

Abstract
Nitric oxide is a gaseous signaling molecule that is well-known for the Nobel prize-winning research that defined nitric oxide as a physiological regulator of blood pressure in the cardiovascular system. Nitric oxide can signal via the classical pathway involving activation of guanylyl cyclase or by a post-translational modification, referred to as S-nitrosylation (SNO) that can occur on cysteine residues of proteins. As proteins with cysteine residues are common, this allows for amplification of the nitric oxide signaling. This review will focus on the possible mechanisms through which SNO can alter protein function in cardiac cells, and the role of SNO occupancy in these mechanisms. The specific mechanisms that regulate protein SNO, including redox-dependent processes, will also be discussed.
1. Introduction
Until recently, the effects of nitric oxide (NO) were thought to result primarily from the activation of the soluble guanylyl cyclase/cGMP/protein kinase G pathway, but NO is also capable of signaling independent of cGMP, namely via post-translational modification (PTM) of protein thiol groups. This labile, redox-sensitive modification is generally referred to as protein S-nitrosylation (SNO). Similar to other post-translational modifications, SNO affects proteins and enzymes in all cellular compartments and in most signaling pathways. In cardiomyocytes, SNO can occur on a large number of mitochondrial proteins [1–4]. Figure 1 shows an analysis of the pathways that are regulated by SNO. As illustrated, SNO has been shown to regulate an increasing number of cellular pathways and signaling molecules (Figure 1A) in the cardiovascular system and SNO has been implicated as a critical regulator of many of the processes (Figure 1B) that govern normal cellular physiology. 2. Formation, regulation and localization of protein SNO:
Figure 1.

Illustrates the chemistry of S-nitrosylation. Abbreviations: NOS, nitric oxide synthase; GSNOR, GSNO reductase; Trx, thioredoxin.
SNO is an NO-dependent modification, and NO is usually generated by NO synthase (NOS) in the myocardium. There are two constitutive NOS isoforms, endothelial NOS (eNOS) and neuronal NOS (nNOS), as well as an inducible isoform (iNOS). In the presence of the appropriate substrates and co-factors (i.e., tetrahydrobiopterin [BH4], L-arginine), eNOS and nNOS are activated by calcium-calmodulin and produce low levels of NO, while iNOS, which is typically only expressed in the myocardium during inflammatory responses, produces much higher amounts of NO independent of calcium. In the case of co-factor depletion, NOS can become uncoupled and this leads to the production of superoxide rather than NO. In addition, NOS activity can be regulated via PTMs. For example, phosphorylation of S1177 via AKT activates both coupled and uncoupled eNOS [5], while SNO of eNOS promotes the inactive monomeric state [6]. NO can also be generated by non-enzymatic mechanisms (i.e., nitrite reduction), particularly under conditions of low pH as occur during ischemia [7].
NO can promote SNO of protein thiols through several different mechanisms, as illustrated in Figure 1. SNO can be generated through the addition of NO by nitrosylating species, such as dinitrogen trioxide (N2O3) or the nitrosonium ion (NO+). Trans-S-nitrosylation represents another major process that leads to protein SNO. In the case of trans-S-nitrosylation, the direct transfer of NO occurs between SNO proteins with the donor protein referred to as a nitrosylase. Nitrosylases serve to propagate protein SNO beyond local NO signaling domains and offer the potential for target specificity, as recent data suggests that specific protein-protein interactions can lead to specific trans-S-nitrosylation reactions [8, 9]. This could explain why there is currently no consensus SNO sequence. Conversely, SNO is removed from proteins by the action of denitrosylases (see figure 1). S-nitrosoglutathione (GSNO) reductase and thioredoxin are two well characterized denitrosylases [10], with NADH and NADPH serving as electron donors to regenerate glutathione and thioredoxin.
NO signaling is also spatially localized [11–13]. eNOS is targeted to caveolae in the sarcolemmal membrane, whereas nNOS is typically localized to the sarcoplasmic reticulum. NOS localization can also change with disease, as nNOS has been reported to translocate to the plasma membrane following ischemia and heart failure [14, 15]. Although NO is highly diffusible, it is also highly reactive and studies have shown that its bioavailability is spatially limited [12]. Thus, the NO generated by spatially localized NOS isoforms regulates distinct protein targets and trans-S-nitrosylation appears to be an important mechanism for amplification of the NO/SNO signal. For example, NOS does not appear to be present in the nucleus, but SNO signaling is transmitted to the nucleus via trans-S-nitrosylation from proteins such as GAPDH[16]. Several other protein trans-S-nitrosylases have also been described, including hemoglobin [17], caspase-3 [18], and thioredoxin [19, 20]. In addition, the existence of a mitochondrial isoform of NOS is controversial, and thus a role for nitrosylases in the transmission of the SNO signal into the mitochondria or other organelles should be considered.
