

Abstract
Rationale: The incidence of pulmonary arterial hypertension is greater in women, suggesting estrogens may play a role in the disease pathogenesis. Experimentally, in males, exogenously administered estrogen can protect against pulmonary hypertension (PH). However, in models that display female susceptibility, estrogens may play a causative role.
Objectives: To clarify the influence of endogenous estrogen and sex in PH and assess the therapeutic potential of a clinically available aromatase inhibitor.
Methods: We interrogated the effect of reduced endogenous estrogen in males and females using the aromatase inhibitor, anastrozole, in two models of PH: the hypoxic mouse and Sugen 5416/hypoxic rat. We also determined the effects of sex on pulmonary expression of aromatase in these models and in lungs from patients with pulmonary arterial hypertension.
Measurements and Main Results: Anastrozole attenuated PH in both models studied, but only in females. To verify this effect was caused by reduced estrogenic activity we confirmed that in hypoxic mice inhibition of estrogen receptor α also has a therapeutic effect specifically in females. Female rodent lung displays increased aromatase and decreased bone morphogenetic protein receptor 2 and Id1 expression compared with male. Anastrozole treatment reversed the impaired bone morphogenetic protein receptor 2 pathway in females. Increased aromatase expression was also detected in female human pulmonary artery smooth muscle cells compared with male.
Conclusions: The unique phenotype of female pulmonary arteries facilitates the therapeutic effects of anastrozole in experimental PH confirming a role for endogenous estrogen in the disease pathogenesis in females and suggests aromatase inhibitors may have therapeutic potential.
Keywords: pulmonary hypertension, estrogen, sex
At a Glance Commentary
Scientific Knowledge on the Subject
Females develop pulmonary arterial hypertension (PAH) more frequently than males. The role of estrogen in this female susceptibility is poorly understood.
What This Study Adds to the Field
Our research shows that inhibition of endogenous estrogen synthesis using an aromatase inhibitor or inhibition of estrogen receptor α has therapeutic effects and restores bone morphogenetic protein receptor 2 expression in female but not male models of PAH. These findings suggest estrogen plays a pathogenic role in the pathology of PAH specifically in females.
Pulmonary arterial hypertension (PAH) is characterized by severe pulmonary arterial remodeling and occlusive pulmonary vascular lesions, leading to right ventricular failure. Epidemiologic studies report a greater incidence of the disease in females; depending on the disease classification the female-to-male ratio can be as great as 4:1 (1, 2). The female predisposition to PAH has given rise to the hypothesis that female sex hormones, primarily estrogens, may play a causative role in the development of the condition. However, the role of estrogen in PAH remains controversial (3).
Most preclinical studies into the role of estrogens in pulmonary hypertension (PH) have used male animals (4–6) and describe protective effects of estrogen when administered exogenously. However, there is compelling evidence that endogenous estrogen may contribute to the pathogenesis of PH. Recently, we have described novel murine models where only female animals develop PH, such as mice overexpressing the serotonin transporter gene (SERT+ mouse) (7) and Mts1 (8). In these models the predominant circulating estrogen 17β-estradiol plays an essential role in the development of the PH phenotype (7–9). Estrogen can also induce proliferation of human pulmonary artery smooth muscle cell (hPASMCs) and may therefore contribute to the pulmonary artery remodeling observed in PAH (7, 10).
One explanation of the current controversies is that there are sex differences in the influence of endogenous estrogens on the pathophysiology of PH. Determining any sex differences in pulmonary responses to estrogen is vitally important for understanding the nature and origins of PAH. To our knowledge, there have been no comprehensive male versus female comparative studies into the role of endogenous estrogen in the pulmonary circulation.
Aromatase (CYP19A1), a member of the cytochrome P-450 superfamily, synthesizes estrogens through the aromatization of androgens. In premenopausal women estrogen synthesis occurs mainly in the ovarian follicles and corpus luteum, but also to a lesser extent in nonglandular tissues, such as adipose tissue and liver. In postmenopausal women and men, adipose tissue is a major source of estrogen (11). Little is known of the role aromatase plays within the pulmonary circulation. Therefore, to characterize the role of endogenous estrogen in PAH, the effects of an aromatase inhibitor were studied in two models of PH in male and female animals. We also assessed aromatase expression in the lung tissue of these animal models and in lung samples from patients with PAH.
Methods
An expanded methods section is available in the online supplement.
Hypoxic Studies
The ability of anastrozole to reduce progression on established PH in hypoxic mice was assessed. Hypoxic PH in C57BL/6 mice was achieved with 14 days hypoxia as described previously (10, 12). Mice were then maintained in hypoxic conditions for a further 14 days during which time the aromatase inhibitor anastrozole was administered (Tocris, Bristol, UK) 0.3 mg ⋅ kg−1 or 3 mg ⋅ kg−1 or vehicle (1% carboxymethylcellulose) daily (subcutaneously). Another cohort of mice was administered with an estrogen receptor (ER) α antagonist, 1,3-bis(4-hydroxyphenyl)-4-methyl-5-(4-[2-piperidinylethoxy]phenol)-1H-pyrazole dihydrochloride (MPP), 2 mg ⋅ kg−1 ⋅ d−1 for 14 days (subcutaneously). Age-matched mice housed in normoxic conditions were studied as control animals. See the online supplement for ethical considerations and housing details.
