

Abstract
Technological advances have enabled the use of DNA sequencing as a flexible tool to characterize genetic variation and to measure the activity of diverse cellular phenomena such as gene isoform expression and transcription factor binding. Extracting biological insight from the experiments enabled by these advances demands the analysis of large, multi-dimensional datasets. This unit describes the use of the BEDTools toolkit for the exploration of high-throughput genomics datasets. I present several protocols for common genomic analyses and demonstrate how simple BEDTools operations may be combined to create bespoke pipelines addressing complex questions.
Keywords: genomics, bioinformatics, genome analysis, genome intervals, genome features
INTRODUCTION
Modern genomics research combines high throughput DNA sequencing with computational analysis to gain insight into genome biology. Both the spectrum of experimental assays that are now possible and the scale of the datasets generated complicate the interpretation of experimental results. Additional complexity comes from the fact that the genomics research community employs multiple file formats, such as BED(Kent et al., 2002), GFF, VCF(Danecek et al., 2011), BAM(Li et al., 2009), and BigWig(Kent et al., 2010), to represent experimental datasets and genome annotations. While these data formats differ in their structure and intended use, they each describe the attributes of one or more genome intervals (a.k.a. genome “features”). A single genome interval represents a consecutive stretch of nucleotides on a chromosome or assembly scaffold. Despite file format differences, most analyses involving multiple sets of genome intervals can be distilled to what I colloquially refer to as “genome arithmetic”: that is, the analysis of sets of genome intervals via multiple comparative operations. For example, quantifying transcript expression in RNA-seq experiments is essentially a process of counting the number of cDNA alignments (i.e., intervals in BAM format) that intersect (overlap) transcript annotations (i.e., intervals in GFF or BED format).
BEDTools is an open source software package comprised of multiple tools for comparing and exploring genomic datasets via fundamental “genome arithmetic” tasks. The individual tools in the BEDTools suite are each focused on a relatively simple operation, such as those illustrated in Figure 1. The goals of this unit are to introduce the basic concepts of genome arithmetic with BEDTools and to demonstrate, via biologically relevant examples, how analytical power is conferred through clever combinations of individual BEDTools operations. This unit is intended to give new users a sense of what is possible with the BEDTools suite. I encourage the reader to subsequently read the BEDTools documentation (bedtools.readthedocs.org), since only the most widely useful subset of the nearly forty individual operations is covered.
Figure 1. Examples of genome arithmetic operations.
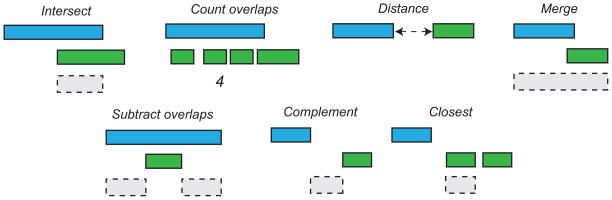
Each tool in the BEDTools suite performs a relatively simple operation on one, a pair, or multiple genome interval datasets. The examples presented here reflect genome arithmetic operations on two genome interval files (green and blue). For example, if blue intervals represent gene annotations and green intervals represent DNA sequence alignments, then the result of the intersect tool (gray interval) represents the genomic interval that is shared between a single gene annotation and sequence alignment.
STRATEGIC PLANNING
Completion of the protocols covered will require a computer with an UNIX, Linux, or Apple OS X operating system. Microsoft Windows users may also complete the unit if they first install Cygwin, but Windows usage is not directly supported. In the following sections, I will describe how to install BEDTools and other required software, as well as provide an overview of basic usage concepts.
Conventions
Throughout this unit, I will demonstrate BEDTools usage via commands issued on the UNIX command line. Such commands will use a different font and appear in bold. Also, the “$” character is merely intended to represent the command prompt and should not be typed.
$ bedtools --help
Additionally, this unit will include commands in the R programming language, primarily as a means for creating plots describing the results of BEDTools analyses. Such commands will use the same font, yet will be preceded by a “>“ to denote the R command prompt, which should likewise not be typed.
> x <- c(1,2,3)
Each protocol will also provide brief comments (preceded with a “#”) that describe the basic intent of the subsequent command. Comment lines can be copied and pasted or typed, but are merely provided to document the purpose of the command.
# The following command invokes the BEDTools help menu from the command line $ bedtools --help
Lastly, a single command will often span multiple lines. In order to function properly, the entire command (except for the leading “$”), spanning multiple lines, must be copied and pasted into one’s command prompt. As an example, below are two different multi-line commands. Each would be copied and pasted separately into the command prompt including each line of each command.
# Multi-line command number 1
$ bedtools intersect \
-a foo.bed \
-b bar.bed \
> foobar.bed
# Multi-line command number 2
$ bedtools intersect \
-a fiz.bed \
-b biz.bed \
> fizbiz.bed
Background knowledge
This unit assumes that the reader has previous experience working on the UNIX command line, as well as a basic understanding of common genomics file formats such as BED(Kent et al., 2002), VCF, GFF, and BAM. If not, I encourage you to first read the BEDTools documentation (bedtools.readthedocs.org), as well as the papers (Danecek et al., 2011; Kent et al., 2002; Li et al., 2009) describing the above formats. There are also many freely available tutorials on the Internet that describe the basics of working on the UNIX command line.
SUPPORT PROTOCOL 1: INSTALLING AND PREPARING TO USE BEDTOOLS
BEDTools is freely available software that is archived and maintained on GitHub. The latest version of BEDTools can be found at the following URL: https://github.com/arq5x/bedtools2/releases. At the time this unit as written, the latest release was 2.19.1. Future readers should check for subsequent releases and adjust the installation commands below accordingly.
Necessary Resources
A C/C++ compiler such as GCC. For OS X users, this typically requires the installation of the Xcode developer tools.
The zlib and zlib-devel compression libraries (installed by default on many systems)
Installation options
The first installation strategy requires that one downloads and compiles the latest version of BEDTools directly from the source code. In this example, we will download and install version 2.19.1.
-
1a
Download the BEDTools source code.
$ curl -OL https://github.com/arq5x/bedtools2/releases/download/v2.19.1/bedtools-2.19.1.tar.gz
-
2a
Extract the source code into a new directory.
$ tar -zxvf bedtools-2.19.1.tar.gz
-
3a
Navigate to the new directory containing the source code.
$ cd bedtools2-2.19.1
-
4a
Compile the source code into executable software.
$ make
-
5a
At this point, you need to make the bedtools executable accessible on your system. Make a new “bin” directory within your home directory (“~”) and copy the executable to the new “bin” directory. Then update your PATH to include the new “bin” directory so that the BEDTools executable code can be found.
$ mkdir ~/bin $ cp bin/bedtools ~/bin $ PATH=PATH:~/bin
-
1b
Alternatively, BEDTools may also be more installed via automatic package management software available on most UNIX systems.
# Fedora / Centos $ yum install bedtools # Debian / Ubuntu $ apt-get install bedtools # OSX (via HomeBrew: http://brew.sh/) $ brew install bedtools
Downloading datasets for this unit
Throughout this unit, you will be using BEDTools to analyze several datasets in various genomics data formats. Therefore, you will first need to download the following files before beginning the analysis protocols that follow.
-
6
Use the ‘curl’ command to download example datasets to your computer.
$ curl -OL http://quinlanlab.cs.virginia.edu/bedtools-protocols/cpg.bed $ curl -OL http://quinlanlab.cs.virginia.edu/bedtools-protocols/exons.bed $ curl -OL http://quinlanlab.cs.virginia.edu/bedtools-protocols/human.hg19.genome
The bedtools help menu
-
7
The BEDTools software package is comprised of many sub-tools. One can be reminded of the tools available and a brief summary of their functionality by typing the following on the command line:
$ bedtools --help
If BEDTools has been installed correctly, on your system, you should see several examples of the BEDTools “subcommands”. If not, please refer back to the installation instructions above.
$ bedtools --help
bedtools: flexible tools for genome arithmetic and DNA sequence analysis.
usage: bedtools <subcommand> [options]
The bedtools sub-commands include:
[ Genome arithmetic ]
intersect Find overlapping intervals in various ways.
window Find overlapping intervals within a window around an interval.
closest Find the closest, potentially non-overlapping interval.
coverage Compute the coverage over defined intervals.
map Apply a function to a column for each overlapping interval.
genomecov Compute the coverage over an entire genome.
merge Combine overlapping/nearby intervals into a single interval.
cluster Cluster (but do not merge) overlapping/nearby intervals.
complement Extract intervals_not_represented by an interval file.
subtract Remove intervals based on overlaps b/w two files.
slop Adjust the size of intervals.
flank Create new intervals from the flanks of existing intervals.
sort Order the intervals in a file.
random Generate random intervals in a genome.
shuffle Randomly redistribute intervals in a genome.
sample Sample random records from file using reservoir sampling.
annotate Annotate coverage of features from multiple files.
-
8
In order to conduct an analysis of genomic intervals with BEDTools, one must employ one or more of the BEDTools subcommands. I will illustrate this with the intersect subcommand (more details will be provided in the first protocol). For example, to intersect BED files representing Alu elements and CpG islands, one would use the following command.
$ bedtools intersect -a alu.bed -b cpg.bed
One may also request a detailed help menu regarding the specifics of each tool as follows.
