 6 days. Gö6976 was injected daily for 5 consecutive days starting from 1 day after the first RANKL administration. On day 7, osteoclast number per millimeter of trabecular bone surface, and the percentage of bone surface covered by osteoclasts (eroded surface) were measured as described previously (Choi et al., 2013; Takayanagi et al., 2000). All mouse experiments were performed under an animal protocol approved by the Animal Care Committee of Ewha Laboratory Animal Genomics Center.
Statistical analysis
Data are expressed as mean ± SD from at least three independent experiments. Statistical analyses were performed using the two-tailed Student’s t-test to analyze differences among groups. P < 0.05 was considered statistically significant.
RESULTS
Effects of PKC inhibitors on osteoclast formation
We first investigated the effects of pharmacologically inhibiting PKCs on osteoclastogenesis. BMMs were stimulated with recombinant RANKL and M-CSF, and the formation of TRAP+ MNCs was analyzed. Inhibition of PKCs by the general PKC inhibitor GF109203X or PKCα/β inhibitor Gö6976, but not the PKCδ inhibitor Rottlerin, effectively suppressed formation of TRAP+ MNCs in a dose-dependent manner (Figs. 1A and 1B), without a marked effect on cell viability (data not shown). These results suggest the importance of PKC family members such as PKCα and/or PKCβ in the differentiation process.
Fig. 1.

Pharmacological inhibition of PKCα and β suppresses osteoclast differentiation and NFATc1 induction. (A) In vitro differentiation of osteoclasts with pharmacological inhibitors of PKCs. BMMs were cultured for 3 days with M-CSF and RANKL in the presence of GF109302X, Gö6976, and Rottlerin, respectively. TRAP staining was performed. Scale bar, 200 μm. (B) TRAP+ MNCs with more than 3 nuclei were counted. Data represent means ± S.D. **P < 0.001 vs. positive control (RANKL). (C) Effect of Gö6976 on NFATc1 induction by RANKL stimulation. BMMs were cultured for 2 days with M-CSF and RANKL in the presence of 500 nM Gö6976. Fractionated nuclear and cytoplasmic lysates were analyzed by Western blotting with specific antibodies as indicated. Anti-tubulin and anti-TBP were used as the loading control for cytoplasmic and nuclear extracts. (D) Inhibition of NFATc1 transcriptional activity by Gö6976. RAW264.7 cells were transfected with an NFATc1 reporter construct and cultured with RANKL (400 ng/ml) and Gö6976 as indicated for 2 days, and then the transcriptional activity of NFATc1 was measured. The results shown are the mean of relative luciferase activity ± S.D. of at least three independent experiments. *P < 0.01 vs. positive control. n.s., not significant.
Inhibiting PKCα/β blocks NFATc1 induction and activation by RANKL
To better understand the role of PKCα and PKCβ in osteoclast differentiation, we further assessed the effect of PKCα/β inhibition on NFATc1 induction in response to RANKL. Notably, the PKCα/β inhibitor Gö6976 significantly decreased NFATc1 level induced by RANKL both in the cytosol and nucleus compared to that of the control (Fig. 1C). We also examined NFAT transcriptional activity with PKCα/β inhibition. For this, a reporter assay with a luciferase reporter plasmid driven by tandem NFAT binding sites in RAW 264.7 cells was performed. Cells treated with Gö6976 showed remarkably downregulated NFAT transcriptional activity in a dose-dependent manner, whereas control cells effectively elevated NFAT transcriptional activity caused by RANKL stimulation (Fig. 1D). These results suggest that PKCα/β may play a role as a positive regulator to induce and activate NFATc1, which is critical for RANKL-induced osteoclastogenesis.
PKCβ but not PKCα is involved in osteoclast differentiation
To investigate a specific isoform necessary for RANKL-mediated osteoclast differentiation, we downregulated PKCα or PKCβ using RNA interference. Primary BMMs were transfected with siRNA targeting PKCα or infected with retrovirus expressing short-hairpin RNA targeting PKCβ. Notably, inhibiting PKCβ by RNA interference significantly suppressed osteoclast formation (Fig. 2A), whereas downregulating PKCα did not change osteoclast formation (Fig. 2B), suggesting that PKCβ is critical for osteoclast differentiation.
Fig. 2.