Sugen 5416 + Hypoxia (Su/Hx) Study
The ability of anastrozole to reduce progression of established PH was also assessed in the rat model of hypoxia + Sugen 5416 (Su) as described in detail in the online supplement. Briefly, Wistar Kyoto rats were given a single injection of Su, 20 mg ⋅ kg−1 (subcutaneously) or 0.9% (subcutaneously) saline and exposed to hypoxia for 14 days then retained in normoxia for 2 weeks during which time they were dosed with anastrozole (0.03, 0.3, or 3 mg ⋅ kg−1 ⋅ d−1) or vehicle (1% carboxymethylcellulose) orally.
Hemodynamic Measurements
Heart rate, right ventricular systolic pressure (RVSP), systemic arterial pressure, and cardiac output were measured and analyzed as previously described (10, 12, 13). See the online supplement for details.
Right Ventricular Hypertrophy
Right ventricular hypertrophy (RVH) was assessed by weighing the right ventricular free wall and left ventricle plus septum, the ratio expressed as RV/LV+S. See the online supplement for details.
Lung Histopathology
Three-micrometer lung sagittal sections were stained with α-smooth muscle actin (<80 μm external diameter) and microscopically assessed for degree of muscularization in a masked fashion, as previously described (14) and in the online supplement.
Quantitative Reverse Transcriptase Polymerase Chain Reaction
mRNA expression was assessed in the lungs of mice by quantitative reverse transcriptase polymerase chain reaction as described previously and in the online supplement (10).
Immunoblotting
Protein expression was assessed by immunoblotting in whole lung and hPASMCs as described previously and in the online supplement (10).
Lung Immunolocalization
Aromatase expression was investigated in murine, rat, and human lung by immunohistochemistry as described previously and in the online supplement (10).
Measurement of Estradiol Concentrations
Circulating estradiol was quantified in plasma by ELISA (Estradiol ELISA, Life Technologies, Paisley, UK).
hPASMCs and PAH-PASMCs
hPASMCs were prepared and cultured as described previously and in the online supplement (10).
Statistics
All data are expressed as mean ± SEM. Data were analyzed using one-way analysis of variance with Dunnett or Bonferroni post hoc analyses and Student unpaired t test (as appropriate and indicated in figure legends) using GraphPad Prism 5 software (GraphPad Prism Inc., San Diego, CA). A P value less than 0.05 was considered statistically significant.
Results
Inhibition of Aromatase Attenuates Experimental PH in Female but Not Male Mice
In female mice anastrozole reduced hypoxia-induced increases in RVSP, RVH, and pulmonary vascular remodeling (PVR) (Figures 1A–1D). However, in male mice, anastrozole when used at the most effective dose (3 mg ⋅ kg−1 ⋅ d−1) had no significant effect on hypoxia-induced elevations in RVSP, RVH, or PVR (Figures 1E and 1H). Anastrozole had no significant effect on mean systemic arterial blood pressure or heart rate (see Figure E1 in the online supplement).
Figure 1.

Inhibition of aromatase attenuates chronic hypoxia-induced pulmonary hypertension (PH) in female mice. Female mice: Effects of the aromatase inhibitor, anastrozole (ANA), 0.3 mg ⋅ kg−1 ⋅ d−1 and 3 mg ⋅ kg−1 ⋅ d−1 for 14 days, on (A) right ventricular systolic pressure (RVSP) (n = 8–10 per group), (B) right ventricular hypertrophy (n = 8–10 per group) (as determined by RV/LV+S ratio), and (C) the % of remodeled pulmonary arteries in normoxic mice and hypoxic female mice with established PH (n = 6 per group). (D) Representative images of pulmonary arteries from normoxic and hypoxic female mice treated with or without anastrozole, 3 mg ⋅ kg−1 ⋅ d−1 (brown = α-smooth muscle actin; scale bar = 20 μm). Male mice: Effects of ANA, 3 mg ⋅ kg−1 ⋅ d−1 for 14 days, on (E) RVSP (n = 8–10 per group), (F) right ventricular hypertrophy (n = 8–10 per group) (as determined by RV/LV+S ratio), and (G) the % of remodeled pulmonary arteries in normoxic mice and hypoxic male mice with established PH (n = 6 per group). (H) Representative images of pulmonary arteries from normoxic and hypoxic male mice treated with or without anastrozole, 3 mg ⋅ kg−1 ⋅ d−1 (brown = α-smooth muscle actin; scale bar = 20 μm). Data displayed as mean ± SEM. **P < 0.01 and ***P < 0.001 as indicated, determined by one-way analysis of variance with Bonferroni post test. RV/LV+S = right ventricle/left ventricle + septum.
To confirm that this effect was not caused by any off-target effects of anastrozole, the effect of MPP, an ERα antagonist, was assessed. MPP was selected because ERα protein levels were found to be significantly elevated in pulmonary arteries from female hypoxic mice, whereas ERβ was significantly reduced. No significant differences in the expression of ERα or ERβ were observed in pulmonary arteries from male mice (see Figure E2). MPP markedly attenuated hypoxia-induced increases in RVSP and PVR in females but had no therapeutic effect in males (see Figure E3).
In female Su/Hx rats anastrozole reduced increases in RVSP, RVH, and PVR and reduced the development of occlusive vascular lesions (Figures 2A–2F). In contrast to females, anastrozole had no significant effect on Su/Hx–induced changes in RVSP, RVH, or PVR in male animals (Figures 3A–3F).
Figure 2.
