

Abstract
Rationale: Emphysema is a heritable trait that occurs in smokers with and without chronic obstructive pulmonary disease. Emphysema occurs in distinct pathologic patterns, but the genetic determinants of these patterns are unknown.
Objectives: To identify genetic loci associated with distinct patterns of emphysema in smokers and investigate the regulatory function of these loci.
Methods: Quantitative measures of distinct emphysema patterns were generated from computed tomography scans from smokers in the COPDGene Study using the local histogram emphysema quantification method. Genome-wide association studies (GWAS) were performed in 9,614 subjects for five emphysema patterns, and the results were referenced against enhancer and DNase I hypersensitive regions from ENCODE and Roadmap Epigenomics cell lines.
Measurements and Main Results: Genome-wide significant associations were identified for seven loci. Two are novel associations (top single-nucleotide polymorphism rs379123 in MYO1D and rs9590614 in VMA8) located within genes that function in cell-cell signaling and cell migration, and five are in loci previously associated with chronic obstructive pulmonary disease susceptibility (HHIP, IREB2/CHRNA3, CYP2A6/ADCK, TGFB2, and MMP12). Five of these seven loci lay within enhancer or DNase I hypersensitivity regions in lung fibroblasts or small airway epithelial cells, respectively. Enhancer enrichment analysis for top GWAS associations (single-nucleotide polymorphisms associated at P < 5 × 10−6) identified multiple cell lines with significant enhancer enrichment among top GWAS loci, including lung fibroblasts.
Conclusions: This study demonstrates for the first time genetic associations with distinct patterns of pulmonary emphysema quantified by computed tomography scan. Enhancer regions are significantly enriched among these GWAS results, with pulmonary fibroblasts among the cell types showing the strongest enrichment.
Keywords: emphysema, COPD, genetics, gene regulation, spiral computed tomography
At a Glance Commentary
Scientific Knowledge on the Subject
Emphysema is a heritable phenotype, and emphysema occurs in distinct pathologic patterns. Previous genome-wide association studies have implicated three loci with emphysema susceptibility.
What This Study Adds to the Field
Using a novel method of computed tomography emphysema quantification, this study identifies novel genetic loci and established chronic obstructive pulmonary disease susceptibility loci that are associated with distinct emphysema patterns. These loci are located in regulatory functional genomic regions.
Chronic obstructive pulmonary disease (COPD) is a heterogeneous disorder likely to result from multiple underlying disease processes (1, 2). Chronic airflow obstruction in patients with COPD results from variable combinations of emphysema and airway disease. Genetic association analysis may be informative for identifying molecular determinants of these different disease processes. Emphysema, defined as parenchymal destruction and enlargement of distal airspaces in the absence of fibrosis, can be present in individuals with COPD and in smokers with normal spirometry. Emphysema is partly determined by genetics, with an estimated heritability of approximately 30% (3); and genome-wide association studies (GWAS) have identified genetic determinants associated with COPD susceptibility (4–6), spirometric measures (7–9), and emphysema (10, 11).
Emphysema assessment is resource-intensive and requires lung computed tomography (CT) data that must be processed by visual assessment or semiautomatic emphysema quantification algorithms. Semiautomatic algorithms to assess lung densitometric data can efficiently generate reproducible emphysema phenotypes, and they have been widely used in COPD studies. Threshold-based quantification approaches, such as the percentage of low-attenuation area less than −950 Hounsfield units (%LAA-950), are the current standard (12), but they are limited because they quantify emphysema as a single measure, despite the fact that distinct patterns of emphysema have been well-described based on pathology and CT (13, 14). Novel quantification methods use regional CT information to quantify distinct emphysema types (15, 16). One of these methods, local histogram-based emphysema (LHE) quantification, has recently been shown to be more strongly associated with COPD-related physiologic and functional measures than %LAA-950 (17). This method analyzes chest CT scans as 24 × 24 mm2 regions of interest (ROIs), classifying each ROI into distinct parenchymal categories and generating continuous measures for each CT that represent the percentage of ROIs classified to each LHE pattern. Details of the LHE approach, comparison with visual assessments, and epidemiologic associations have been previously described (16, 17).