PKCβ is positively regulates osteoclast formation. (A) Effect of siRNA against PKCα on RANKL-induced osteoclastogenesis. Nonspecific duplex was used as the negative control (s.c). Knockdown of PKCα was confirmed by Western blot analysis. (B) As in (A), except that BMMs were infected with shRNA specifically targeting PKCβ. PKCβ knockdown was confirmed by Western blot analysis. Relative band intensities of PKC and PKC were quantified and normalized based on the intensities of GAPDH and are indicated below each set of blots (in arbitrary unit). TRAP+ MNCs with more than 3 nuclei were counted. Data represent means ± S.D. *P <0.001. n.s., not significant. Scale bar, 200 μm.
Induction and phosphorylation of PKCβ during RANKL-induced osteoclastogenesis
To further confirm the role of PKCβ in osteoclast differentiation, we assessed the time course of PKCβ induction and phosphorylation in response to RANKL. As shown in Fig. 3A, PKCβ mRNA remarkably increased compared to that of other PKCs (Supplementary Fig. S1), suggesting the importance of PKCβ in RANKL-induced osteoclastogenesis. Moreover, phosphorylation of PKCβ on the Ser660 residue, a carboxy-terminal hydrophobic site that is phosphorylated upon activation of cell surface receptors, increased following RANKL stimulation in BMMs (Fig. 3B). Interestingly, phosphorylation of PKCβ was highly increased on day 2 after RANKL stimulation as the protein level of PKCβ increased gradually during osteoclast differentiation. Taken together, these results suggest that the induction and phosphorylation of PKCβ may be regulated by RANKL.
Fig. 3.

PKCβ inactivates GSK-3β and induces NFATc1 during RANKL signaling. (A) Induction of PKCβ during RANKL-induced osteoclastogenesis. BMMs were cultured with M-CSF and RANKL for the indicated time periods and expression level of PKCβ was analyzed for total mRNA by real-time PCR. GAPDH was used as an internal control for normalization. Data are means ± S.D. of PKC isoform/GAPDH. *P < 0.01 (B) As in (A), except that total cell lysates were subjected to Western blotting with specific antibodies as indicated. The β-actin was used as the loading control. Band intensity of p-PKCβ and PKCβ was normalized and quantified using densitometry. (C) Effect of siRNA against PKCβ on NFATc1 induction during osteoclastogenesis. BMMs were transfected with 100 nM scrambled or PKCβ targeting siRNA and cultured with 100 ng/ml RANKL for 3 days. Whole cell lysates were analyzed by Western blotting using specific antibodies for PKCβ, p-GSK-3β, GSK-3β, and NFATc1. Anti-tubulin was used as the loading control. (D) Effects of Gö6976 (Gö) on GSK-3β phosphorylation and NFATc1 induction. BMMs were cultured with M-CSF and RANKL in the presence of DMSO (Con) or 50 nM Gö6976 for the indicated time periods. Total cell extracts were analyzed by Western blotting using specific antibodies as indicated. Anti-tubulin was used as the loading control. Relative band intensities (C, D) were quantified and normalized based on the intensities of tubulin and are indicated below each set of blots (in arbitrary units).
Inactivation of GSK-3β by PKCβ regulates osteoclast differentiation
We have reported that inactivating GSK-3β through RANKL signaling is required for osteoclastogenesis (Jang et al., 2011). We investigated the signaling link between PKCβ and GSK-3β upon RANKL stimulation. As shown in Fig. 3C, downregulating PKCβ by RNA interference significantly decreased GSK-3β phosphorylation and NFATc1 induction caused by RANKL stimulation. Inhibiting PKCβ with Gö6976 also suppressed phosphorylation of GSK-3β as well as induction of NFATc1 and Atp6v0d2 upon RANKL treatment (Fig. 3D). To further confirm the signaling link between PKCs and GSK-3β, we examined the effects of PKCs inhibitors, including GF109203X, Gö6976, and Rottlerin on GSK-3β phosphorylation. Consistent with Fig. 1, phosphorylation of GSK-3β by RANKL was remarkably inhibited by GF109203X or Gö6976, but not by Rottlerin, in a dose-dependent manner (Supplementary Fig. S2). Taken together, these data suggest that PKCβ regulates RANKL-induced osteoclast differentiation by inactivating GSK-3β.