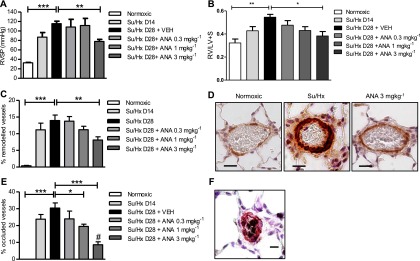
Inhibition of aromatase attenuates Su5416/hypoxia (Su/Hx)-induced pulmonary hypertension (PH) in female rats. (A) Right ventricular systolic pressure (RVSP) (n = 5–8 per group), (B) right ventricular hypertrophy (n = 5–8 per group) (as determined by RV/LV+S), and (C) the % remodeled pulmonary arteries (n = 5–8 per group) were assessed on Day 14 (D14) and Day 28 (D28) following administration of Su/Hx in female rats treated with or without anastrozole (ANA), 0.3 mg ⋅ kg−1 ⋅ d−1, 1 mg ⋅ kg−1 ⋅ d−1, or 3 mg ⋅ kg−1 ⋅ d−1, for 14 days in female rats (from D14–28). Representative images showing (D) α-smooth muscle actin (α-SMA) staining in pulmonary arteries from Su/Hx female rats treated with or without ANA (α-SMA = brown; scale bar = 20 μm). (E) The percentage of pulmonary arteries that are fully occluded in female rats treated with or without ANA (n = 5–8) and (F) representative image of an occluded pulmonary artery (α-SMA = pink, von Willebrand factor = black; scale bar = 20 μm). Data displayed as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 as indicated, #P < 0.01 versus D14 Su/Hx as determined by one-way analysis of variance with Dunnett post test.
Figure 3.
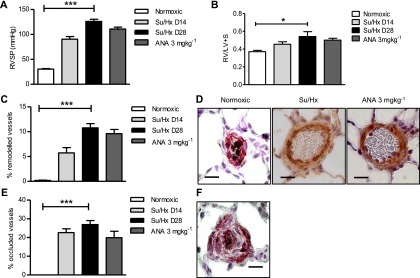
Inhibition of aromatase does not attenuate Su5416/hypoxia (Su/Hx)-induced pulmonary hypertension (PH) in male rats. (A) Right ventricular systolic pressure (RVSP) (n = 5–8 per group), (B) right ventricular hypertrophy (n = 5–8 per group), and (C) the % remodeled pulmonary arteries (n = 5–8 per group) were assessed on Day 14 (D14) and Day 28 (D28) following administration of Su/Hx in male rats treated with or without anastrozole (ANA), 3 mg ⋅ kg−1 ⋅ d−1 (from D14 to D28). Representative images showing (D) α-smooth muscle actin (α-SMA) staining in pulmonary arteries from SU/Hx male rats treated with or without anastrozole (α-SMA = brown; scale bar = 20 μm). (E) The percentage of pulmonary arteries that are fully occluded (n = 5–8 per group) in male rats treated with or without anastrozole, 3 mg ⋅ kg−1 ⋅ d−1, and (F) representative image of an occluded pulmonary artery (α-SMA = pink; von Willebrand factor = black ; scale bar = 10 μm). Data displayed as mean ± SEM. *P < 0.05 and ***P < 0.001 as indicated, determined by one-way analysis of variance with Dunnett post test.
Cardiac and pulmonary functions were assessed by echocardiography. In female rats anastrozole had no effect on cardiac output, but did slightly restore decreased pulmonary artery acceleration time (see Figure E4). In Su/Hx male rats anastrozole had no effect on cardiac output or pulmonary artery acceleration time (see Figure E4).
Aromatase Expression in Hypoxic and Su/Hx–induced PH
Weak aromatase expression was observed in pulmonary arteries from normoxic mice, whereas expression was abundant in hypoxic mice, localizing to the smooth muscle layer (Figure 4A). Analysis of whole-lung tissue by immunoblotting showed that female mice express significantly higher levels of aromatase protein in whole lung compared with male in both normoxic and hypoxic conditions (Figure 4B). Similarly, in pulmonary arteries from normoxic rats negligible to weak aromatase staining was observed, whereas expression was abundant in arteries from Su/Hx rats, localizing within the vascular smooth muscle (Figure 5A). Aromatase staining was absent from the endothelial layer of rat pulmonary arteries (Figure 5B). In addition, aromatase was absent from endothelial cells within the small occlusive vascular lesions observed in Su/Hx rat lung (Figure 5C). Male rats also express significantly lower aromatase protein levels in the whole lung compared with female under both normoxic and Su/Hx conditions (Figure 5D). Exposure to Su/Hx had no effect on aromatase expression in female or male rat lung when compared with normoxic control (Figure 5D).
Figure 4.
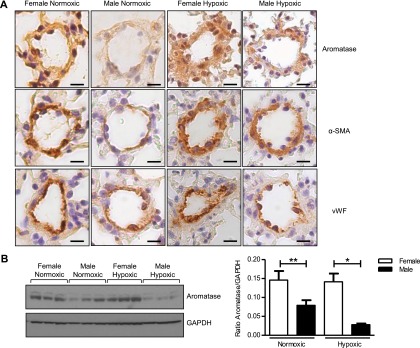
Effect of chronic hypoxia on aromatase expression in mouse pulmonary artery and whole lung. Representative images showing (A) aromatase immunolocalization in pulmonary arteries (scale bar = 20 μm) with 3 μm consecutive sections showing α-smooth muscle actin (α-SMA) and von Willebrand factor (vWF) (representative of n = 4 per group, brown staining). For IgG control see Figure E5. (B) Representative immunoblot and quantification of aromatase protein expression in whole lung from normoxic and hypoxic, female and male mice (n = 5–6 per group). Data displayed as mean ± SEM. *P < 0.05 and **P < 0.01 as indicated, determined by one-way analysis of variance with Bonferroni post test. GAPDH = glyceraldehyde phosphate dehydrogenase.