3. Modification of protein function
SNO has been suggested to modify cellular function by regulating protein or enzyme activity, mediating protein localization, changing binding partners, competing with other PTMs, and shielding critical cysteine residue(s) from irreversible oxidation and altering protein stability. These mechanisms are not mutually exclusive and many act in concert. For example, SNO can alter protein-protein interaction, which can in turn promote a shift in protein localization. Each of these scenarios will be discussed separately and examples will be provided.
a) SNO can alter protein or enzyme activity
Like most PTMs, SNO can alter protein conformation, thus altering activity [4]. The active sites of many enzymes contain cysteine residues, and modification of these residues can induce a change in enzyme activity [21]. SNO has been reported to increase or decrease protein activity, depending on the target. Because of the presence of cysteine residues in the active site of many enzymes there is a tendency for SNO to inhibit rather than activate enzyme activity, although SNO occurs on many regulatory sites leading to activation. In many cases the effect of SNO on activity were confirmed by demonstrating that the addition of an NO donor can alter both activity and protein SNO. For some of these proteins, the modified SNO cysteine residue has been identified, and when this cysteine was mutated, the activity of the protein was no longer altered by NO donors [22, 23]. Although mutagenesis is a common approach to demonstrate a role for PTMs in protein function, appropriate controls need to be performed in order to assure that the mutation (i.e., cysteine to serine or alanine conversion) per se, does not lead to altered conformation or the loss of activity.
For a list of enzyme activities that are increased or decreased by SNO please see our previous review [24]. We will only highlight a few recent examples. SNO of acyl CoA dehydrogenase has been shown recently to decrease the Km of the enzyme, thereby improving catalytic efficiency [25].
A large number of contractile proteins have also been found to be S-nitrosylated [1, 3], although the functional consequence of the modification is just beginning to be explored. Sips et al reported that SNO of myofilament proteins alters calcium sensitivity, thus resulting in an increase in contractility without a concomitant change in the level of calcium [26].
b) SNO can mediate protein localization
Recent data have suggested that PTMs can alter function by changing protein localization. One of the first examples showing that SNO can alter protein localization was from studies in neuronal cells. For example, SNO of GAPDH leads to an interaction with Siah1, thereby stabilizing Siah1 and inducing the nuclear translocation of both proteins[27]. While in the nucleus Siah1, an E3 ubiquitin ligase, selectively degrades nuclear proteins, such as the nuclear receptor co-repressor (N-CoR). GAPDH has also been shown to trans-S-nitrosylate many different proteins while in the nucleus, including SIRT1 and HDAC2, and is therefore referred to as a SIRT1 or HDAC2 trans-S-nitrosylase [8]. Additionally, SNO of β-arrestin 2 has been shown to promote its localization with the clathrin heavy chain/β-adaptin complex, thus leading to internalization into endosomal vesicles [28]. Transglutaminase crosslinks extracellular matrix proteins promoting aortic stiffness; SNO of transglutaminase restricts its localization to the cytosol and reduces the crosslinking of matrix proteins [29]. SNO of apurinic-apyrimidinic endonuclease 1, a major DNA repair enzyme, triggers the export of the enzyme from the nucleus to the cytosol [30]. SNO has also been shown to play a role in the activation and translocation of nuclear factor (erythroid-derived)-related factor 2, which is a transcription factor that plays a role in protecting the cell against oxidative stress [31, 32].
c) SNO can change binding partners
Alterations in protein localization occurring as a result of SNO often act in concert with SNO-induced changes in binding partners. As discussed previously, SNO of β-arrestin 2 promotes dissociation from eNOS in the caveolae and its association with the clathrin heavy chain/β-adaptin complex, thereby altering localization [28]. SNO of GOSPEL (GAPDH’s competitor of Siah1 Protein Enhances Life) leads to an interaction with GAPDH in the cytosol, and this inhibits the nuclear translocation of GAPDH by decreasing the interaction with Siah1 [33]. In addition, SNO of MLC1 increases the binding of matrix metalloproteinase-2, which increases the degradation of MLC1 [34].
d) SNO can compete with other post-translational modifications and shield cysteine residues from irreversible oxidation
In addition to oxidation, there are other modifications, including acylation, prenylation and palmitylation, which can occur on cysteine residues, and these modifications can compete with one another. For example, Snyder’s group has shown that palmitylation targets postsynaptic density protein 95 to the synaptosome and SNO can compete with palmitylation thereby altering the localization of the protein [35]. Furthermore, as discussed in detail in a previous review [36], SNO can also serve as an intermediate in the generation of other modifications such as glutathionylation, sulfinic acid and sulfonic acid.
SNO also can compete with higher order oxidative modifications. Van Eyk and coworkers [37] showed that C294 of the alpha subunit of the F1-F0-ATPase can form a disulfide bond, or undergo SNO or S-glutathionylation depending on the disease state of the heart. In hearts subjected to dyssynchronous heart failure, C294 forms a disulfide bond with another cysteine. With cardiac resynchronization therapy, however this disulfide bond is replaced with an SNO moiety. The disulfide bond was also shown to promote inhibition of the F1-F0-ATPase compared to SNO, suggesting that this PTM change could have important functional implications for the heart. These data are consistent with studies by Sun et al who showed that SNO of the F1-F0-ATPase occurs during preconditioning and is associated with cardioprotection [4].