Protective effects of PKCβ inhibition on RANKL-induced bone loss in mice
To investigate the effects of pharmacologically inhibiting PKCβ in the RANKL-induced bone loss model, we injected RANKL or PBS together with Gö6976 subperiosteally into the midline calvaria of 6-week-old mice. The extent of bone erosion was notably reduced in Gö6976-treated mice, and the formation of TRAP+ MNCs was greatly suppressed in a dose-dependent manner (Figs. 4A and 4B), suggesting that Gö6976 inhibits RANKL-induced bone loss in vivo. Thus, these results suggest that inhibiting PKCβ can provide a new therapeutic strategy targeting osteoclasts for treatment of bone diseases.
Fig. 4.
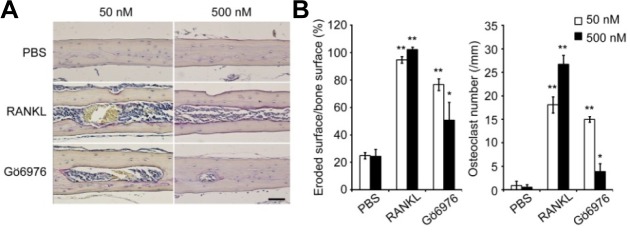
Effects of Gö6976 on RANKL-induced bone loss in mice. RANKL administered locally on calvarial bone of 6-week-old male mice induces bone destruction. Injecting Gö6976 inhibited RANKL-induced bone resorption by osteoclasts which increased in number in the RANKL-administered group compared to that in the control group (PBS). (A) Histology of the calvarial bone injected with PBS, RANKL, or two different doses of Gö6976 (TRAP and hematoxylin staining). Scale bar, 50 μm. (B) Eroded surface and the number of osteoclasts on calvarial bone section were measured and analyzed using Osteomeasure XP. Data represent means ± S.D. n = 5. *P < 0.05, **P < 0.01.
DISCUSSION
Our study focused on elucidating the regulatory role of PKCβ in RANKL-induced osteoclastogenesis. Through the use of cellular and pharmacological approaches, we demonstrated a novel role for PKCβ as a positive regulator of osteoclast differentiation by inactivating GSK-3β, which is a negative regulator of NFATc1.
To examine the involvement of PKCs in osteoclastogenesis, we used the general PKC inhibitor GF109203X, the PKCα/β inhibitor Gö6976, and the PKCδ inhibitor Rottlerin. As shown in Fig. 1, both GF109203X and Gö6976, but not Rottlerin, effectively suppressed osteoclast formation, suggesting that PKCα and/or PKCβ are involved in the differentiation process. We found that Gö6976 was able to block NFATc1 induction that is critical step for optimal differentiation of osteoclasts (Wang et al., 2006a). Downregulation of PKCβ by RNA interference also resulted in a reduced number of osteoclasts consistent with Gö6976 treatment. However, downregulation of PKCα did not change osteoclast differentiation. Taken together, these findings suggest that among PKCs, PKCβ plays a role in RANKL-induced osteoclastogenesis.
The involvement of PKCβ in osteoclast differentiation was supported by our finding that PKCβ mRNA levels increased during osteoclast differentiation caused by RANKL compared to those of other PKC isoforms. Similarly, induction of PKCβ after RANKL stimulation was confirmed at the protein level. Interestingly, PKCβ phosphorylation significantly increased on day 2 of RANKL treatment, suggesting its activation in response to RANKL. Notably, the phosphorylation pattern coincided with the timing of RANKL-stimulated NFATc1 induction. It is plausible that PKCβ signaling in response to RANKL may contribute to NFATc1 induction, thereby promoting osteoclastogenic potential.
How does PKCβ signaling link to NFATc1? Our data prove that activation of PKCβ inactivates GSK-3β during osteoclast formation. In agreement with our findings, previous studies have reported that GSK-3β is the substrate for some specific PKC isoforms (Goode et al., 1992), including PKCα, PKCβ, and PKCγ. Vilimek and Duronio (2006) reported that phosphorylation of GSK-3β is blocked by PKC inhibitors in response to cytokines such as interleukin-3 and granulocyte-macrophage colony-stimulating factor. In addition, a natural phospholipid, lysophosphatic acid utilizes a PKC-dependent pathway to modulate GSK-3β (Fang et al., 2002).