Figure 5.

Effect of Su5416/hypoxia (Su/Hx) on aromatase expression in rat pulmonary artery and whole lung. Representative images showing (A) aromatase immunolocalization in pulmonary arteries (scale bar = 20 μm) with 3 μm consecutive sections showing α-smooth muscle actin (α-SMA) and von Willebrand factor (vWF) (representative of n = 4 per group, brown staining) (for IgG control see Figure E5). (B) Representative image showing the absence of aromatase immunolocalization in the endothelial layer of rat pulmonary artery (scale bar = 50 μm [×400 magnification]) with 3 μm consecutive sections showing α-SMA and vWF (brown staining). (C) Representative image showing aromatase immunolocalization in small occlusive vascular lesions from SuHx rat (scale bar = 20 μm) with 3 μm consecutive sections showing α-SMA and vWF (brown staining). (D) Representative immunoblot and quantification of aromatase protein expression in whole lung from normoxic and hypoxic, female and male rats (n = 5–6 per group). Data displayed as mean ± SEM. *P < 0.05 and **P < 0.01 as indicated, determined by one-way analysis of variance with Bonferroni post test. GAPDH = glyceraldehyde phosphate dehydrogenase.
Aromatase Expression in Human Lung
Aromatase was found to be expressed in pulmonary arteries of control subjects and female and male patients with PAH localizing mainly within the smooth muscle layer (Figure 6A). Aromatase immunostaining was also present in vascular lesions from patients with PAH (Figure 6B) also localizing to the smooth muscle layer. Aromatase immunoreactivity was absent in the endothelium of most patients with PAH regardless of bone morphogenetic protein receptor 2 (BMPR2) status (Figure 6B; see Figure E5B). In addition, there was no evidence of aromatase expression in human microvascular pulmonary artery endothelial cells (see Figure E5C). PASMCs isolated from control postmenopausal females express significantly higher levels of aromatase than those from males (Figure 6C); however, no significant difference in aromatase expression in PASMCs from female control subjects versus female patients with PAH was observed (Figure 6D).
Figure 6.

Aromatase (AROM) expression in human pulmonary arterial hypertension (PAH). (A) Representative images showing AROM immunolocalization in control and female and male patients with PAH. (B) Representative images showing examples of AROM immunolocalization in vascular lesions from patients with PAH (AROM = pink; α-smooth muscle actin [α-SMA] and von Willebrand factor [vWF] = brown; scale bar = 100 μm; for IgG control see Figure E5). AROM protein expression was also assessed by immunoblotting using human pulmonary artery smooth muscle cells. (C) Representative immunoblots and graph showing quantification comparing AROM expression in human pulmonary artery smooth muscle cells isolated from male and female control subjects (n = 4 samples per group) and (D) female control and female patients with PAH (n = 4 samples per group). Data displayed as mean ± SEM. ***P < 0.001 as indicated, determined by two-tailed, unpaired t test. 1–11, a, e, i, and j correspond to patient information on human tissues and cells referred to in Table E3. GAPDH = glyceraldehyde phosphate dehydrogenase.
Effect of Anastrozole on Circulating Estrogen Levels
Anastrozole, 0.3 mg ⋅ kg−1 and 3 mg ⋅ kg−1, decreased levels of circulating estradiol, the major bioactive estrogen in female mice and had no effect on circulating estrogen levels in the male mice. Furthermore, hypoxia alone had no effect of circulating estrogen levels in either female or male mice (Figure 7A).
Figure 7.
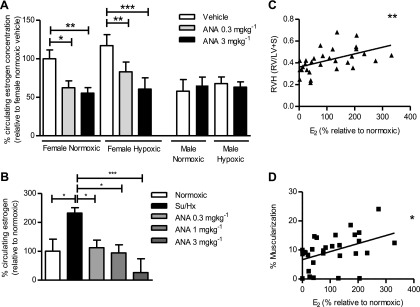
Effect of aromatase inhibition on circulating estradiol (E2) levels in models of pulmonary hypertension. (A) Circulating plasma E2 levels in normoxic and hypoxic female and male mice treated with or without anastrozole (ANA) for 14 days (n = 5 per group). Data displayed as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 as indicated, determined by one-way analysis of variance with Bonferroni post test. (B) Circulating E2 levels in female Su/Hx rats treated with or without 0.3 mg ⋅ kg−1 ⋅ d−1, 1 mg ⋅ kg−1 ⋅ d−1, or 3 mg ⋅ kg−1 ⋅ d−1 ANA (n = 4–5 per group). Data displayed as mean ± SEM. *P < 0.05 and ***P < 0.001 as indicated, determined by one-way analysis of variance with Dunnett post test. Plasma E2 levels from female Su/Hx rats were used to determine if there was any correlation with disease severity using Pearson coefficient. Significant correlation between plasma E2 levels and (C) right ventricular hypertrophy (RVH) (as determined by RV/LV+S) (n = 30), and (D) the percentage of muscularized pulmonary arteries (n = 30). *P < 0.05 and **P < 0.01 as indicated. All E2 concentrations are expressed as a percentage relative to normoxic set at 100%.