$ bedtools intersect -h
All other subcommands follow the same basic convention.
$ bedtools [SUBCOMMAND] [OPTIONS]
Working with “genome-sorted” datasets
-
9
The default algorithm that BEDTools leverages to detect intersections loads one of the two files into a tree data structure based on the UCSC binning algorithm(Kent et al., 2002). While fast, it can consume substantial memory, especially for very large files. For this reason, we provide an alternative, yet very fast and memory efficient algorithm that requires one’s input files to be “genome-sorted”: that is, sorted first by chromosome and then by start position. When both input files are genome-sorted, the algorithm can “sweep” through the data and detect overlaps on the fly in a manner much like the way database systems join two tables. As an example, I demonstrate how to sort a BED file in this manner below (the first column of the BED format represents the chromosome and the second column represents the start coordinate.
$ sort -k1,1 –k2,2n fileA.bed > fileA.sorted.bed
This algorithm is invoked via the -sorted option and its use is demonstrated in subsequent sections. The substantial performance gains conferred through the use of “genome-sorted” datasets are illustrated in Figure 2.
Figure 2. BEDTools scalability.

The runtime in seconds (left panel) and memory usage (right panel) are compared when using either unsorted genome intervals (dashed red) or genome intervals that have been pre-sorted in genome order (solid red). As a basis of comparison, the BEDTools performance is compared to the BEDOPS toolset, both with (solid gray) and without (dashed gray) automatic error checking.
Genome files
-
10
In order to function correctly, some of the BEDTools subcommands need to be informed of the length of each chromosome in the organism you are studying. These “genome” files must be tab delimited; the first column must be the chromosome label and the second column must be the length of the chromosome. For example, below is an example “genome” file for build 37 (a.k.a “hg19”) of the human genome.
$ head -5 human.hg19.genome chr1 249250621 chr2 243199373 chr3 198022430 chr4 191154276 chr5 180915260
BASIC PROTOCOL 1: FINDING INTERSECTIONS BETWEEN GENOME INTERVAL FILES
One of the most common questions asked of two sets of genomic intervals is whether or not any of the intervals in the two sets “overlap” with one another. This is known as interval intersection. Given its broad utility, the intersect command is the most widely-used utility in the BEDTools suite. By default, intersect reports the subset of intervals that are common to your two files. The “A” file is considered the “query” file, whereas the “B” file is considered the “database” file. To demonstrate the basic functionality of the intersect utility, we will use the BED files we downloaded in the Strategic Planning section to identify CpG islands that overlap exons in the human genome.
Necessary Resources
See Support Protocol 1
-
1
Display the first five BED intervals reflecting CpG islands.
$ head -n 5 cpg.bed chr1 28735 29810 CpG:_116 chr1 135124 135563 CpG:_30 chr1 327790 328229 CpG:_29 chr1 437151 438164 CpG:_84 chr1 449273 450544 CpG:_99
-
2
Display the first five BED intervals reflecting exons.
$ head -n 5 exons.bed chr1 11873 12227 NR_046018_exon0 chr1 12612 12721 NR_046018_exon1 chr1 13220 14409 NR_046018_exon2 chr1 14361 14829 NR_024540_exon0 chr1 14969 15038 NR_024540_exon1
-
3a
Identify the CpG (file ‘A’) coordinates that overlap exons (file ‘B’).
$ bedtools intersect -a cpg.bed -b exons.bed | head -n 5 chr1 29320 29370 CpG:_116 chr1 135124 135563 CpG:_30 chr1 327790 328229 CpG:_29 chr1 327790 328229 CpG:_29 chr1 327790 328229 CpG:_29
In this example, the BED file representing CpG islands is treated as the “query” file, and as such, the reported intervals reflect the portion of each original CpG island that overlap one or more exons in the “database” file. For example, the first CpG island above contained 75 base pairs (29810-28735, where the start coordinate 28735 is zero-based), yet the interval that the intersect tool reports reflects the subset of 50 base pairs that actually overlapped an exon.
Rather than report solely the intersecting intervals, it is often desirable to instead report the original intervals that intersected from both files. For each intersection between the two input files, the “write A” and “write B” options (-wa and –wb) report the original interval from the “A” and the “B” file, respectively.
-
3b
Alternative: show overlaps with both CpG and exon coordinates (-wa, -wb).
$ bedtools intersect -a cpg.bed -b exons.bed -wa -wb | head -n 5 chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon10 chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon0 chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon2 chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon2 chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon3
While this demonstrates how the -wa and –wb options allow us to understand which exact intervals from each file intersected, it is not immediately apparent (without squinting) how many base pairs of overlap exist between the intersecting features. The “write overlap” (-wo) option addresses this by reporting the original intervals followed by the number of overlapping bases observed between each interval pair.
-
3c
Alternative: show the overlap with both CpG and exon coordinates (-wo).
$ bedtools intersect -a cpg.bed -b exons.bed -wo | head –n 5 chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon10 50 chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon0 439 chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon2 439 chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon2 439 chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon3 439
In many situations, one is less concerned with enumerating every single overlapping interval between two files. Using the -c option, one can simply count the number of intervals that intersect each “query” interval.
-
3d
Alternative: show the count of exons that overlap CpG islands (-c).
$ bedtools intersect -a cpg.bed -b exons.bed -c | head –n 5 chr1 28735 29810 CpG:_116 1 chr1 135124 135563 CpG:_30 1 chr1 327790 328229 CpG:_29 3 chr1 437151 438164 CpG:_84 0 chr1 449273 450544 CpG:_99 0
Similarly, the -v option allows one to focus solely on the CpG islands that do not overlap exons.
-
3e
Alternative: show those CpG islands that do not overlap exons (-v).
$ bedtools intersect -a cpg.bed -b exons.bed -v | head –n 5 chr1 437151 438164 CpG:_84 chr1 449273 450544 CpG:_99 chr1 533219 534114 CpG:_94 chr1 544738 546649 CpG:_171 chr1 801975 802338 CpG:_24
The examples presented thus far have demonstrated intersections that require a single base pair of overlap in order to be reported as output. There are many cases, however, where the biological question at hand demands stricter criteria. For example, if one is interested in studying exons that have a role in transcript regulation, one could begin by using the –f 0.5 option to identify CpG islands where at least half of the DNA content is comprised of coding exons.
-
4
Display CpG islands with >= 50% of the interval overlapped by an exon (-f 0.50).
$ bedtools intersect -a cpg.bed -b exons.bed –f 0.50 -wo | head –n 5 chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon0 439 chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon2 439 chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon2 439 chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon3 439 chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047519_exon5 348
BASIC PROTOCOL 2: MEASURING COVERAGE IN WHOLE-GENOME DNA SEQUENCING EXPERIMENTS
The fundamental utility of whole genome sequencing is the ability to characterize the full spectrum of genetic variation present in the individual’s genome. However, the power to detect genetic variation is a function of the number of times a given nucleotide is independently sampled by the sequencing experiment. For example, if a diploid individual is heterozygous at a given position in the genome, then a single aligned sequence from modern DNA sequencing technologies (e.g., Illumina) will sample only one of the two inherited alleles. Therefore, the more that each nucleotide is sampled, the more likely it is that both inherited alleles will be detected. While coverage is typically modeled as a Poisson distribution, biases such as GC content and the number of PCR cycles used to amplify DNA prior to sequencing prevent uniform genome coverage to a degree that is greater than would be expected by a Poisson distribution. Given known biases and the importance of coverage to quality of the experiment, it is standard practice to assess the empirical coverage distribution as a quality control measure. The BEDTools genomecov tool is designed for precisely this purpose. The following example computes a histogram of sequence coverage for each chromosome as well as for the entire genome. Specifically, this histogram measures the fraction of each chromosome (and the genome as a whole) that is sampled by 0, 1, 2,…N independent sequence alignments.
Before beginning this protocol, you must first download an example dataset from the 1000 Genomes Project (Durbin et al., 2010). This dataset represents whole genome sequencing of a Yoruban individual (NA19146) using the Illumina sequencing platform. The DNA sequences have been aligned to build 37 of the human reference genome and the resulting alignments are stored in BAM format. You should anticipate the download to require 15 to 30 minutes.
Necessary Resources
See Support Protocol 1
-
1
Download a BAM alignment file for NA19146 from the 1000 Genomes website.
$ KGFTP=ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/data/NA19146/alignment $ curl -0 $KGFTP/NA19146.mapped.ILLUMINA.bwa.YRI.low_coverage.20130415.bam
-
2
Create a symbolic link to the file for brevity.
$ ln -s NA19146.mapped.ILLUMINA.bwa.YRI.low_coverage.20130415.bam NA19146.bam
Now that we have downloaded a whole-genome sequencing dataset for the N19146 individual, we can use the genomecov tool to compute a histogram of aligned DNA sequence coverage observed throughout the genome of this sample.
-
3
Use the genomecov tool to compute a genome-wide histogram of sequence coverage.
$ bedtools genomecov -ibam NA19146.bam > NA19146.coverage.hist.txt # The output format of the resulting file is a tab-delimited file with # the following columns: # column 1 = chromosome # column 2 = depth # column 3 = number of base pairs with depth=column2 # column 4 = size of the chromosome # column 5 = fraction of base pairs with depth=column2
-
4
Display the first 11 lines of the coverage histogram for chromosome 1.