It has been suggested that SNO can shield critical cysteine residues from the damaging effects of irreversible oxidation [38]. If SNO occurs on critical cysteine residues prior to oxidative stress as occurs during ischemia and reperfusion, these residues can be protected from irreversible oxidation, and because SNO is a transient, reversible modification, normal protein function quickly resumes upon the reversal of SNO. With oxidative stress, proteins can become irreversibly oxidized and dysfunctional, and these proteins then need to be degraded and re-synthesized in order to regain normal function.
To determine if cardioprotection-induced SNO decreases irreversible cysteine oxidation at the same site following ischemia-reperfusion injury, Kohr et al examined a well-established cardioprotective mechanism known as ischemic preconditioning (IPC), which increases protein SNO. Kohr et al reported that ~70% of the cysteine residues that were SNO with IPC showed a concomitant reduction in higher order oxidation at the same site following 5 min of reperfusion [38]. As a proof of concept, it was also shown that with ischemia followed by 5 minutes of reperfusion, GAPDH activity was inhibited due to an increase in higher order oxidation, with no change in SNO. GAPDH is modified via SNO following IPC (prior to sustained ischemia), which also led to the inhibition of activity. However, if IPC hearts were then subjected to sustained ischemia followed by 5 minutes of reperfusion, GAPDH exhibited neither SNO nor oxidation, and GAPDH activity tended toward normal. The IPC-induced SNO was quickly degraded upon reperfusion, but was present long enough to shield the critical cysteine residues of GAPDH from irreversible oxidation. Kohr also found that IPC increased SNO of C203 of cyclophilin D and that this cysteine was also found to exhibit an increase in irreversible oxidation during I/R in the absence of IPC. Nguyen examined the role of SNO of C203 of cyclophilin D and found that it was critical for cyclophilin D mediated activation of the mitochondrial permeability transition pore opening [23]. It is important to emphasize the transient nature of SNO, as it can be rapidly reversed. SNO can have a similar inhibitory effect on the enzyme activity compared to higher order oxidation, but because the modification is quickly lost on reperfusion, it does not have a lasting effect on cardiac function. Thus, cardioprotective mechanisms result in an increase in NO and NOS prior to ischemia and appear to be able to shield some critical cysteine residues from irreversible oxidation. Consistent with this concept, Yakushev et al showed that during hypoxia there is a decrease in SNO, which leads to an increase in glutathionylation of the Na,K-ATPase and this contributes to the inhibition of the Na,K-ATPase that is observed with hypoxia [39].
The studies by Kohr et al discussed above, suggest that the increase in SNO that occurs during IPC is needed for cardioprotection. This raises the question as to whether NO/SNO increases during ischemia in the absence of IPC. Kohr et al found that protein SNO at 5 minutes of reperfusion following ischemia was lower than the level of SNO at baseline, prior to ischemia or the level at 5 min of reperfusion following IPC and ischemia [38]. Thus SNO levels are reduced following ischemia and 5 minutes of reperfusion. On one hand this might be expected since NOS requires oxygen which is limited during global ischemia. Furthermore NOS becomes uncoupled during ischemia due to oxidation of its cofactor BH4 [40] and glutathionylation [41, 42] of eNOS. When NOS is uncoupled it generates superoxide rather than NO, and thus during ischemia and early reperfusion, one would expect NO production and basal SNO to decrease.. However, Zweier et al used electron paramagnetic resonance spectroscopy to measure NO during ischemia and reported a 10-fold increase in NO after 30 minutes of ischemia [43]. Somewhat surprisingly, this generation of NO increased with longer durations of ischemia and a significant percentage of the NO generated during ischemia was blocked by addition of the NOS inhibitor L-NAME. It is particularly surprising that L-NAME blocked the increase in NO during late ischemia, as Zweier and colleagues also reported that NOS is uncoupled during ischemia [40]. Thus, additional studies may be needed to address the relative role of NO/SNO during ischemia.
e) SNO can alter protein stability
SNO appears to be capable of altering the half-life of target proteins. SNO of myosin light chain 1 (MLC1) increases tyrosine nitration, which increases matrix metalloproteinase-2 (MMP2) binding and leads to the degradation of MLC1 [34]. However, the relative role of SNO versus tyrosine nitration in the enhancement of MMP2 binding is unclear.