In the Su/Hx rat model, estradiol levels were undetectable in male rats. However, in female rats, circulating estradiol levels were found to be significantly elevated in the Su/Hx rats compared with normoxic control animals (Figure 7B). Anastrozole reduced circulating estradiol levels in a dose-dependent fashion. Analysis across the female Su/Hx treatment groups revealed a significant correlation between circulating estradiol concentrations and RVH (Figure 7C) and the percentage of muscularized pulmonary arteries (Figure 7D).
Effects of Anastrozole on Hypoxia and Su/Hx–mediated Changes in BMPR2
In normoxic conditions lung transcript levels of BMPR2 were significantly lower in female mice than male. Administration of anastrozole resulted in a significant up-regulation of BMPR2 in female lung, restoring levels to that of males while having no effect on BMPR2 levels in male lung (Figure 8A). In hypoxia, BMPR2 transcript and protein levels were significantly reduced in both male and female lung. Anastrozole treatment restored the hypoxia-mediated down-regulation in BMPR2 in females (Figures 8A and 8B) but not males (Figures 8A and 8C). In the Su/Hx rat model, BMPR2 transcript was also found to be significantly decreased in both male and female lung compared with normoxic control animals (Figures 8D–8F). This effect was reversed in female rats treated with anastrozole, 3 mg ⋅ kg−1 ⋅ d−1 (Figures 8D and 8E), but not male rats (Figure 8F). The male rats had significantly higher normoxic transcript levels of BMPR2 compared with females, consistent with our observations in mice (Figure 8D).
Figure 8.
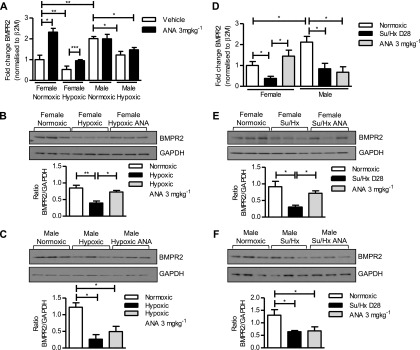
Effects of aromatase inhibition on hypoxia and Su/Hx–induced changes in bone morphogenetic protein receptor 2 (BMPR2) expression. (A–C) Hypoxic mice. (A) Relative gene expression levels of BMPR2 in male and female normoxic and hypoxic mouse lung treated with or without anastrozole (ANA), 3 mg ⋅ kg−1 ⋅ d−1 (n = 6 per group). Representative immunoblot and quantification showing effects of ANA, 3 mg ⋅ kg−1 ⋅ d−1, on BMPR2 protein expression in (B) female and (C) male normoxic and hypoxic mouse lung (n = 6 per group). (D–F) Su/Hx rats. (D) Relative gene expression levels of BMPR2 in female and male normoxic and Su/Hx rat lung treated with or without ANA, 3 mg ⋅ kg−1 ⋅ d−1 for 14 days (n = 5–6 per group). Representative immunoblot and quantification showing effects of ANA, 3 mg ⋅ kg−1 ⋅ d−1, on BMPR2 protein expression in (E) female and (F) male lung from normoxic and Su/Hx rats treated with or without ANA, 3 mg ⋅ kg−1 ⋅ d−1 for 14 days (n = 5–6 per group). Gene expression levels are normalized to β2-microglobulin (β2M). Data displayed as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 as indicated, determined by one-way analysis of variance with Bonferroni post test. GAPDH = glyceraldehyde phosphate dehydrogenase.
Female normoxic mouse and rat lung demonstrated significantly lower levels of Id1 and Id3 (see Figure E6) than males. In females anastrozole significantly elevated Id1 and Id3 to levels similar to that observed in males but had no effect on Id1 and Id3 expression in male lung. Id1 expression was significantly reduced in both male and female disease models, whereas Id3 was specifically down-regulated in males. The PH-mediated decreases in Id1 were rescued by administration of anastrozole in female animals but not male (see Figure E6). ERα antagonist MPP also restored hypoxia-mediated reductions in BMPR2 and Id1 (see Figure E7).
Discussion
Increased synthesis of estrogen has been clinically associated with portopulmonary hypertension (15) and estrogen is causative in female susceptible models of PH (7–9). Studies into the role of estrogen in PAH have failed to reach a consensus, mainly because of the variety of experimental approaches adopted. Indeed, many experimental studies have demonstrated a protective effect of estrogen in male animals (4–6). These valuable studies examined the influence of estrogen administered to males where estrogen levels are normally extremely low or undetectable. In addition, these studies use intact males, hence the presence of high endogenous testosterone combined with high circulating estrogen levels (because of the exogenously added estradiol) is not a state that would normally occur physiologically and may influence interpretation of results. In the instance of monocrotaline-induced PH the beneficial effects of estrogen may be caused by the fact that monocrotaline is a toxin reported to cause gonadal toxicity and reduce estrogen levels (16). In our experimental design we wished to compare males and females and address a different question: “what is the role of endogenous estrogen and is it different in intact males and females?”
The data presented in this study explain some of these current controversies by providing several unique insights into the influence of sex and endogenous estrogen in the development of PH. We demonstrate that endogenous estrogen contributes to the pathophysiology of PH in females and that there is potential for local estrogen synthesis in PASMCs. Given the previously demonstrated mitogenic effects of estrogen in PASMCs (7), we also describe a unique proproliferative phenotype in female PASMCs caused by elevated aromatase and reduced BMPR2 and Id1 expression.