$ head -n 11 NA19146.coverage.hist.txt 1 0 29172804 249250621 0.117042 1 1 7196069 249250621 0.0288708 1 2 9698769 249250621 0.0389117 1 3 11608275 249250621 0.0465727 1 4 12960686 249250621 0.0519986 1 5 13769135 249250621 0.0552421 1 6 14188765 249250621 0.0569257 1 7 14176958 249250621 0.0568783 1 8 13950378 249250621 0.0559693 1 9 13432208 249250621 0.0538904 1 10 12732827 249250621 0.0510844
-
5
Display the first 11 lines of the coverage histogram for the entire genome.
$ grep ^genome NA19146.coverage.hist.txt | head –n 11 genome 0 321856083 3137454505 0.102585 genome 1 117285058 3137454505 0.0373822 genome 2 152949464 3137454505 0.0487495 genome 3 176290526 3137454505 0.056189 genome 4 189358028 3137454505 0.060354 genome 5 194473449 3137454505 0.0619845 genome 6 193704084 3137454505 0.0617392 genome 7 188548984 3137454505 0.0600962 genome 8 180274956 3137454505 0.057459 genome 9 169595911 3137454505 0.0540553 genome 10 157422837 3137454505 0.0501753
The output of the genomecov histogram consists of the chromosome (column 1), the observed aligned sequence depth (column 2), the number of base pairs with this observed sequencing depth (column 3), the size of the chromosome (column 4), and the fraction of base pairs with this observed sequencing depth (column 5). This output allows us to observe that 11.7% of NA19146’s chromosome 1 was not sampled by a single aligned sequence, and by extension, 88.3% of chromosome 1 had one or more aligned sequences. Similarly, we see that 10.3% of the entire genome lacked a single aligned sequence and therefore 89.7% of the genome was sampled by one or more aligned sequences. Using the following R code, one can convert the text histogram produced by the genomecov tool into a plot (the result is presented in Figure 3) that describes the distribution of genome-wide sequencing coverage. Plotting the sequence coverage distribution allows one to observe that, on average, this individual’s genome was sampled by ~5 independent sequences alignments. This reflects the fact that this dataset was generated as part of the 1000 Genome Projects “low coverage” study.
Figure 3.

Histogram of genome-wide sequencing coverage for NA19146.
-
6
Invoke R from the command line.
$ R
-
7
Plot a histogram of genome-wide coverage for NA19146. The following R commands will produce a plot identical to Figure 3.
# 7a. Load the output of genomecov into an R data frame > cov = read.table(‘NA19146.coverage.hist.txt’) # 7b. Extract the genome-wide histogram entries > gcov = cov[cov[,1] == ‘genome’,] # 7c. Plot the histogram > plot(gcov[1:51,2], gcov[1:51,5], type=‘h’, col=‘darkgreen’, lwd=3, xlab=‘Depth’, ylab=‘Fraction of genome at depth’) # 7d. Add axis labels > axis(1,at=c(1,5,10,15,20,25,30,35,40,45,50))
ALTERNATE PROTOCOL 2: IDENTIFYING SPECIFIC GENOMIC REGIONS WITH HIGH OR LOW SEQUENCE COVERAGE
While Basic Protocol 2 demonstrates a strategy for measuring genome wide coverage statistics, it merely provides a summary of coverage without cataloging the observed coverage at each base pair in the genome. Such details are necessary in order to identify specific genomic regions (e.g., genes) where insufficient coverage is available and to identify regions where excessively high coverage was observed, perhaps owing to technical artifacts such as biases in GC content. Through the use of the -bga option, the genomecov tool produces BEDGRAPH output that describes the sequence coverage observed at discrete genome intervals.
Necessary Resources
See Support Protocol 1
-
1
Calculate a genome-wide BEDGRAPH of coverage.
$ bedtools genomecov \
-ibam NA19146.bam \
-bga \
> NA19146.coverage.bedgraph
-
2
Display the first ten lines of the output.
$ head –n 10 NA19146.coverage.bedgraph 1 0 9994 0 1 9994 9996 1 1 9996 9999 2 1 9999 10000 4 1 10000 10001 42 1 10001 10002 82 1 10002 10003 112 1 10003 10004 145 1 10004 10005 184 1 10005 10006 233
At this point, we have created a BEDGRAPH representing the coverage through the entire genome of NA19146. The first three columns represent each genome interval in BED format and the fourth column represents the number of aligned sequences observed at each interval. The UNIX awk utility can be used to extract intervals with insufficient coverage. For example, based on the distribution shown in Figure 3, one might wish to identify genomic regions with less than five aligned sequences.
-
3a
Identify regions with less than 5 reads.
$ awk ‘$4 < 5’ NA19146.coverage.bedgraph \
| head -n 10
1 0 9994 0
1 9994 9996 1
1 9996 9999 2
1 9999 10000 4
1 10525 10526 4
1 10526 10534 3
1 10534 10550 2
1 10550 10576 1
1 10576 10589 2
1 10589 10617 1
Similarly, we can identify regions with excessive coverage, which based on the distribution described in Figure 3, we will define as intervals with more than 25 aligned sequences.
-
3b
Identify regions with more than 25 reads.
$ awk ‘$4 > 25’ NA19146.coverage.bedgraph \
| head -n 10
1 10000 10001 42
1 10001 10002 82
1 10002 10003 112
1 10003 10004 145
1 10004 10005 184
1 10005 10006 233
1 10006 10007 250
1 10007 10008 258
1 10008 10009 306
1 10009 10010 313
Notice that many of the intervals with excessive coverage (yet differing individual coverage measurements) are adjacent to one another. In such cases, we may wish to combine these adjacent bases into single, consecutive, high-coverage intervals. This can easily be accomplished with the BEDTools merge tool. The following example demonstrates how multiple BEDTools operations can be combined to conduct more sophisticated analyses. The use of “-i -” follows the UNIX convention of specifying to the merge tool that the input be being passed directly from the output of the awk command, rather than from a file.
-
3c
Merge coordinates with high coverage. This is an example of how one may refine analyses by passing the output from one BEDTools command as input to another command. Note that we specify to the merge tool that input is being passed directly (as opposed to a proper file) from the awk command via the use of ‘-’ as the input.
$ awk ‘$4 > 25’ NA19146.coverage.bedgraph \
| bedtools merge -i - \
| head -n 10
1 10000 10287
1 10337 10465
1 11740 11741
1 11742 11822
1 11834 13924
1 13931 15266
1 15305 15308
1 15310 15311
1 15317 15528
1 15568 15576
BASIC PROTOCOL 3: MEASURING COVERAGE IN TARGETED DNA SEQUENCING EXPERIMENTS
By focusing the fixed sequencing “budget” (that is, a fixed number of sequences at a given cost) of current sequencing platforms on a subset of the genome, targeted DNA sequencing strategies are used to confer greater power to detect genetic variation in the genomic regions of interest. For example, exome capture kits focus the sequence budget on the 1–2% of the genome that encodes protein coding genes and UTRs(Ng et al., 2010). Consequently, many more sequences are aligned to these regions than would be possible with whole genome sequencing, thus allowing more sensitive detection of heterozygous loci in the sequenced individual.
However, current targeted capture assays require DNA hybridization and have biases in GC content. This often leads to extensive variability in the sequence coverage observed at each targeted genomic region. As a result, it is crucial to measure the uniformity of coverage at the targeted regions in order to assess overall discovery potential, since those regions with lower coverage are inherently less empowered for the discovery of genetic variation.
Necessary Resources
Before beginning this protocol, you must first download another example dataset from the 1000 Genomes Project. This dataset represents exome sequencing of a CEU individual (NA12891) using the Illumina sequencing platform. The DNA sequences have been aligned to build 37 of the human reference genome and the resulting alignments are stored in BAM format. In addition, we must also download a BED file whose genomic intervals represent the DNA probes that were used to selectively hybridize DNA from coding exons. You should anticipate the file downloads to require 15 to 30 minutes.
-
1
Make a shortcut to the 1000 Genomes FTP site.
$ KGFTP=ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/phase3_EX_or_LC_only_alignment/data/NA12891/exome_alignment/
-
2
Download the NA19146 BAM alignment file from the 1000 Genomes website.
$ curl -O $KGFTP/NA12891.mapped.ILLUMINA.bwa.CEU.exome.20121211.bam
-
3
Create a symbolic link to the long filename for brevity.
$ ln -s NA12891.mapped.ILLUMINA.bwa.CEU.exome.20121211.bam NA12891.exome.bam
-
4
Download the exome capture targets.
$ KGFTP=ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference $ curl -O $KGFTP/exome_pull_down_targets/20130108.exome.targets.bed
-
5
Use “sed” to remove “chr” from the chromosome labels so that they match the BAM file.
sed -e ‘s/chr//’ 20130108.exome.targets.bed \ > targets.numeric.chroms.bed
The following command illustrates how to use the BEDTools coverage tool to investigate the uniformity of coverage for targeted DNA sequencing experiments. This command computes, for each targeted interval in the BED file (-b), a histogram of coverage observed among the aligned sequences from the BAM file. The output of the coverage histogram for each interval consists of the chromosome (column 1), start (column 2) and end (column 3) of the targeted interval, followed by the observed aligned sequence depth (column 5), the number of base pairs in the interval with this observed sequencing depth (column 6), the size of the interval (column 6), and finally, the fraction of base pairs in the interval with this observed sequencing depth (column 7). In addition, the report contains a summary of the coverage histogram among all of the targeted intervals. The output of this overall summary is structured slightly differently and is described in the example below.