The role of SNO in regulating protein stability has been studied more extensively in other tissues and several examples are worth considering, as they are likely to provide some future directions in cardiovascular studies. SNO of phosphatase and tensin homolog (PTEN) at C83 is reported to increase degradation through the ubiquitin-proteosomal pathway [44]. In contrast, SNO of Bcl-2 at C158 and C329 decreases degradation by the ubiquitin-proteosomal pathway [45]. Additionally, SNO of HIF-1α has been reported to increase protein stability [46]. In fact, SNO and normoxic stabilization of HIF-1α was shown to occur in mice lacking the denitrosylase, GSNO reductase, and this increase in HIF-1α levels was shown to reduce myocardial infarct size [47]. SNO can also indirectly affect protein stability by modifying E3 ubiquitin ligases that target specific compartments. In 2004, two groups reported that SNO of E3 ubiquitin ligase parkin modulated its activity, although they differed somewhat in the details. Chung et al [48] reported that SNO inhibited parkin, whereas Yao et al [49] reported that SNO initially activated parkin followed by a gradual decrease in activity. In a follow-up report, Chung et al agreed that there was an initial activation of parkin with SNO. Recently Ozawa et al [50] reported that SNO of parkin at C323, results in its activation. These findings are interesting in light of the recent publication detailing the structure of parkin, which shows that the protein is auto-inhibited [51]. The authors mention that auto-inhibition is likely to be regulated by a cysteine modification. Taken together it appears that SNO can lead to activation of parkin, likely by relieving the auto-inhibition. However, SNO of the cysteines involved in zinc coordination would likely lead to inhibition of parkin. The mechanisms of SNO regulation of parkin can likely be applied to other proteins sharing similar structures or motifs.
We recently studied the ability of SNO to alter the stability of the membrane repair protein tripartite motif-containing protein (TRIM72) or mitsugumin-53 (MG53). Several groups have shown that oxidation of TRIM72 at C242 leads to oligomerization and translocation to the plasma membrane, where TRIM72 can participate in membrane repair [52, 53]. We found that C144 of TRIM 72 can undergo SNO [1], and examined the functional effects of this modification at C144. We found that under conditions of oxidative stress, TRIM72 exhibits higher order oxidation and is degraded through a proteosomal-dependent mechanism. However, SNO of C144 or cysteine mutagenesis (cysteine to serine conversion) blocks the oxidative stress-mediated decrease in TRIM72 levels. Taken together these data support the concept that SNO of TRIM72 at C144 contributes to cytoprotection by blocking irreversible oxidation and subsequent degradation of TRIM72. These data further show that SNO can shield critical cysteines from irreversible oxidation and alter protein stability.
4. SNO occupancy
A large number of sites of SNO have been identified in previous studies [1]; these proteins showed SNO at baseline without any stimulus. Figure 1 shows the pathways that contain SNO proteins, which are present at baseline. SNO has been suggested to modify cellular function by altering protein stability, regulating protein or enzyme activity, mediating protein localization, changing binding partners, competing with other PTMs, and shielding critical cysteine residues from irreversible oxidation. If hearts are perfused or homogenates are treated with an NO donor, such as GSNO, there is a large increase in the number of proteins that exhibit potential sites of SNO [1]. A limitation of some of the studies that sought to identify sites of SNO, is that a high dose of an NO donor was often used to induce protein SNO. This leads to the loss of any localized or compartmentalized signaling, and this, coupled with the high dose of an NO donor, can lead to SNO of protein sites that do not occur in physiological or pathophysiological conditions. It is therefore important to verify that these sites occur with physiologic or pathophysiologic stimuli.
In the phosphorylation literature there is an ongoing discussion as to whether all sites of phosphorylation result in functional consequences [54, 55]. It has been suggested that some of these PTMs may be neutral and have no functional consequence. The issue of functional consequences is often raised for PTMs that occur on only a small fraction of a protein. Occupancy is a term used to express the percentage of a protein that is modified at a specific residue. Clearly, a PTM on a small fraction of a protein can have large functional consequences when this PTM occurs on a signaling kinase or phosphatase leading to activation or inhibition of a signaling cascade. For example, a PTM that activates only a small percentage of a signaling molecule such as Akt can have a major impact on cellular function. PTM-mediated inhibition of a small percentage of the sarcolemmal NaK-ATPase at the start of reperfusion could drastically slow the recovery of ion gradients. Similarly, a PTM on a small fraction of a protein which alters the localization, binding partners or stability of the target protein can also have important functional consequences. However, occupancy becomes an important issue when considering the role of SNO in regulating the activity of an enzyme in a metabolic pathway or shielding cysteine residues from irreversible oxidation. For example if SNO leads to inhibition of a metabolic enzyme, such as GAPDH and if only 5% of the enzyme becomes SNO with a treatment, for most metabolic pathways, a transient inhibition of 5% is unlikely to have a major functional consequence. The case of a PTM activation of enzyme activity is more complicated. For example, if the PTM leads to an order of magnitude activation of enzyme activity, then a PTM on 5% of the protein could still have a major effect on the flux through the pathway. A PTM could change substrates or products of a reaction and this could have important functional effects. As discussed previously, S-glutathionylation of eNOS uncouples the enzyme so that it now produces superoxide rather than NO [41]. S-glutathionylation and uncoupling of a small percentage of eNOS could have important function consequences; it could increase reactive oxygen species (ROS) and lead to additional eNOS uncoupling. A PTM of a metabolic enzyme could also alter the localization of the enzyme which could in turn alter the local concentration of a metabolite and this may have functional consequences (e.g hexokinase targeting to the mitochondria). It is also possible that the re-targeted metabolic enzyme could take on a new function in the cell (e.g. moonlighting), as is the case for GAPDH serving as a nuclear trans-nitrosylase. GAPDH is a well-established protein in glycolysis, but GAPDH is also involved with many other cellular signaling pathways. Recently, GAPDH has been suggested to play a role in regulating heme insertion into enzymes such as NOS, and SNO of GAPDH at C152 has been reported to inhibit heme insertion [56]. Therefore, it is possible that these moonlighting functions are also altered by SNO.