Using anastrozole we inhibited the enzyme aromatase, which is responsible for estrogen synthesis, to determine the role of endogenous estrogen on established PH in females and males. Anastrozole reduced plasma estrogen and attenuated hypoxia and Su/Hx–induced changes in RVSP, RVH, and PVR in females. Furthermore, in the female Su/Hx model a positive correlation between circulating estrogen concentrations and disease severity was established, suggesting the therapeutic effects of anastrozole were related to a decrease in plasma estrogen levels. In the males, there was no therapeutic effect of anastrozole; plasma estrogen was below the level of detection and unaffected by anastrozole.
We also demonstrated that there is a dysregulation in the expression of ERs in hypoxic female mice but not male, with ERα expression significantly increased and ERβ decreased in female pulmonary artery, whereas both receptors remain unaffected in males. Furthermore, we show that an ERα antagonist, MPP, has selective therapeutic effect in female hypoxic mice, not male. This corroborates our hypothesis that endogenous estrogen is pathogenic in female models of PH. Anastrozole is a third-generation highly selective competitive inhibitor of aromatase and as such has few off-target actions. Preclinical studies show that even when used up to doses of 10 mg ⋅ kg−1 in rats no reported disturbances in adrenal steroidogenesis were observed (17).
Estrogen is widely described to be cardioprotective because of its direct action on the heart. Epidemiologic evidence shows that premenopausal women have a lower risk for mortality from cardiovascular diseases than men (18–20). Given the cardioprotective effects of estrogen there is concern that treatment with aromatase inhibitors may facilitate right ventricular dysfunction. Hence, we interrogated the influence of anastrozole on heart function in the Su/Hx rat model by echocardiography. Anastrozole had no detrimental effects on cardiac output or pulmonary artery acceleration time in rats. These findings suggest that depletion of estrogen does not have detrimental effects on the heart that might limit the use of anastrozole in the treatment of PAH. Aromatase inhibitors are currently widely prescribed to patients with ER-sensitive breast cancer and many systemic side effects have been investigated. Available data do not support an association between aromatase inhibitors and an increased risk of cardiovascular disease, PAH, or a deleterious effect on lipid metabolism in humans (21).
Aromatase was expressed in small pulmonary arteries of both female and male rodents localizing within the smooth muscle layer. However, aromatase expression was significantly higher in the lungs from female rats and mice than males. This may partially explain the increased therapeutic effect of anastrozole in the females. We also verified that aromatase is expressed in the smooth muscle of pulmonary arteries in human lung, demonstrating that aromatase is abundantly expressed in vascular smooth muscle from control non-PAH lung sections and in complex vascular lesions. This coupled with the elevated aromatase expression observed in female PASMCs suggests female PASMCs have the ability to synthesis higher levels of estradiol than male. This may contribute to the female susceptibility to PAH given the mitogenic properties of estradiol.
This is the first study to report that there is the potential for local estrogen production in pulmonary arteries. We could find no evidence for aromatase expression in the endothelium of rats, mice, or humans in our studies regardless of their disease status. Likewise, human microvascular pulmonary artery endothelial cells do not express aromatase. This suggests that estrogen produced by PASMCs exerts a paracrine proliferative effect on adjacent PASMCs. Indeed, we have previously demonstrated that estrogen induces proliferation in human PASMCs (7, 8). Estrogen synthesized within extragonadal compartments has been postulated to act at a local tissue level in a paracrine fashion (22). Thus, the total amount of estrogen synthesized by these extragonadal sites may be small but the local tissue concentrations achieved high enough to exert significant biologic influence locally (11). Given the expression of aromatase in the smooth muscle layer of the pulmonary artery the local concentration of estrogen in the pulmonary artery may be much greater than circulating concentrations. Estrogen levels are also affected by metabolism. We have previously shown that expression of cytochrome P-450 1B1 (CYP1B1), an estrogen-metabolizing enzyme, is dysregulated in the Su/Hx mouse model of PH (10). Differences in estrogen metabolism between the hypoxic mouse model and Su/Hx rat model of PH may explain why circulating estrogen levels are elevated in Su/Hx rats but not hypoxic mice.
Loss of function associated with BMPR2 mutations in PAH results in reduction of the growth-inhibitory effects of BMPs, facilitating the proliferation of PASMCs and contributing to PVR (23). BMPR2 is also often observed to be down-regulated in animal models of PAH (24, 25). Here we showed that expression of BMPR2 and its downstream mediator Id1 are significantly decreased in the lungs of normoxic female rodents compared with males. The significantly lower levels of BMPR2 and Id1 in females can be restored to levels similar to that observed in males by anastrozole, suggesting estrogen may be responsible for the suppressed BMPR2 signaling axis in females. Furthermore, anastrozole treatment restored hypoxic and Su/Hx–mediated reductions in BMPR2 mRNA and protein levels in female rodents while having no effect on males. These observations provide one further explanation for the selective therapeutic effect of anastrozole on the development of PH in female models (i.e., endogenous estrogen in the lungs of females is greater because of increased aromatase expression); this combined with the effects of hypoxia or Su/Hx, decreases expression of BMPR2, which is already significantly reduced in females. Consequently anastrozole, by decreasing endogenous estrogen levels, has a selective therapeutic effect in females.