-
1
Compute a histogram of coverage for each target and for all targets as a whole.
$ bedtools coverage \
-hist \
-abam NA12891.exome.bam \
-b targets.numeric.chroms.bed \
> NA12891.exome.coverage.hist.txt
# The output format of the resulting file is a tab-delimited file with
# the following columns:
# column 1 = chromosome
# column 2 = start coordinate of the interval
# column 3 = end coordinate of the interval
# column 4 = depth
# column 5 = number of base pairs in the interval with depth=column 4
# column 6 = size of the interval
# column 7 = fraction of base pairs in the interval with depth=column 4
-
2
Display the first 10 lines of the coverage histogram for one of the intervals.
$ head -n 10 NA12891.exome.coverage.hist.txt 1 39845860 39846100 41 2 240 0.0083333 1 39845860 39846100 43 2 240 0.0083333 1 39845860 39846100 45 1 240 0.0041667 1 39845860 39846100 47 1 240 0.0041667 1 39845860 39846100 48 2 240 0.0083333 1 39845860 39846100 49 1 240 0.0041667 1 39845860 39846100 50 1 240 0.0041667 1 39845860 39846100 51 4 240 0.0166667 1 39845860 39846100 52 4 240 0.0166667 1 39845860 39846100 53 3 240 0.0125000
-
3
Display the first 10 lines of the coverage histogram among all intervals.
$ grep ^all NA12891.exome.coverage.hist.txt | head -n 10 all 0 329870 46492725 0.0070951 all 1 252695 46492725 0.0054352 all 2 256906 46492725 0.0055257 all 3 259382 46492725 0.0055790 all 4 261672 46492725 0.0056282 all 5 264214 46492725 0.0056829 all 6 266291 46492725 0.0057276 all 7 263126 46492725 0.0056595 all 8 264020 46492725 0.0056787 all 9 262028 46492725 0.0056359
-
4
Create a subset reflecting solely the coverage observed across all targets
$ grep ^all NA12891.exome.coverage.hist.txt > NA12891.exome.coverage.all.txt
By examining the observed coverage among all targeted intervals, we can quickly see that a mere 329,870 base pairs, or 0.7% of the targeted bases had no detectable sequence coverage. Plotting a cumulative coverage distribution based upon these results allows one to assess the fraction of targeted bases with various levels of coverage (and thus power to detect genetic variation).
-
5
Load the output of ‘coverage’ into an R data frame.
> cov = read.table(‘NA12891.exome.coverage.all.txt’)
-
6
Create a cumulative distribution from the “raw” histogram.
# Note: we will truncate the x axis at depth >=400) > allcov_cumul = 1 - cumsum(cov[,5])
-
7
Create a plot of the cumulative density function of the target coverage. The code below will result in a plot identical to Figure 4.
Figure 4.

Cumulative distribution of sequencing coverage observed among all exome-targeted bases for NA12891.
> plot(cov[2:401,2], allcov_cumul[1:400], col=‘darkred’, type=‘l’, lwd=2, xlab=“Depth”, ylab=“Fraction of capture target bases >= depth”, ylim=c(0,1.0)) # 7a. Add gridlines to the plot > abline(v = 20, col = “gray60”) > abline(v = 50, col = “gray60”) > abline(v = 80, col = “gray60”) > abline(v = 100, col = “gray60”) > abline(h = 0.50, col = “gray60”) > abline(h = 0.90, col = “gray60”) # 7b. Add axis labels to the plot > axis(1, at=c(20,50,80), labels=c(20,50,80)) > axis(2, at=c(0.90), labels=c(0.90)) > axis(2, at=c(0.50), labels=c(0.50))
Figure 4 demonstrates that, for this sample from the 1000 Genomes Project, just under 90% of the targeted bases had at least 20 aligned sequences and over half the targeted bases had at least 80 aligned sequences.
ALTERNATE PROTOCOL 3: IDENTIFYING SPECIFIC TARGETED INTERVALS THAT LACKED COVERAGE
While the previous protocol measured the overall fraction of targeted bases having sufficient coverage for genetic discovery, it did not reveal specific intervals that, despite being targeted, completely lacked any sequence coverage. It could be important to identify such intervals, as they may prove to be problematic (perhaps owing to a lack of sequence complexity) for all samples studied and could therefore be candidates for new capture probe design. The following alternative protocol illustrates how identify such intervals. It combines the BEDGRAPH output of the genomecov tool with both awk and the intersect tool to first identify genomic intervals with zero coverage and then intersect such zero-coverage intervals with the targeted genomic intervals to reveal targeted intervals (or portions thereof) that lack coverage.
-
1
Create a BEDGRAPH of coverage genome wide but use awk to filter solely for intervals having zero coverage.
$ bedtools genomecov \
-ibam NA12891.exome.bam \
-bga \
awk ‘$4 == 0’ \
> NA12891.uncovered.bedg
-
2
Intersect intervals having 0 coverage against the original targeted intervals to reveal targeted intervals having zero coverage.
$ bedtools intersect \
-a targets.numeric.chroms.bed \
-b NA12891.uncovered.bedg \
-sorted \
> unsequenced.exome.target.intervals.bed
-
3
Display the first ten intervals that completely lacked coverage.
$ head unsequenced.exome.target.intervals.bed 1 15903 15930 1 16714 16719 1 69551 69560 1 129150 129152 1 877795 877813 1 878015 878017 1 878068 878123 1 896053 896180 1 896634 896659 1 899929 899939 1 896053 896180 1 896634 896659 1 899929 899939
BASIC PROTOCOL 4: MEASURING TRANSCRIPTION FACTOR OCCUPANCY AT TRANSCRIPTION START SITES
The goal of this protocol is to demonstrate how create plots that describe global occupancy profiles of transcription factors at transcription start sites (TSS) throughout the human genome.
Necessary Resources
To demonstrate the use of BEDTools for TSS occupancy profiles, we will use ChIP-seq (in BigWig format) datasets for the Sp1 transcription factor, as well as a reverse cross-linked control, both from the ENCODE project (Dunham et al., 2012).
-
1
Create a shortcut to the ENCODE data hosting site.
$ ENCODE=http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/
-
2
Download a BigWig file for the Sp1 ChIP-seq assay in H1hesc cells.
$ wget $ENCODE/wgEncodeHaibTfbs/wgEncodeHaibTfbsH1hescSp1Pcr1xRawRep1.bigWig
-
3
Download a BigWig file for the reversed crosslink control.
$ wget $ENCODE/wgEncodeHaibTfbs/wgEncodeHaibTfbsH1hescRxlchPcr1xRawRep1.bigWig
Next, we must download a BED file of the transcription start sites for every gene in the UCSC Genome Browser’s “knownGene” track; I have pre-computed this file and it can be directly downloaded with the following command.
-
4
Download a BED file of transcription start sites (TSS).
$ curl -OL http://quinlanlab.cs.virginia.edu/bedtools-protocols/tss.bed
-
5
Display the first 5 lines of the TSS BED file.
$ head -5 tss.bed chr1 11873 11874 DDX11L1 1 + chr1 16764 16765 WASH7P 1 - chr1 17750 17751 WASH7P 1 - chr1 18060 18061 WASH7P 1 - chr1 19758 19759 WASH7P 1 -
Since we are interested in transcription factor occupancy both upstream and downstream of the TSS, we must extend the single base pair of each TSS by, for example, 1000 base pairs upstream and downstream of the TSS. To do this, we will use the BEDTools slop command.
-
6
Add 1000 base pairs both (-b) upstream and downstream of each TSS BED record.
$ bedtools slop \
-b 1000 \
-i tss.bed \
-g hg19.chromsizes \
> tss.plusminus.1000bp.bed
-
7
Display the first 5 lines of the “expanded” (by 2000 bp) TSS BED file.
$ head -n 5 tss.plusminus.1000bp.bed chr1 10873 12874 DDX11L1 1 + chr1 15764 17765 WASH7P 1 - chr1 16750 18751 WASH7P 1 - chr1 17060 19061 WASH7P 1 - chr1 18758 20759 WASH7P 1 -
To provide greater resolution to the plot we will produce, we will break up each 2000bp (TSS +/− 1000bp) interval flanking each TSS into 400, 5bp “sub-windows”. One can easily do this with the BEDTools makewindows command.
-
8
Create 5 base pair BED “sub-records” across each 2 kilobase TSS and its flanks.
# The ‘tr’ command makes the window number the 5th column. This will be used
# to summarize the coverage observed at each of the 2000 bases flanking the
# TSS across all TSSs. The ‘sort’ command ensures that each sub-window remains
# in “genome” order (that is, in ascending order by start position).
$ bedtools makewindows \
-b tss.plusminus.1000bp.bed \
-w 5 \
-i srcwinnum \
| sort -k1,1 -k2,2n \
| tr “_” “\t” \
> tss.plusminus.1000bp.5bp.windows.bed
Lastly, at the time this unit was written, BEDTools did not have native support for BigWig files. Since the Sp1 and control ChIP-seq datasets downloaded from ENCODE are in BigWig format, we need to download the bigWigToBedGraph utility from the UCSC Genome Browser in order to convert the BigWig files to the BEDGRAPH format that BEDTools supports.