However, other signaling pathways regulating GSK-3β might be involved as well. It is well known that insulin and peptide growth factors such as insulin-like growth factor 1 and platelet derived growth factor regulate GSK-3β activity via the PI3K-Akt pathway (Doble and Woodgett, 2003; Fang et al., 2002; Schiaffino and Mammucari, 2011). We have shown that this mode of GSK-3β regulation is required for osteoclast formation (Jang et al., 2013; Moon et al., 2012). This pathway acts through activating PI3K and Akt, phosphorylating GSK-3β, and downregulating NFATc1 induction. Therefore, it is likely that RANKL control GSK-3β through the PI3K-Akt module and through an alternative or redundant PKCβ-dependent pathway. The reason for the presence of the two independent pathways to inhibit GSK-3β is uncertain. Additional studies are required to determine more precisely the mechanism of how the two pathways coordinate to work together for optimal osteoclast formation.
Given the ubiquitous expression throughout various cell types and its role in multiple cellular functions (Roffey et al., 2009; Tan and Parker, 2003), PKCs have been implicated in various diseases such as cardiovascular diseases, Alzheimer’s disease, tumors, and autoimmune diseases (Geraldes and King, 2010; Kawakami et al., 2002). In our experiments, we showed the protective effects of PKC inhibitor, Gö6976 on RANKL-induced bone destruction in mice, suggesting the possibility of PKCβ as a therapeutic target for bone diseases. However, more studies on tissue-specific inhibition of PKCβ in skeletal tissues will be required because of the possible side effects on the immune system due to its importance in T-cell or B-cell mediated immune responses (Tan and Parker, 2003).
In summary, we demonstrated herein that PKCβ functions as a positive regulator in RANKL-induced osteoclast formation, a mechanism by which PKCβ functions occur through GSK3β-NFATc1 axis, and provides a rationale for therapeutic intervention of PKC in bone-related diseases.
Supplementary data
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIP) (No. 2013R1A2A1A05005153; No. 2012R1A5A1048236; No. 2012M3A9C5048708).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Anderson D.M., Maraskovsky E., Billingsley W.L., Dougall W.C., Tometsko M.E., Roux E.R., Teepe M.C., DuBose R.F., Cosman D., Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Boyle W.J., Simonet W.S., Lacey D.L. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Choi H.K., Kang H.R., Jung E., Kim T.E., Lin J.J., Lee S.Y. Early estrogen-induced gene 1, a novel RANK signaling component, is essential for osteoclastogenesis. Cell Res. 2013;23:524–536. doi: 10.1038/cr.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G.R., Olson E.N. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Doble B.W., Woodgett J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Yu S., Tanyi J.L., Lu Y., Woodgett J.R., Mills G.B. Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol. Cell. Biol. 2002;22:2099–2110. doi: 10.1128/MCB.22.7.2099-2110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldes P., King G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode N., Hughes K., Woodgett J.R., Parker P.J. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J. Biol. Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- Hogan P.G., Chen L., Nardone J., Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Jang H.D., Shin J.H., Park D.R., Hong J.H., Yoon K., Ko R., Ko C.Y., Kim H.S., Jeong D., Kim N., et al. Inactivation of glycogen synthase kinase-3beta is required for osteoclast differentiation. J. Biol. Chem. 2011;286:39043–39050. doi: 10.1074/jbc.M111.256768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H.D., Noh J.Y., Shin J.H., Lin J.J., Lee S.Y. PTEN regulation by the Akt/GSK-3beta axis during RANKL signaling. Bone. 2013;55:126–131. doi: 10.1016/j.bone.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Kawakami T., Kawakami Y., Kitaura J. Protein kinase C beta (PKC beta).: normal functions and diseases. J. Biochem. 2002;132:677–682. doi: 10.1093/oxfordjournals.jbchem.a003273. [DOI] [PubMed] [Google Scholar]