To better evaluate these issues, it was important to develop methods to measure SNO occupancy. Kohr et al developed a method using different isobaric tags to label free and SNO cysteine residues [57]. Other have developed similar methods for estimating SNO occupancy [58, 59]. Using these methods, basal SNO occupancy was determined to be low for most protein targets (usually less than 5–10%) as expected, but these occupancy levels can be enhanced by 3 to 4 fold or more with an increase in NOS activity. IPC-mediated cardioprotection results in an increase in SNO of several enzymes to occupancy levels in the range of 20 to 30%. These proteins include hexokinase-1, short-chain acyl CoA dehydrogenase and glycine cleavage system H protein. These data suggest that the SNO occupancies occurring in vivo are sufficient to regulate cellular function.
5. Redox regulation of NO signaling and dysregulation in cardiovascular disease
SNO is a redox-dependent PTM, and changes in cellular redox state can alter the generation of NO, the lifetime or bioavailability of NO, and the reactions that lead to protein SNO and/or denitrosylation. An increase in oxidative stress leads to a decrease in protein SNO, thereby altering the balance between NO/SNO and ROS. ROS leads to the consumption of NO, and thus cardiac specific overexpression of superoxide dismutase leads to an increase in NO bioavailability [60]. SNO can also alter ROS generation. For example the mitochondrial targeted NO donors lead to selective SNO of cysteine 39 on the subunit ND3 of the mitochondrial complex I [58]. NO donors added at the start of reperfusion slow the reactivation of complex I, and thereby reduce ROS production by complex I. Another mechanism by which an increase in oxidative stress reduces NO/SNO signaling is through the uncoupling of NOS. Normally, NOS converts L-arginine to L-citrulline, generating NO in the process. This reaction requires oxygen and the cofactor BH4, which can become oxidized during ischemia [40]. In the absence of BH4, NOS becomes uncoupled to NO generation and primarily produces superoxide, rather than NO. NOS can also become uncoupled if L-arginine levels become limiting. Alterations in NOS signaling have been proposed as a predisposition for cardiovascular disease [61]. Oxidation of BH4 and NOS uncoupling both occur in mouse models of transaortic constriction (TAC), and treatment with BH4 has been shown to improve cardiac function measured after 6 weeks of TAC [62]. Uncoupled NOS has also been reported to contribute to diastolic dysfunction [63], and recent studies have demonstrated that BH4 improves diastolic dysfunction by reversing changes in myofibrillar proteins [64]. This has led to the suggestion that BH4 administration could be beneficial in the treatment of heart failure [65]. However, the initial excitement was tempered by data showing that there was a very narrow beneficial range for dosing with BH4 [66], and a recent randomized control trial of BH4 administration showed no beneficial effect, likely due to the lack of an improvement in the ratio of BH4/BH2 (BH2 represents the oxidized form of BH4) [67].
In addition, recent studies report that glutathionylation of eNOS leads to NOS uncoupling [41]. Crabtree et al. also showed an interaction between eNOS uncoupling due to BH4 oxidation and glutathionylation [42]. More specifically, they reported that a decrease in GSH/GSSG led to glutathionylation and uncoupling of eNOS, which in turn promoted the oxidation of BH4. Similarly, the loss or oxidation of BH4 enhances the glutathionylation of eNOS [42]. Interestingly, nNOS has been shown to regulate the activity of xanthine oxidoreductase, such that the loss of nNOS results in an increase in superoxide, which subsequently leads to an increase in the S-glutathionylation and uncoupling of eNOS [68]. The lack of benefit with BH4 administration may be due to the continued cycle of NOS uncoupling (due to glutathionylation) and ROS generation, leading to the oxidation of BH4. Thus, a more comprehensive approach is needed in which BH4, as well as other mechanisms (glutathionylation, L-arginine levels) of NOS uncoupling, are targeted.
Cardiovascular disease is associated with alterations in the genes that regulate many of the major NO signaling pathways. There are several single nucleotide polymorphisms in the eNOS gene that are associated with cardiomyopathy and heart failure [69–71]. In addition to alterations in NOS levels, alterations in NOS localization or targeting could have important effects on cardiovascular function and disease. As illustrated by the increase in ROS due to loss of nNOS, alterations in NO signaling could have profound effects on SNO and ROS signaling and could play an important role in cardiovascular disease.