-
10
Create a shortcut to the UCSC tools website.
$ UCSC=http://hgdownload.cse.ucsc.edu/admin/exe/
-
11a
Download and install ‘bigWigToBedGraph’ for LINUX operating systems.
$ curl -O $UCSC/linux.x86_64/bigWigToBedGraph $ chmod a+x bigWigToBedGraph $ cp bigWigToBedGraph ~/bin
-
11b
Download and install ‘bigWigToBedGraph’ for OS X operating systems.
$ curl -O $UCSC/macOSX.x86_64/bigWigToBedGraph $ chmod a+x bigWigToBedGraph $ cp bigWigToBedGraph ~/bin
Now that we have downloaded and created the appropriate datasets, we must compare the ChIP-seq peaks for both the Sp1 transcription factor and the negative control to the BED file representing the 2,000 base pair intervals flanking (1,000 bp on each side) each TSS. Recall that we created 5 bp “windows” across each 2,000 bp interval in order to measure transcription factor occupancy at greater resolution than a single statistic count for the entire 2kb interval. Therefore, there are 400 such windows in BED format for each TSS, and our goal is to tabulate the average observed ChIP-seq alignment coverage from the ENCODE BigWig files for each of the 5 base pair windows. As illustrated below, we see that one complication is that there may be multiple discrete BigWig signals that overlap the same (or multiple) 5bp window from a given TSS.
-
12
Peek at the BigWig file (converted to BEDGRAPH format) for Sp1
# Note that the last four records are for four different base pairs and will
# thus overlap the same 5bp TSS window.
$ bigWigToBedGraph wgEncodeHaibTfbsH1hescSp1Pcr1xRawRep1.bigWig stdout /
| head -n 5
chr1 10159 10174 0.25732
chr1 10240 10241 0.32165
chr1 10241 10243 0.38598
chr1 10243 10244 0.45031
chr1 10244 10255 0.51464
In such cases, we will need to summarize each individual observation from the BigWig file to produce a single measure for each 5 bp window. The BEDTools map tool allows one to summarize data from overlapping features in a “B” file onto features in an “A” file by applying summary statistics to specific columns in the B file (Figure 5). In this case we will use map to compute the average BigWig intensity for each ChIP-seq interval from the BigWig file. However, in order to do this, we will first have to convert the BigWig file to BedGraph format for use with BEDTools. Once converted, we will compute the mean of the score (4th) column, which represents the normalized ChIP-seq intensity observed for each interval.
Figure 5. Schematic of the BEDTools map tool’s functionality.

The map tool summarizes the overlaps observed between two interval files, A and B. The result is a summary of all intervals in B that overlapped each interval in A. The summary is computed based on operations such as mean or max, which compute the average or maximum value for a given column from all of the intersecting B intervals.
First, we will use the map tool to summarize both the Sp1 and negative control ChIP-seq intensities. Note that the BigWig file is converted to BedGraph “on the fly” via a UNIX FIFO so that we don’t have to store the data redundantly as both a BigWig and a BedGraph file. Secondly, if there are no overlaps between a given 5 bp window and the BigWig file, we default that window’s value to 0 using the -null option.
-
13
Map the Sp1 transcription factor to the 5bp windows flanking TSS loci.
# Notes:
# (a) ‘-c 4 -o mean’: get the mean of the coverage
# (b) ‘-null 0’: if no overlap with bigwig, set to zero
$ bedtools map \
-a tss.plusminus.1000bp.5bp.windows.bed \
-b <(bigWigToBedGraph wgEncodeHaibTfbsH1hescSp1Pcr1xRawRep1.bigWig stdout) \
-c 4 \
-o mean \
-null 0 \
> sp1.tss.window.coverage.bedg
-
14
Map the Rx1 reverse x-linked control to the 5bp windows flanking TSS loci
$ bedtools map \
-a tss.plusminus.1000bp.5bp.windows.bed \
-b <(bigWigToBedGraph wgEncodeHaibTfbsH1hescRxlchPcr1xRawRep1.bigWig stdout) \
-c 4 \
-o mean \
-null 0 \
> rxl.tss.window.coverage.bedg
At this point, we have summarized the ChIP-seq signal for both Sp1 and the negative control for each 5bp interval flanking each transcript’s TSS. Our goal, however, is to summarize the transcription factor occupancy across all TSS in the entire genome. Recall that when creating the 5bp windows for each TSS, we added a 5th column reflecting which of the 400 distinct 5bp windows each interval represents. We can now take advantage of this by using the BEDTools groupby tool, which will group input data by a particular column (or columns) and for each distinct group, it will summarize the values observed in other columns for each group. In this case, we want to calculate the sum of all mean ChiP-seq intensity values observed for each of the 400, 5bp windows measured for each TSS. The window number is the 5th column and represents our “grouping” column, and the mean ChiP-seq intensity values are in the 6th column and reflects the column that we want to summarize for each group (i.e., 5bp window number). In order to group the data efficiently, the groupby tool requires that the input data be pre-sorted by the grouping column.
-
15
Sum the Sp1 ChIP-seq signal observed for each 5bp window at each TSS start site.
# Note: sort by the window number -t$’\t’ to specify that TABS
# should be used as the delimiter
$ sort -t$’\t’ -k5,5n sp1.tss.window.coverage.bedg \
| bedtools groupby \
-i - \
-g 5 \
-c 6 \
-o sum \
> sp1.tss.window.counts.txt
-
16
Sum the reverse cross-linked control ChIP-seq signal observed for each 5bp window at each TSS start site.
$ sort -t$’\t’ -k5,5n rxl.tss.window.coverage.bedg \
| bedtools groupby \
-i - \
-g 5 \
-c 6 \
-o sum \
> rxl.tss.window.counts.txt
At this point, the resulting files will contain the 401 5bp windows describing summary of all 2000bp intervals surrounding (and including) every TSS. Since the TSS themselves will always be window number 200, we can compare the ChIP-seq signal at the TSS from both the Sp1 and negative control experiments.
-
17
Display the Sp1 signal observed at the TSS (col 1 = 200) and the five 5bp windows upstream and downstream of the TSS.
$ grep -C 5 ^200 sp1.tss.window.counts.txt 195 2510.08048816667860592 196 2526.00322200000437078 197 2551.78026433334161993 198 2575.32502633334479469 199 2587.95086900001206232 200 2580.09832033333896106 201 2556.6253755000034289 202 2543.30477683333856476 203 2520.3936473333401409 204 2477.16924816667051346 205 2462.13425500001221735
-
18
Display the control signal observed at the TSS (col 1 = 200) and the five distinct 5bp windows upstream and downstream of the TSS.
$ grep -C 5 ^200 rxl.tss.window.counts.txt 195 140.054976083333343695 196 136.446768416666657231 197 140.504492583333330913 198 143.949013100000001941 199 147.90348491666665609 200 155.239274333333298728 201 160.626802333333330353 202 154.804520766666655618 203 146.191233833333342318 204 141.775147250000031818 205 133.537657333333356746
As expected, the ChIP-seq signal closest to the TSS is substantial stronger for Sp1 (a well-characterized transcription factor), than for the negative control. Using the following R script, we can visualize the occupancy of these two experiments relative to the 2000 bp intervals flanking all TSS.
-
19
Create a plot of the cumulative density function of the target coverage. The result of the following commands will produce a plot identical to Figure 6.
Figure 6. Transcription factor binding occupancy at transcription start sites.

Depicted are the consensus binding profiles of the Sp1 transcription factor (red) and a reverse cross-linked negative control (gray) as observed among all transcription start sites (TSS) in the human genome.
# 19a. Load the TSS profiles for Sp1 and the negative control into data frames. > sp1 <- read.table(‘sp1.tss.window.counts.txt’) > rxl <- read.table(‘rxl.tss.window.counts.txt’) # 19b. Plot the SP1 profile. > plot(sp1[,1], sp1[,2], col=‘darkred’, xaxt = “n”, xlab=“Distance from TSS”, ylab=“Depth”) # 19c. Plot the negative control profile. > points(rxl[,1], rxl[,2], col=‘darkgrey’) # 19d. Adjust labels based on distance to TSS. # Recall that the window size is 5 base pairs > axis(1, at=seq(0,400,40), labels=seq(-1000,1000,200)) # 19e. Add a vertical line at the TSS. > abline(v = 200, col = “gray60”, lwd=3, lty=3) # 19f. Add a legend. > legend(‘topright’, c(“SP1 Trans. factor”,”Reverse cross-link Control”), lty=1, col=c(‘darkred’, ‘darkgrey’), bty=‘n’)
BASIC PROTOCOL 5: COMPARING INTERVALS AMONG MANY DATASETS
The protocols described thus far have described how to compare pairs of genome interval files with BEDTools. This unit will introduce the multiinter and unionbedg tools, which are each designed to facilitate the comparison of intervals among many distinct files.
Necessary Resources
To demonstrate the utility of the multiinter tool, we will download files representing Dnase I hypersensitivity sites observed by Maurano et al among multiple human tissues (Maurano et al., 2012). I have selected Dnase I hypersensitivity sites from a random subset of 20 fetal tissue samples. Before beginning this protocol, we must first download and extract the 20 individual files.