In hPAH families, penetrance of PAH in BMPR2 mutation carriers is low, suggesting other risk factors must influence the emergence of the PAH phenotype. Indeed, further predisposing genes, such as KCNK3 and TOPBP4, have been recently identified (26, 27). Although increased aromatase expression combined with decreased BMPR2 signaling may predispose susceptible females to PAH it is unlikely that these factors alone are responsible for the clinical presentation of disease in all females; and clearly males develop PAH, displaying poorer survival rates than females (1). Our results suggest that once the disease is established, the increased influence of both circulating and locally produced estrogen in women results in an enhanced pathogenic effect on the pulmonary circulation compared with males. Consistent with this, female patients with PAH have 2.8-fold higher number of plexiform lesions compared with their male counterparts (20).
In some patient subgroups including PAH associated with HIV, sleep apnea, and portopulmonary hypertension the prevalence of PAH is greater in males (1). However, these primary conditions occur more frequently in men, potentially influencing the male-to-female ratio of those developing PAH (e.g., [28–30]). In addition, estrogen may contribute to the disease pathophysiology in males within these subgroups. For instance, in HIV dysregulation in sex hormone concentrations has been reported in both sexes. In one study estradiol was reported to significantly increase over an 18-month period in male patients with HIV (31). Furthermore, obstructive sleep apnea is most common in obese men (29), in which elevated circulating estrogen levels are common because of the high expression and activity of aromatase within adipose tissue (32, 33). Polymorphisms in the aromatase gene have also been associated with increased risk of portopulmonary hypertension in patients with liver disease. These polymorphisms are associated with increased estradiol production, supporting a functional effect of aromatase activity in both male and female patients (15). Thus, elevated estrogen is observed in the males in these PAH subgroups. This is not incompatible with the suggestion that when elevated, endogenous estrogen may contribute to the pathobiology of PAH in males and females.
The results of this study also suggest that nonestrogenic contraceptives be recommended to premenopausal patients with PAH, although these are already contraindicated for patients with PAH because of increased risk of venous thromboembolic disease (34).
In summary, we have demonstrated that endogenous estrogen plays a causative role in the development of experimental PH in female animal models of the disease. Inhibition of aromatase with anastrozole reduces moderate and severe experimental PH in female animals via reduction in endogenous estrogen. The reason for the sexual dimorphism in the therapeutic effects of anastrozole may be caused by a unique phenotype of female pulmonary arteries. We propose that increased capability of female PASMCs to produce estrogen locally via aromatase contributes to a reduction in the BMPR2 signaling axis and may contribute to the pathology and increased incidence of the disease in females. The results partly explain the “estrogen paradox” and suggest that aromatase inhibitors may have therapeutic potential in the treatment of PAH in females.
Acknowledgments
Acknowledgment
The authors are grateful to Professor N. W. Morrell (University of Cambridge) for providing human lung tissue and Valerie Khambata (Novartis Institutes of BioMedical Research) for technical assistance.
Footnotes
Supported by a British Heart Foundation program grant (RG/11/7/28916). A.F.W. was a British Heart Foundation funded doctoral student.
Author Contributions: Involvement in the conception, hypothesis delineation, and design of the study, K.M.M., N.D., D.J.R., M.J.H., A.F.W., M.T., and M.R.M. Acquisition of the data or the analysis and interpretation of such information, K.M.M., N.D., S.R., J.F., M.N., L.L., A.F.W., and M.R.M. Writing the article or substantial involvement in its revision before submission, K.M.M., M.T., and M.R.M.
Originally Published in Press as DOI: 10.1164/rccm.201403-0483OC on June 23, 2014
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141:363–373. doi: 10.1378/chest.10-3114. [DOI] [PubMed] [Google Scholar]
- 2.McGoon MD, Benza RL, Escribano-Subias P, Jiang X, Miller DP, Peacock AJ, Pepke-Zaba J, Pulido T, Rich S, Rosenkranz S, et al. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol. 2013;62(Suppl. 25):D51–D59. doi: 10.1016/j.jacc.2013.10.023. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, Brown MB, Van Demark M, Trulock KM, et al. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–980. doi: 10.1164/rccm.201107-1293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, van der Laarse A, Eghbali M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184:715–723. doi: 10.1164/rccm.201101-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu DQ, Luo Y, Liu Y, Wang J, Zhang B, Xu M, Wang YX, Dong HY, Dong MQ, Zhao PT, et al. Beta-estradiol attenuates hypoxic pulmonary hypertension by stabilizing the expression of p27kip1 in rats. Respir Res. 2010;11:182. doi: 10.1186/1465-9921-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res. 2011;90:373–382. doi: 10.1093/cvr/cvq408. [DOI] [PubMed] [Google Scholar]