- Kim H., Choi H.K., Shin J.H., Kim K.H., Huh J.Y., Lee S.A., Ko C.Y., Kim H.S., Shin H.I., Lee H.J., et al. Selective inhibition of RANK blocks osteoclast maturation and function and prevents bone loss in mice. J. Clin. Invest. 2009;119:813–825. doi: 10.1172/JCI36809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Kim K., Youn B.U., Jin H.M., Kim N. MHC class II transactivator negatively regulates RANKL-mediated osteoclast differentiation by downregulating NFATc1 and OSCAR. Cell. Signal. 2010;22:1341–1349. doi: 10.1016/j.cellsig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Kobayashi N., Kadono Y., Naito A., Matsumoto K., Yamamoto T., Tanaka S., Inoue J. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001;20:1271–1280. doi: 10.1093/emboj/20.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey D.L., Timms E., Tan H.L., Kelley M.J., Dunstan C.R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Lee J., Kim M.S. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract. 2007;77(Suppl 1):S49–57. doi: 10.1016/j.diabres.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Lee S.W., Kwak H.B., Chung W.J., Cheong H., Kim H.H., Lee Z.H. Participation of protein kinase C beta in osteoclast differentiation and function. Bone. 2003;32:217–227. doi: 10.1016/s8756-3282(02)00976-6. [DOI] [PubMed] [Google Scholar]
- Moon J.B., Kim J.H., Kim K., Youn B.U., Ko A., Lee S.Y., Kim N. Akt induces osteoclast differentiation through regulating the GSK3beta/NFATc1 signaling cascade. J. Immunol. 2012;188:163–169. doi: 10.4049/jimmunol.1101254. [DOI] [PubMed] [Google Scholar]
- Roffey J., Rosse C., Linch M., Hibbert A., McDonald N.Q., Parker P.J. Protein kinase C intervention: the state of play. Curr. Opin. Cell Biol. 2009;21:268–279. doi: 10.1016/j.ceb.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Saito N., Kikkawa U., Nishizuka Y. The family of protein kinase C and membrane lipid mediators. J. Diabetes Complications. 2002;16:4–8. doi: 10.1016/s1056-8727(01)00200-8. [DOI] [PubMed] [Google Scholar]
- Schiaffino S., Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet. Muscle. 2011;1:4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T., Jimi E., Nakamura I., Takahashi N. Role of 1 alpha,25-dihydroxyvitamin D3 in osteoclast differentiation and function. Methods Enzymol. 1997;282:223–235. doi: 10.1016/s0076-6879(97)82110-6. [DOI] [PubMed] [Google Scholar]
- Suda T., Takahashi N., Udagawa N., Jimi E., Gillespie M.T., Martin T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- Takayanagi H., Ogasawara K., Hida S., Chiba T., Murata S., Sato K., Takaoka A., Yokochi T., Oda H., Tanaka K., et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- Takayanagi H., Kim S., Koga T., Nishina H., Isshiki M., Yoshida H., Saiura A., Isobe M., Yokochi T., Inoue J., et al. Induction and activation of the transcription factor NFATc1 (NFAT2). integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Tan S.L., Parker P.J. Emerging and diverse roles of protein kinase C in immune cell signalling. Biochem. J. 2003;376:545–552. doi: 10.1042/BJ20031406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilimek D., Duronio V. Cytokine-stimulated phosphorylation of GSK-3 is primarily dependent upon PKCs, not PKB. Biochem. Cell Biol. 2006;84:20–29. doi: 10.1139/o05-154. [DOI] [PubMed] [Google Scholar]
- Wang J., Wang N., Xie J., Walton S.C., McKown R.L., Raab R.W., Ma P., Beck S.L., Coffman G.L., Hussaini I.M., et al. Restricted epithelial proliferation by lacritin via PKCalpha-dependent NFAT and mTOR pathways. J. Cell Biol. 2006a;174:689–700. doi: 10.1083/jcb.200605140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhou Y., Evers B.M. Neurotensin phosphorylates GSK-3alpha/beta through the activation of PKC in human colon cancer cells. Neoplasia. 2006b;8:781–787. doi: 10.1593/neo.06259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B.R., Rho J., Arron J., Robinson E., Orlinick J., Chao M., Kalachikov S., Cayani E., Bartlett F.S., 3rd, Frankel W.N., et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A., et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T., Tsuji T., Chen C. Roles of PKC isoforms in the induction of apoptosis elicited by aberrant Ras. Oncogene. 2010;29:1050–1061. doi: 10.1038/onc.2009.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.