-
1
Download an archive of Dnase I hypersensitivity sites from 20 cells.
$ curl -O http://quinlanlab.cs.virginia.edu/bedtools-protocols/maurano.dnaseI.tgz
-
2
Extract the 20 individual files.
$ tar -zxvf maurano.dnaseI.tgz
At this point, your directory should now contain 20 new BED files, which reflect Dnase I hypersensitivity sites measured in twenty different fetal tissue samples from the brain, heart, intestine, kidney, lung, muscle, skin, and stomach.
-
3
List the 20 BED files of Dnase I hypersensitivity sites.
$ ls *fdr0.05.merge.bed fBrain-DS14718.hotspot.twopass.fdr0.05.merge.bed fBrain-DS16302.hotspot.twopass.fdr0.05.merge.bed fHeart-DS15643.hotspot.twopass.fdr0.05.merge.bed fHeart-DS15839.hotspot.twopass.fdr0.05.merge.bed fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed fIntestine_Sm-DS16559.hotspot.twopass.fdr0.05.merge.bed fIntestine_Sm-DS16712.hg19.hotspot.twopass.fdr0.05.merge.bed fIntestine_Sm-DS16822.hotspot.twopass.fdr0.05.merge.bed fIntestine_Sm-DS17808.hg19.hotspot.twopass.fdr0.05.merge.bed fIntestine_Sm-DS18495.hg19.hotspot.twopass.fdr0.05.merge.bed fKidney_renal_cortex_L-DS17550.hg19.hotspot.twopass.fdr0.05.merge.bed fLung_L-DS17154.hg19.hotspot.twopass.fdr0.05.merge.bed fLung_L-DS18421.hg19.hotspot.twopass.fdr0.05.merge.bed fLung_R-DS15632.hotspot.twopass.fdr0.05.merge.bed fMuscle_arm-DS19053.hg19.hotspot.twopass.fdr0.05.merge.bed fMuscle_back-DS18454.hg19.hotspot.twopass.fdr0.05.merge.bed fMuscle_leg-DS19115.hg19.hotspot.twopass.fdr0.05.merge.bed fMuscle_leg-DS19158.hg19.hotspot.twopass.fdr0.05.merge.bed fSkin_fibro_bicep_R-DS19745.hg19.hotspot.twopass.fdr0.05.merge.bed fStomach-DS17659.hg19.hotspot.twopass.fdr0.05.merge.bed
By inspecting the BedGraph file for a single sample, we see that each interval represents a site of Dnase I hypersensitivity and the value (column 4) reflects the “signal” of hypersensitivity reflected in the normalized number of aligned reads.
-
4
Display the first five lines of one of the Dnase I hypersensitivity BED files.
$ head -n 5 fBrain-DS14718.hotspot.twopass.fdr0.05.merge.bed chr1 10152 10318 15.5324 chr1 16220 16278 5.45397 chr1 237727 237784 8.80646 chr1 521559 521614 16.5749 chr1 569891 569982 82.3494
Given that Dnase I hypersensitivity sites reflect putative regulatory elements, a natural analysis is the identification of hypersensitivity sites that are either private to a single cell type or common to many cell types. By simultaneously detecting intersections among multiple files, the multiinter tool will report, for every interval observed among the input files, the number and identity of those files having a interval that overlaps with the reported interval. For example, below we use the multiinter tool to report intersecting hypersensitivity sites among the five fetal intestine files. The first three columns reflect the interval in question, followed by the number of input files having intersections at the interval, a list of the file labels (provide by the -names parameter). Lastly, there are five additional columns reflecting whether each file had (value=1) or lacked (value=0) an intersection with the interval in question.
-
5
Report the intervals that are common to 1 or more of the sets of Dnase I hypersensitivity sites from the five fetal intestine samples.
$ bedtools multiinter -i fIntestine*.bed \
-header \
-names DS16559 DS16712 DS16822 DS17808 DS18495 \
| head
chrom start end num list DS16559 DS16712 DS16822 DS17808 DS18495
chr1 10148 10150 2 DS16712,DS17808 0 1 0 1 0
chr1 10150 10151 3 DS16712,DS16822,DS17808 0 1 1 1 0
chr1 10151 10284 4 DS16559,DS16712,DS16822,DS17808 1 1 1 1 0
chr1 10284 10315 3 DS16559,DS16712,DS17808 1 1 0 1 0
chr1 10315 10353 2 DS16712,DS17808 0 1 0 1 0
chr1 237719 237721 1 DS16822 0 0 1 0 0
chr1 237721 237728 3 DS16712,DS16822,DS17808 0 1 1 1 0
chr1 237728 237783 4 DS16559,DS16712,DS16822,DS17808 1 1 1 1 0
chr1 237783 237784 3 DS16559,DS16712,DS16822 1 1 1 0 0
By inspecting the fourth column, one can identify hypersensitivity sites that were exclusive to a single sample, or common to all five intestinal samples.
-
6
Find hypersensitivity sites that are private to a single sample.
$ bedtools multiinter -i fIntestine*.bed \
-header \
-names DS16559 DS16712 DS16822 DS17808 DS18495 \
| awk ‘$4 == 1’ \
| head -n 5
chr1 237719 237721 1 DS16822 0 0 1 0 0
chr1 237787 237788 1 DS16822 0 0 1 0 0
chr1 521474 521559 1 DS18495 0 0 0 0 1
chr1 521571 521614 1 DS16712 0 1 0 0 0
chr1 567615 567712 1 DS16822 0 0 1 0 0
-
7
Find hypersensitivity sites that are common to all samples.
$ bedtools multiinter -i fIntestine*.bed \
-header \
-names DS16559 DS16712 DS16822 DS17808 DS18495 \
| awk ‘$4 == 5’ \
| head -n 5
chr1 569819 569961 5 DS16559,DS16712,DS16822,DS17808,DS18495 1 1 1 1 1
chr1 713936 714345 5 DS16559,DS16712,DS16822,DS17808,DS18495 1 1 1 1 1
chr1 762672 763140 5 DS16559,DS16712,DS16822,DS17808,DS18495 1 1 1 1 1
chr1 840068 840231 5 DS16559,DS16712,DS16822,DS17808,DS18495 1 1 1 1 1
chr1 840681 840910 5 DS16559,DS16712,DS16822,DS17808,DS18495 1 1 1 1 1
Similarly, we can combine the multiinter tool with the groupby tool to measure the proportion of hypersensitive bases that are common to the 20 tissue samples assayed.
-
8
Measure how many bases were observed to be hypersensitive in 1,2,…20 samples.
$ bedtools multiinter -i *.bed \
| awk ‘{print $4”\t”$3-$2}’ \
| sort -k1,1n \
| bedtools groupby -g 1 -c 2 -o sum \
> dnase.occupancy.dist.txt
-
9
Display the number of bases common to 1,2,3,4 and 5 samples.
# Column 1 = Number of cell types # Column 2 = Number of base pairs $ head -n 5 dnase.occupancy.dist.txt 1 172639699 2 70626095 3 51945770 4 35992709 5 27751090
-
10
As before, we will use R to plot the distribution of hypersensitive base pairs common to the 20 samples. The resulting plot (Figure 7) demonstrates that among the assayed tissues, the majority of hypersensitive bases are exclusive to a single cell.
Figure 7.

Histogram of the fraction of Dnase I hypersensitivity sites common to the 20 cell types compared.
# 10a. Load the Dnase I hypersensitivity distribution into an R dataframe. > dnase_occ <- read.table(‘dnase.occupancy.dist.txt’) # 10b. Plot the fraction of bases hypersensitive in 1,2,...20 assayed cells. > plot(dnase_occ[,1], dnase_occ[,2] / sum(dnase_occ[,2]), ‘h’, col=“darkred”, lwd=4, xlab=“No. of assayed cells”, ylab=“Fraction of bases”)
ALTERNATE PROTOCOL 5: COMPARING QUANTITATIVE MEASURES AMONG MULTIPLE BEDGRAPH FILES
We demonstrated how the multiinter tool can be used to identify specific intervals that were either private to a single file or common to one or more genomic interval files. However, the output of the multiinter tool solely reports whether or not a given file had an interval that intersected the reported interval; it does not report the value associated with the BedGraph interval from each file. The unionbedg tool provides the ability to report overlapping intervals from multiple BedGraph files while also reporting the scores observed in each file.
$ bedtools unionbedg -i fIntestine*.bed \
-header \
-names DS16559 DS16712 DS16822 DS17808 DS18495 \
| head
chrom start end DS16559 DS16712 DS16822 DS17808 DS18495
chr1 10148 10150 0 7.76326 0 12.6573 0
chr1 10150 10151 0 7.76326 9.704 12.6573 0
chr1 10151 10284 9.71568 7.76326 9.704 12.6573 0
chr1 10284 10315 9.71568 7.76326 0 12.6573 0
chr1 10315 10353 0 7.76326 0 12.6573 0
chr1 237719 237721 0 0 7.36415 0 0
chr1 237721 237728 0 11.4351 7.36415 7.88268 0
chr1 237728 237783 8.57969 11.4351 7.36415 7.88268 0
chr1 237783 237784 8.57969 11.4351 7.36415 0 0
BASIC PROTOCOL 6: STATISTICS FOR MEASURING DATASET SIMILARITY
As we demonstrated in the previous protocol, the multiinter and unionbedg tools can be used to examine the individual intervals (and their respective scores) that are shared among multiple genome interval files. However, genome datasets are typically comprised of many thousands or millions of individual intervals; this scale complicates simple measurements reflecting the overall similarity of two or more datasets. In this protocol, we will use the jaccard tool to provide a simple similarity metric for pairs of datasets and to facilitate assessments of the similarity of many datasets.