6. Summary and Future Directions
SNO can alter protein and enzyme function by a variety of mechanisms as reviewed herein. For discussion, we have classified SNO action into categories such as altering binding partners, altering protein stability, competing with other cysteine PTMs etc. These are likely to be artificial distinctions. For example, the stability of a protein can be altered by changing its binding partners or by altering other PTMs. Thus these different mechanisms synergize to modulate protein function.
There has been a tendency to classify SNO as either beneficial or detrimental. It is likely that the effect of SNO depends on the context of the cell. Thus the level and duration of the NO/SNO signal, the localization of the NO/NOS signal, the redox state of the cell and activity of other signaling pathways, can all influence the role of SNO. In certain settings, such as acute I/R injury, low levels of SNO appear to be beneficial, likely by reducing [58] or ameliorating the increase in ROS associated with early reperfusion [38]. It is also clear that prolonged dysregulation of NO/SNO signaling can contribute to disease.
An important area for future studies is to understand how the cell integrates SNO signaling with other signaling pathways and PTMs [36]. It is also important to better characterize the role of compartmentalization in signaling. eNOS and nNOS are compartmentalized and their activation leads to localized signaling. SNO can also lead to translocation of proteins which can transmit the signal to a different, but still localized compartment. These challenges will likely require the development of novel methodology for studying compartmentalization and for understanding how multiple PTMs are integrated by the cell.
Figure 2.
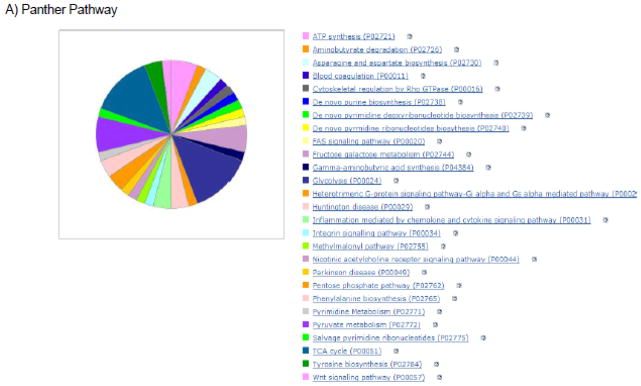
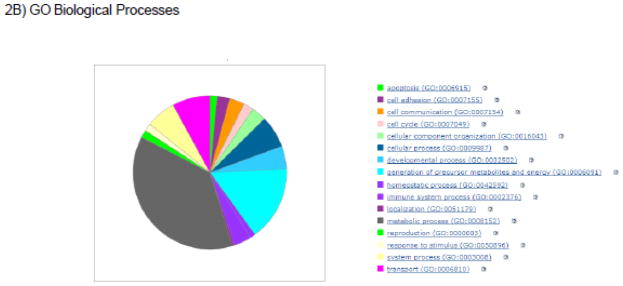
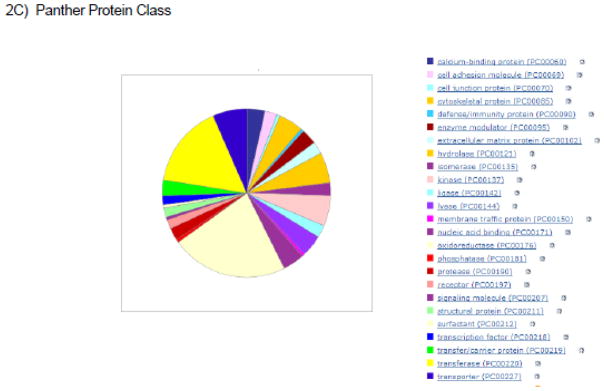
Illustrations of pathways, biological processes, and protein classes that are targeted and/or regulated by SNO. The proteins exhibiting baseline SNO as measured with SNORAC in reference 1 were used for this analysis.
HIGHLIGHTS.