- 8.Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, Rabinovitch M, MacLean MR. Development of pulmonary arterial hypertension in mice over-expressing s100a4/mts1 is specific to females. Respir Res. 2011;12 doi: 10.1186/1465-9921-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dempsie Y, MacRitchie NA, White K, Morecroft I, Wright AF, Nilsen M, Loughlin L, Mair KM, MacLean MR. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res. 2013;99:24–34. doi: 10.1093/cvr/cvt064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, Morecroft I, Loughlin L, McClure JD, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–1098. doi: 10.1161/CIRCULATIONAHA.111.062927. [DOI] [PubMed] [Google Scholar]
- 11.Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase—a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- 12.Dempsie Y, Morecroft I, Welsh DJ, MacRitchie NA, Herold N, Loughlin L, Nilsen M, Peacock AJ, Harmar A, Bader M, et al. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation. 2008;117:2928–2937. doi: 10.1161/CIRCULATIONAHA.108.767558. [DOI] [PubMed] [Google Scholar]
- 13.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–1182. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- 14.Keegan A, Morecroft I, Smillie D, Hicks MN, MacLean MR. Contribution of the 5-HT(1B) receptor to hypoxia-induced pulmonary hypertension: converging evidence using 5-HT(1B)-receptor knockout mice and the 5-HT(1B/1D)-receptor antagonist GR127935. Circ Res. 2001;89:1231–1239. doi: 10.1161/hh2401.100426. [DOI] [PubMed] [Google Scholar]
- 15.Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, Tighiouart H, Knowles JA, Rabinowitz D, Benza RL, et al. Pulmonary Vascular Complications of Liver Disease Study Group. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835–842. doi: 10.1164/rccm.200809-1472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tofovic SP, Jackson EK. Complexities of oestradiol pharmacology in pulmonary arterial hypertension. Eur Respir J. 2013;41:1465–1466. doi: 10.1183/09031936.00164912. [DOI] [PubMed] [Google Scholar]
- 17.Dukes M, Edwards PN, Large M, Smith IK, Boyle T. The preclinical pharmacology of “Arimidex” (anastrozole; ZD1033)—a potent, selective aromatase inhibitor. J Steroid Biochem Mol Biol. 1996;58:439–445. doi: 10.1016/0960-0760(96)00064-7. [DOI] [PubMed] [Google Scholar]
- 18.Barrett-Connor E. Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys lecture. Circulation. 1997;95:252–264. doi: 10.1161/01.cir.95.1.252. [DOI] [PubMed] [Google Scholar]
- 19.Silber DH. Heart failure in women. Curr Womens Health Rep. 2003;3:104–109. [PubMed] [Google Scholar]
- 20.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–272. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez EA. Safety profiles of tamoxifen and the aromatase inhibitors in adjuvant therapy of hormone-responsive early breast cancer. Ann Oncol. 2007;18(Suppl 8):26–35. doi: 10.1093/annonc/mdm263. [DOI] [PubMed] [Google Scholar]
- 22.Labrie F, Luu-The V, Lin SX, Labrie C, Simard J, Breton R, Bélanger A. The key role of 17 beta-hydroxysteroid dehydrogenases in sex steroid biology. Steroids. 1997;62:148–158. doi: 10.1016/s0039-128x(96)00174-2. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Davies RJ, Southwood M, Long L, Yang X, Sobolewski A, Upton PD, Trembath RC, Morrell NW. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ Res. 2008;102:1212–1221. doi: 10.1161/CIRCRESAHA.108.173567. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, Fukuchi Y. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;290:L450–L458. doi: 10.1152/ajplung.00206.2005. [DOI] [PubMed] [Google Scholar]
- 25.Long L, Crosby A, Yang X, Southwood M, Upton PD, Kim DK, Morrell NW. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119:566–576. doi: 10.1161/CIRCULATIONAHA.108.821504. [DOI] [PubMed] [Google Scholar]
- 26.de Jesus Perez VA, Yuan K, Lyuksyutova MA, Dewey F, Orcholski ME, Shuffle EM, Mathur M, Yancy L, Jr, Rojas V, Li CG, et al. Whole exome sequencing reveals TOPBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:1260–1272. doi: 10.1164/rccm.201310-1749OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girerd B, Perros F, Antigny F, Humbert M, Montani D.KCNK3: new gene target for pulmonary hypertension? Expert Rev Respir Med(In press) [DOI] [PubMed] [Google Scholar]
- 28.Popovic RM, White DP. Upper airway muscle activity in normal women: influence of hormonal status. J Appl Physiol (1985) 1998;84:1055–1062. doi: 10.1152/jappl.1998.84.3.1055. [DOI] [PubMed] [Google Scholar]
- 29.Muxfeldt ES, Margallo VS, Guimarães GM, Salles GF.Prevalence and associated factors of obstructive sleep apnea in patients with resistant hypertension Am J Hypertens(In press) [DOI] [PubMed] [Google Scholar]
- 30.Presanis AM, Gill ON, Chadborn TR, Hill C, Hope V, Logan L, Rice BD, Delpech VC, Ades AE, De Angelis D. Insights into the rise in HIV infections, 2001 to 2008: a Bayesian synthesis of prevalence evidence. AIDS. 2010;24:2849–2858. doi: 10.1097/QAD.0b013e32834021ed. [DOI] [PubMed] [Google Scholar]
- 31.Teichmann J, Schmidt A, Lange U, Stracke H, Discher T, Friese G, Lohmeyer J, Bretzel RG. Longitudinal evaluation of serum estradiol and estrone in male patients infected with the human immunodeficiency virus. Eur J Med Res. 2003;8:77–80. [PubMed] [Google Scholar]
- 32.Schneider G, Kirschner MA, Berkowitz R, Ertel NH. Increased estrogen production in obese men. J Clin Endocrinol Metab. 1979;48:633–638. doi: 10.1210/jcem-48-4-633. [DOI] [PubMed] [Google Scholar]
- 33.Zumoff B, Strain GW, Kream J, O’Connor J, Levin J, Fukushima DK. Obese young men have elevated plasma estrogen levels but obese premenopausal women do not. Metabolism. 1981;30:1011–1014. doi: 10.1016/0026-0495(81)90102-5. [DOI] [PubMed] [Google Scholar]
- 34.Plu-Bureau G, Hugon-Rodin J, Maitrot-Mantelet L, Canonico M. Hormonal contraceptives and arterial disease: an epidemiological update. Best Pract Res Clin Endocrinol Metab. 2013;27:35–45. doi: 10.1016/j.beem.2012.11.003. [DOI] [PubMed] [Google Scholar]