The Jaccard similarity coefficient is a standard metric from set theory that measures the ratio of the size of the intersection of two sets to the size of the union of the two sets. Favorov et al introduced the use of the Jaccard statistic to reflect the similarity of two genome interval sets(Favorov et al., 2012). The authors proposed the metric as the ratio of the total number of intersecting base pairs to the total number of base pairs represented by the intervals in the two sets. As such, the more overlap, the higher the ratio. BEDTools instead calculates the jaccard statistic as the ratio of intersecting base pairs to the union of the two interval sets (in base pairs) minus the intersecting base pairs (Figure 8). This allows the score to range from 0 (no interval overlap) to 1 instead of 0 to 0.5, where the metric is equal to 1 (not 0.5) when the sets are identical.
Figure 8. Schematic of the BEDTools jaccard tool’s functionality.
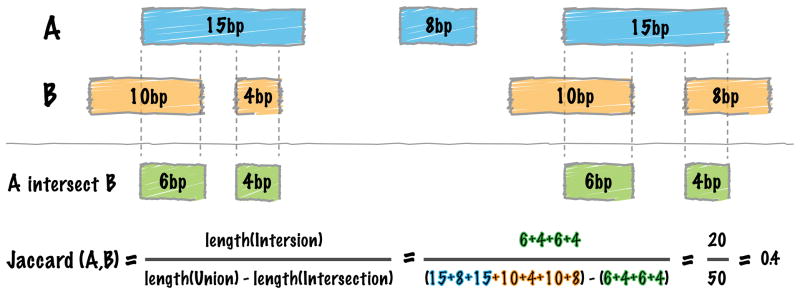
See main text for details.
To demonstrate, we will compute the Jaccard metric for two Dnase I hypersensitivity intervals from independent samples of the same tissue type. The output reports: 1) the total number of intersecting base pairs, 2) the total number of base pairs in the two sets minus the intersecting bases, 3) the Jaccard index, and 4) the total number of interval intersections observed between the two sets. In this example, the Jaccard index exceeds 0.50, reflecting the high similarity of the putative regulatory elements identified in two different fetal heart samples.
-
1
Compute the intersection, union-intersection, Jaccard statistic and the total number of intersections between two fetal heart samples.
$ bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fHeart-DS15839.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
81269248 160493950 0.50637 130852
Intuitively, however, the Jaccard index is substantially lower when comparing the overall similarity of regulatory elements observed in a fetal heart and a fetal skin sample.
-
2
Compute the intersection, union-intersection, Jaccard statistic and the total number of intersections between two different tissue samples.
$ bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fHeart-DS15839.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
28076951 164197278 0.170995 73261
This demonstrates how the jaccard tool can be used to produce a simple statistic to reduce the dimensionality associated with comparing two large (e.g., often containing thousands or millions of intervals) genomic datasets.
We will now extend this analysis to leverage the Jaccard statistic to measure the pairwise similarities among all 20 fetal tissue samples. We will use BASH loops to automatically compute the Jaccard statistic for all 400 (20*20) pairwise comparisons.
-
3
Construct a list of shorter, more reasonable file labels.
$ file_labels=`ls *.bed | \
sed -e ‘s/.hotspot.twopass.fdr0.05.merge.bed//g’ -e ‘s/.hg19//g’`
-
4
Use the short labels as a header for the output file.
$ echo name” “$file_labels ≫ pairwise_jaccard.txt
-
5
Make a matrix of the Jaccard statistic for all 400 (20*20) file comparisons.
$ for file1 in `ls *.bed`
do
file1_short=`echo $file1 \
| sed -e ‘s/.hotspot.twopass.fdr0.05.merge.bed//g’ \
-e ‘s/.hg19//g’`
echo -n $file1_short ≫ pairwise_jaccard.txt
for file2 in `ls *.bed`;
do
jaccard=`bedtools jaccard \
-a $file1 \
-b $file2 \
-valueOnly`
echo -n “ “$jaccard ≫ pairwise_jaccard.txt
done
echo ≫ pairwise_jaccard.txt
done
The result is a text representing a matrix of the 400 pairwise Jaccard metrics observed.
-
6
Display the first 10 tens of the Jaccard matrix.
$ head pairwise_jaccard.txt name fBrain-DS14718 fBrain-DS16302 fHeart-DS15643 fHeart-DS15839 fHeart-DS16621 fIntestine_Sm-DS16559 fIntestine_Sm-DS16712 fIntestine_Sm-DS16822 fIntestine_Sm- DS17808 fIntestine_Sm-DS18495 fKidney_renal_cortex_L-DS17550 fLung_L-DS17154 fLung_L- DS18421 fLung_R-DS15632 fMuscle_arm-DS19053 fMuscle_back-DS18454 fMuscle_leg-DS19115 fMuscle_leg-DS19158 fSkin_fibro_bicep_R-DS19745 fStomach-DS17659 fBrain-DS14718 1 0.354341 0.240543 0.220058 0.227235 0.208477 0.206108 0.201616 0.215246 0.190694 0.260935 0.254287 0.254171 0.260759 0.263694 0.265446 0.266905 0.244176 0.156081 0.248627 fBrain-DS16302 0.354341 1 0.231572 0.209814 0.226111 0.194993 0.178761 0.183642 0.191958 0.175087 0.237877 0.258712 0.251358 0.259236 0.252318 0.24471 0.249426 0.244333 0.135481 0.215611 fHeart-DS15643 0.240543 0.231572 1 0.501461 0.612504 0.274962 0.252228 0.259867 0.279572 0.247334 0.305129 0.321958 0.323752 0.327154 0.322472 0.327793 0.321603 0.297134 0.18551 0.295738 fHeart-DS15839 0.220057 0.209813 0.501456 1 0.50637 0.253843 0.234683 0.236505 0.262172 0.220866 0.278114 0.289965 0.285643 0.280371 0.273637 0.273879 0.265811 0.250537 0.155952 0.2491 fHeart-DS16621 0.227235 0.226111 0.612502 0.50637 1 0.270273 0.226441 0.248787 0.262197 0.23503 0.296984 0.319891 0.317823 0.309777 0.305358 0.309559 0.301319 0.291388 0.170995 0.267889 fIntestine_Sm-DS16559 0.208473 0.194993 0.274958 0.253843 0.270273 1 0.451537 0.607371 0.57613 0.554788 0.313097 0.313202 0.325796 0.310454 0.27922 0.276469 0.273837 0.264617 0.177436 0.347187 fIntestine_Sm-DS16712 0.206104 0.17876 0.252223 0.234683 0.226441 0.451538 1 0.481841 0.523854 0.480868 0.269365 0.251144 0.26833 0.273746 0.242071 0.241538 0.240317 0.210362 0.190258 0.344375 fIntestine_Sm-DS16822 0.201612 0.183641 0.259862 0.236505 0.248787 0.607371 0.481841 1 0.59865 0.569733 0.289532 0.28457 0.300228 0.289781 0.256284 0.258686 0.255764 0.238165 0.18352 0.355715 fIntestine_Sm-DS17808 0.215237 0.191957 0.279561 0.262172 0.262196 0.57613 0.523853 0.59865 1 0.538442 0.311739 0.304329 0.315083 0.306872 0.274056 0.27464 0.272167 0.24807 0.184359 0.361087
This is obviously too much information to discern a sense of the overall patterns observed among the 20 datasets. Yet since the Jaccard statistic serves as a measure of the similarity of two datasets, we may graphically convey the overall similarity of all 20 Dnase I hypersensitivity patterns using a heatmap.
-
7
Plot the Jaccard matrix as a heatmap using R. The result will be identical to Figure 9.
Figure 9.

Heatmap of Jaccard similarities observed for 20 fetal tissue samples based upon Dnase I hypersensitivity profiles.
# 7a. Install RColorBrewer package if missing.
> if (!require(“RColorBrewer”)) { install.packages(“RColorBrewer”);
library(RColorBrewer)}
# 7b. Install heatmap2 package if missing.
> if (!require(“gplots”)) { install.packages(“gplots”); library(glots)}
# 7c. Load the Jaccard matrix into an R data frame.
> jaccard_table <- read.table(‘pairwise_jaccard.txt’, header=TRUE)
# 7d. Add dataset labels.
> row.names(jaccard_table) <- jaccard_table$name
# 7e. Strip the label from the first column
> jaccard_table <- jaccard_table[, -1]
# 7f. Convert the data frame to a matrix for use with the heatmap.2 function.
> jaccard_matrix <- as.matrix(jaccard_table)
# 7g. Plot the Jaccard matrix as a heatmap.
> heatmap.2(jaccard_matrix, col=brewer.pal(9,”Blues”), margins = c(14, 14),
density.info = “none”, lhei = c(2, 8), trace”=none”)
This demonstrates that independent samples of the same fetal tissue have the high degree of similarity among their putative regulatory elements. Moreover, there are, as expected, several examples cases where similar tissue types (e.g., fetal muscle and fetal lung) also exhibit substantial similarity.