S-nitrosylation can modify cellular function by regulating protein or enzyme activity
S-nitrosylation can mediating protein localization or modulate binding partners
S-nitrosylation can competing with other PTMs
S-nitrosylation can shielding critical cysteine residue(s) from irreversible oxidation
S-nitrosylation can altering protein stability
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, et al. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol. 2011;300(4):H1327–35. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, et al. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010 Sep 28;107(39):16958–63. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray CI, Kane LA, Uhrigshardt H, Wang SB, Van Eyk JE. Site-mapping of in vitro S-nitrosation in cardiac mitochondria: implications for cardioprotection. Mol Cell Proteomics. 2011 Mar;10(3):M110 004721. doi: 10.1074/mcp.M110.004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007 Nov 26;101(11):1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 5.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008 Oct 3;283(40):27038–47. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A. 2004 Feb 24;101(8):2619–24. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med. 1995 Aug;1(8):804–9. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 8.Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med (Berl) 2012 Mar;90(3):233–44. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C, Parrott AM, Fu C, Liu T, Marino SM, Gladyshev VN, et al. Thioredoxin 1-mediated post-translational modifications: reduction, transnitrosylation, denitrosylation, and related proteomics methodologies. Antioxid Redox Signal. 2012 Nov 1;15(9):2565–604. doi: 10.1089/ars.2010.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009 Oct;10(10):721–32. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 11.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002 Mar 21;416(6878):337–9. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 12.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006 Dec 26;103(52):19777–82. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol. 2012 Feb;52(2):341–50. doi: 10.1016/j.yjmcc.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Damy T, Ratajczak P, Robidel E, Bendall JK, Oliviero P, Boczkowski J, et al. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J. 2003 Oct;17(13):1934–6. doi: 10.1096/fj.02-1208fje. [DOI] [PubMed] [Google Scholar]
- 15.Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004 Apr 24;363(9418):1365–7. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 16.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, et al. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010 Nov;12(11):1094–100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409(6820):622–6. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, et al. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39(2):184–95. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1(3):154–8. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 20.Wu C, Liu T, Chen W, Oka S, Fu C, Jain MR, et al. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol. 2010;9(10):2262–75. doi: 10.1074/mcp.M110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer PM, Buga GM, Fukuto JM, Pegg AE, Ignarro LJ. Nitric oxide inhibits ornithine decarboxylase via S-nitrosylation of cysteine 360 in the active site of the enzyme. J Biol Chem. 2001 Sep 14;276(37):34458–64. doi: 10.1074/jbc.M105219200. [DOI] [PubMed] [Google Scholar]
- 22.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, et al. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007 May 4;129(3):511–22. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem. 2011 Nov 18;286(46):40184–92. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy E, Kohr M, Sun J, Nguyen T, Steenbergen C. S-nitrosylation: a radical way to protect the heart. J Mol Cell Cardiol. 2011 Mar;52(3):568–77. doi: 10.1016/j.yjmcc.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal. 2013 Jan 1;6(256):rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sips PY, Irie T, Zou L, Shinozaki S, Sakai M, Shimizu N, et al. Reduction of cardiomyocyte S-nitrosylation by S-nitrosoglutathione reductase protects against sepsis-induced myocardial depression. Am J Physiol Heart Circ Physiol. 2013 Apr 15;304(8):H1134–46. doi: 10.1152/ajpheart.00887.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005 Jul;7(7):665–74. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 28.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, et al. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008 Aug 8;31(3):395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung SM, Jandu S, Steppan J, Belkin A, An SS, Pak A, et al. Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am J Physiol Heart Circ Physiol. 2013 Sep;305(6):H803–10. doi: 10.1152/ajpheart.00103.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35(8):2522–32. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Um HC, Jang JH, Kim DH, Lee C, Surh YJ. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric. 2011;25(2):161–8. doi: 10.1016/j.niox.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A. 2006;103(41):15091–6. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sen N, Hara MR, Ahmad AS, Cascio MB, Kamiya A, Ehmsen JT, et al. GOSPEL: a neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron. 2009;63(1):81–91. doi: 10.1016/j.neuron.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polewicz D, Cadete VJ, Doroszko A, Hunter BE, Sawicka J, Szczesna-Cordary D, et al. Ischemia induced peroxynitrite dependent modifications of cardiomyocyte MLC1 increases its degradation by MMP-2 leading to contractile dysfunction. J Cell Mol Med. 2010 May;15(5):1136–47. doi: 10.1111/j.1582-4934.2010.01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho GP, Selvakumar B, Mukai J, Hester LD, Wang Y, Gogos JA, et al. S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron. 2011 Jul 14;71(1):131–41. doi: 10.1016/j.neuron.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evangelista AM, Kohr MJ, Murphy E. S-nitrosylation: specificity, occupancy, and interaction with other post-translational modifications. Antioxid Redox Signal. 2012 Oct 10;19(11):1209–19. doi: 10.1089/ars.2012.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang SB, Foster DB, Rucker J, O’Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011 Sep 16;109(7):750–7. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011 Feb 18;108(4):418–26. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yakushev S, Band M, Tissot van Patot MC, Gassmann M, Avivi A, Bogdanova A. Cross talk between S-nitrosylation and S-glutathionylation in control of the Na,K-ATPase regulation in hypoxic heart. Am J Physiol Heart Circ Physiol. 2012 Dec 1;303(11):H1332–43. doi: 10.1152/ajpheart.00145.2012. [DOI] [PubMed] [Google Scholar]
- 40.Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, et al. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007 Sep 18;104(38):15081–6. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010 Dec 23;468(7327):1115–8. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crabtree MJ, Brixey R, Batchelor H, Hale AB, Channon KM. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J Biol Chem. 2013 Jan 4;288(1):561–9. doi: 10.1074/jbc.M112.415992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zweier JL, Wang P, Kuppusamy P. Direct measurement of nitric oxide generation in the ischemic heart using electron paramagnetic resonance spectroscopy. J Biol Chem. 1995 Jan 6;270(1):304–7. doi: 10.1074/jbc.270.1.304. [DOI] [PubMed] [Google Scholar]
- 44.Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, et al. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener. 2010;5:49. doi: 10.1186/1750-1326-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, et al. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J Biol Chem. 2006 Nov 10;281(45):34124–34. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- 46.Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, et al. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007 Apr 13;26(1):63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009 Apr 14;106(15):6297–302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, et al. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004 May 28;304(5675):1328–31. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 49.Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004 Jul 20;101(29):10810–4. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ozawa K, Komatsubara AT, Nishimura Y, Sawada T, Kawafune H, Tsumoto H, et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep. 2013;3:2202. doi: 10.1038/srep02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013 Jun 21;340(6139):1451–5. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- 52.Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. 2010;121(23):2565–74. doi: 10.1161/CIRCULATIONAHA.110.954628. [DOI] [PubMed] [Google Scholar]
- 53.Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11(1):56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Covian R, Balaban RS. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am J Physiol Heart Circ Physiol. 2012 Oct 15;303(8):H940–66. doi: 10.1152/ajpheart.00077.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solaro RJ, van der Velden J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J Mol Cell Cardiol. 2010 May;48(5):810–6. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chakravarti R, Aulak KS, Fox PL, Stuehr DJ. GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc Natl Acad Sci U S A. 2010 Oct 19;107(42):18004–9. doi: 10.1073/pnas.1008133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohr MJ, Aponte A, Sun J, Gucek M, Steenbergen C, Murphy E. Measurement of S-nitrosylation occupancy in the myocardium with cysteine-reactive tandem mass tags: short communication. Circ Res. 2012;111(10):1308–12. doi: 10.1161/CIRCRESAHA.112.271320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013 Jun;19(6):753–9. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doulias PT, Tenopoulou M, Raju K, Spruce LA, Seeholzer SH, Ischiropoulos H. Site specific identification of endogenous S-nitrosocysteine proteomes. J Proteomics. 2013 Jun 5; doi: 10.1016/j.jprot.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Obal D, Dai S, Keith R, Dimova N, Kingery J, Zheng YT, et al. Cardiomyocyte-restricted overexpression of extracellular superoxide dismutase increases nitric oxide bioavailability and reduces infarct size after ischemia/reperfusion. Basic Res Cardiol. 2012;107(6):305. doi: 10.1007/s00395-012-0305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005 Mar;115(3):509–17. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008 May 20;117(20):2626–36. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD, et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010 Feb 2;121(4):519–28. doi: 10.1161/CIRCULATIONAHA.109.883777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeong EM, Monasky MM, Gu L, Taglieri DM, Patel BG, Liu H, et al. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol. 2012 Mar;56:44–54. doi: 10.1016/j.yjmcc.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moens AL, Kietadisorn R, Lin JY, Kass D. Targeting endothelial and myocardial dysfunction with tetrahydrobiopterin. J Mol Cell Cardiol. 2011 Oct;51(4):559–63. doi: 10.1016/j.yjmcc.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 66.Moens AL, Ketner EA, Takimoto E, Schmidt TS, O’Neill CA, Wolin MS, et al. Bi-modal dose-dependent cardiac response to tetrahydrobiopterin in pressure-overload induced hypertrophy and heart failure. J Mol Cell Cardiol. 2011 Oct;51(4):564–9. doi: 10.1016/j.yjmcc.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cunnington C, Van Assche T, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, et al. Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation. 2012 Mar 20;125(11):1356–66. doi: 10.1161/CIRCULATIONAHA.111.038919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Idigo WO, Reilly S, Zhang MH, Zhang YH, Jayaram R, Carnicer R, et al. Regulation of endothelial nitric-oxide synthase (NOS) S-glutathionylation by neuronal NOS: evidence of a functional interaction between myocardial constitutive NOS isoforms. J Biol Chem. 2012 Dec 21;287(52):43665–73. doi: 10.1074/jbc.M112.412031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doshi AA, Ziolo MT, Wang H, Burke E, Lesinski A, Binkley P. A promoter polymorphism of the endothelial nitric oxide synthase gene is associated with reduced mRNA and protein expression in failing human myocardium. J Card Fail. 2010 Apr;16(4):314–9. doi: 10.1016/j.cardfail.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cattaruzza M, Guzik TJ, Slodowski W, Pelvan A, Becker J, Halle M, et al. Shear stress insensitivity of endothelial nitric oxide synthase expression as a genetic risk factor for coronary heart disease. Circ Res. 2004 Oct 15;95(8):841–7. doi: 10.1161/01.RES.0000145359.47708.2f. [DOI] [PubMed] [Google Scholar]
- 71.Matsa LS, Rangaraju A, Vengaldas V, Latifi M, Jahromi HM, Ananthapur V, et al. Haplotypes of NOS3 gene polymorphisms in dilated cardiomyopathy. PLoS One. 2013;8(7):e70523. doi: 10.1371/journal.pone.0070523. [DOI] [PMC free article] [PubMed] [Google Scholar]