GUIDELINES FOR UNDERSTANDING RESULTS
The protocols presented in this unit demonstrate how to compare, explore, and interpret genomics datasets with the BEDTools suite of command line utilities. Each protocol is designed such that the results presented will exactly match those obtained by the reader if the instructions are followed precisely. The results of each protocol will either be a generated plot that should be identical to the relevant plot in the text or an output file whose contents should match the contents presented. If problems arise with individual BEDTools commands, an error message should be generated indicating the source of the error. In the case of errors in the creating of figures using the R statistical package, the reader will be aware that an error has occurred when a plot is not generated that matches the figure in the text.
COMMENTARY
TROUBLESHOOTING
Readers may receive support in the use of BEDTools via an active email mailing list (https://groups.google.com/forum/#!forum/bedtools-discuss), an extensive documentation site (http://bedtools.readthedocs.org/), and the Biostars bioinformatics question and answer website (http://www.biostarts.org).
Acknowledgments
BEDTools has greatly benefited from Neil Kindlon’s programming expertise and dedication. In addition, the BEDTools user community has been instrumental in maintaining and improving the software through bug fixes, the development of new functionality, and the support of new users. In particular, Assaf Gordon, Ryan Layer, Royden Clark, Ryan Dale, Brent Pedersen, and John Marshall have made important contributions. Work in this unit was supported by a grant to ARQ from the National Human Genome Research Institute: R01HG006693-01 (New algorithms and tools for large-scale genomic analysis).
LITERATURE CITED
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham, Kundaje I, Aldred A, Collins SF, Davis PJ, Doyle CA, Epstein F, Frietze CB, Harrow S, Kaul J, Khatun R, Lajoie J, Landt BR, Lee SG, Pauli BK, Rosenbloom F, Sabo KR, Safi P, Sanyal A, Shoresh A, Simon N, Song JM, Trinklein L, Altshuler ND, Birney RC, Brown E, Cheng JB, Djebali C, Dong S, Ernst X, Furey J, Gerstein TS, Giardine M, Greven B, Hardison M, Harris RC, Herrero RS, Hoffman J, Iyer MM, Kelllis S, Kheradpour M, Lassmann P, Li T, Lin Q, Marinov X, Merkel GK, Mortazavi A, Parker A, Reddy SC, Rozowsky TE, Schlesinger J, Thurman F, Wang RE, Ward J, Whitfield LD, Wilder TW, Wu SP, Xi W, Yip HS, Zhuang KY, Bernstein J, Green BE, Gunter ED, Snyder C, Pazin M, Lowdon MJ, Dillon RF, Adams LA, Kelly LB, Zhang CJ, Wexler J, Good JR, Feingold PJ, Crawford EA, Dekker GE, Elinitski J, Farnham L, Giddings PJ, Gingeras MC, Guigo TR, Hubbard R, Kellis TJ, Kent M, Lieb WJ, Margulies JD, Myers EH, Starnatoyannopoulos RM, Tennebaum JA, Weng SA, White Z, Wold KP, Yu B, Wrobel Y, Risk J, Gunawardena BA, Kuiper HP, Maier HC, Xie CW, Chen L, Mikkelsen X, Gillespie TS, Goren S, Ram A, Zhang O, Wang X, Issner L, Coyne R, Durham MJ, Ku T, Truong M, Eaton T, Dobin ML, Tanzer A, Lagarde A, Lin J, Xue W, Williams C, Zaleski BA, Roder C, Kokocinski M, Abdelhamid F, Alioto RF, Antoshechkin T, Baer I, Batut MT, Bell P, Bell I, Chakrabortty K, Chrast S, Curado J, Derrien J, Drenkow T, Dumais J, Dumais E, Duttagupta J, Fastuca R, Fejes-Toth M, Ferreira K, Foissac P, Fullwood S, Gao MJ, Gonzalez H, Gordon D, Howald A, Jha C, Johnson S, Kapranov R, King P, Kingswood B, Li C, Luo G, Park OJ, Preall E, Presaud JB, Ribeca K, Robyr P, Ruan D, Sammeth X, Sandu M, Schaeffer KS, See L, Shahab LH, Skancke A, Suzuki J, Takahashi AM, Tilgner H, Trout H, Walters D, Wang N, Hayashizaki H, Reymond Y, Antonarakis A, Hannon SE, Ruan GJ, Carninci Y, Sloan P, Learned CA, Malladi K, Wong VS, Barber MC, Cline GP, Dreszer MS, Heitner TR, Karolchik SG, Kirkup D, Meyer VM, Long LR, Maddren JC, Raney M, Grasfeder BJ, Giresi LL, Battenhouse PG, Sheffield A, Showers NC, London KA, Bhinge D, Shestak AA, Schaner C, Kim MR, Zhang SK, Mieczkowski ZZ, Mieczkowska PA, Liu JO, McDaniell Z, Ni RM, Rashid Y, Kim NU, Adar MJ, Zhang S, Wang Z, Winter T, Keefe D, Iyer D, Sandhu VR, Zheng KS, Wang M, Gertz P, Vielmetter J, Partridge J, Varley EC, Gasper KE, Bansal C, Pepke A, Jain S, Amrhein P, Bowling H, Anaya KM, Cross M, Muratet MK, Newberry MA, McCue KM, Nesmith K, Fisher-Aylor AS, Pusey KI, DeSalvo B, Parker G, Balasubramanian SL, Davis S, Meadows NS, Eggleston SK, Newberry T, Levy JS, Absher SE, Wong DM, Blow WH, Visel MJ, Pennachio A, Elnitski LA, Petrykowska L, Abyzov HM, Aken A, Barrell B, Barson D, Berry G, Bignell A, Boychenko A, Bussotti V, Davidson G, Despacio-Reyes C, Diekhans G, Ezkurdia M, Frankish I, Gilbert A, Gonzalez J, Griffiths JM, Harte E, Hendrix R, Hunt DA, Jungreis T, Kay I, Khurana M, Leng E, Lin J, Loveland MF, Lu J, Manthravadi Z, Mariotti D, Mudge M, Mukherjee J, Notredame G, Pei C, Rodriguez B, Saunders JM, Sboner G, Searle A, Sisu S, Snow C, Steward C, Tapanari C, Tress E, van Baren ML, Washieti MJ, Wilming S, Zadissa L, Zhengdong A, Brent Z, Haussler M, Valencia D, Raymond A, Addleman A, Alexander N, Auerbach RP, Bettinger RK, Bhardwaj K, Boyle N, Cao AP, Cayting AR, Charos P, Cheng A, Eastman Y, Euskirchen C, Fleming G, Grubert JD, Habegger F, Hariharan L, Harmanci M, Iyenger A, Jin S, Karczewski VX, Kasowski KJ, Lacroute M, Lam P, Larnarre-Vincent H, Lian N, Lindahl-Allen J, Min M, Miotto R, Monahan B, Moqtaderi H, Mu Z, O’Geen XJ, Ouyang H, Patacsil Z, Raha D, Ramirez D, Reed L, Shi B, Slifer M, Witt T, Wu H, Xu L, Yan X, Yang KK, Struhl X, Weissman K, Tenebaum SM, Penalva SA, Karmakar LO, Bhanvadia S, Choudhury RR, Domanus A, Ma M, Moran L, Victorsen J, Auer A, Centarin T, Eichenlaub L, Gruhl M, Heerman F, Hoeckendorf S, Inoue B, Kellner D, Kirchmaier T, Mueller S, Reinhardt C, Schertel R, Schneider L, Sinn S, Wittbrodt R, Wittbrodt B, Jain J, Balasundaram G, Bates G, Byron DL, Canfield R, Diegel TK, Dunn MJ, Ebersol D, Frum AK, Garg T, Gist K, Hansen E, Boatman RS, Haugen L, Humbert E, Johnson R, Johnson AK, Kutyavin EM, Lee TM, Lotakis K, Maurano D, Neph MT, Neri SJ, Nguyen FV, Qu ED, Reynolds H, Roach AP, Rynes V, Sanchez E, Sandstrom ME, Shafer RS, Stergachis AO, Thomas AB, Vernot S, Vierstra B, Vong J, Weaver S, Yan MA, Zhang Y, Akey M, Bender JA, Dorschner M, Groudine MO, MacCoss M, Navas MJ, Stamatoyannopoulos P, Stamatoyannopoulos G, Beal JA, Brazma K, Flicek A, Johnson P, Lukk N, Luscombe M, Sobral NM, Vaquerizas D, Batzoglou JM, Sidow S, Hussami A, Kyriazopoulou-Panagiotopoulou N, Libbrecht S, Schaub MW, Miller MA, Bickel W, Banfai PJ, Boley B, Huang NP, Li H, Noble JJ, Bilmes WS, Buske JA, Sahu OJ, Kharchenko AO, Park PV, Baker PJ, Taylor D, Lochovsky JL. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favorov A, Mularoni L, Cope LM, Medvedeva Y, Mironov AA, Makeev VJ, Wheelan SJ. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput Biol. 2012;8:e1002529. doi: 10.1371/journal.pcbi.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26:2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]