We hypothesized that GWAS of LHE phenotypes would identify genetic determinants associated with distinct patterns of emphysema on lung CT. Using LHE quantification (17) within the COPDGene Study, a large sample of smokers with genome-wide single-nucleotide polymorphism (SNP) and CT scan data, we performed GWAS in non-Hispanic white (NHW) and African American (AA) subjects to identify genetic determinants associated with distinct patterns of CT emphysema. Some of these results have been previously reported as an abstract (18).
Methods
Subject Enrollment
COPDGene is a multicenter, longitudinal study designed to investigate the genetic and epidemiologic characteristics of COPD and other smoking-related lung diseases. The design of the study has been reported previously (19). Briefly, 10,192 smokers with a wide range of lung function were recruited into the COPDGene Study from 2007 to 2011. NHW and AA subjects between the ages of 45 and 80 with at least a 10 pack-year smoking history were enrolled. Exclusion criteria included pregnancy, history of other lung diseases except asthma, prior lobectomy or lung volume reduction surgery, active cancer undergoing treatment, or known or suspected lung cancer. Volumetric CT scans of the chest were obtained at full inflation and relaxed exhalation. Spirometry was performed with an NDD Easy-One TM Spirometer (Zurich, Switzerland) in accordance with American Thoracic Society/European Respiratory Society recommendations (20).
LHE Quantification
Local histogram-based measures of emphysema were generated from all available inspiratory chest CT scans passing quality control review. Details of the development of the local histogram classifier and training method have been previously described (16, 17). The local histogram method divides the lung CT into 24 mm3 ROIs and produces six quantitative phenotypes for each scan representing the percentage of the ROI falling into each category. The categories are nonemphysematous (or “normal”) lung, mild centrilobular (emphysema with preservation of overall architecture of the secondary lobule), moderate centrilobular (confluent emphysema with preservation of bronchovascular bundle), severe centrilobular (obliteration of the bronchovascular bundle with preservation of septa), panlobular (complete effacement of secondary lobule), and pleural-based emphysema (emphysema abutting the pleural surface). The first five phenotypes were analyzed by GWAS. The pleural-based pattern, meant to capture paraseptal emphysema, poses unique challenges because of the importance of regional information for optimal classification. Methods to further optimize this measure are under development, and this pattern was not included in the current genetic analysis.
Genotyping, Quality Control, and Imputation
The genotyping and quality control procedures for COPDGene have been described (5). COPDGene subjects were genotyped using the Illumina Human Omni Express chip (San Diego, CA). After quality control, 645,914 and 701,491 markers remained in the NHW and AA samples, respectively. Genotype imputation was performed using MaCH (21) and minimac (22) using 1,000 Genomes (23) Phase I v3 European (EUR) and cosmopolitan reference panels for the NHW and AA subjects. The number of variants imputed at R2 greater than 0.3 was 8,117,871 and 14,215,846, respectively. In total, 6,942,916 SNPs present in both NHW and AAs at a minor allele frequency greater than 1% were analyzed. After quality control, genotype data were available for 6,678 NHW and 3,300 AA individuals.
Genome-Wide Association Analysis
GWAS was performed separately in NHWs and AAs for each of the five analyzed quantitative LHE phenotypes using linear regression in plink 1.07 (24) adjusting for age, sex, pack-years of cigarette smoking, and principal components of genetic ancestry. Ancestry principal components were calculated separately for NHWs and AAs using EIGENSOFT 2.0 (25), and adjustment was performed for the first five and six principal components in each racial group, respectively. GWAS results among NHWs and AAs were combined via fixed effects metaanalysis using METAL (version 2010–08–01) (26). The genome-wide significance threshold used for these analyses was P less than 5 × 10−8.
For significant association signals identified in the analysis of all subjects, genetic association was also tested in moderate to very severe COPD cases only (i.e., subjects with Global Initiative for Chronic Obstructive Lung Disease spirometry grade 2–4 disease). Because some LHE phenotypes have distinctly nonnormal distributions, top results were confirmed by performing ordinal logistic regression with the additively coded SNP as the response, and the emphysema phenotype as an independent predictor, adjusting for these same covariates. As additional confirmation, top genetic associations were tested in two logistic regression analyses in which the response was LHE patterns dichotomized at the 50th or 90th percentile, respectively. These analyses were performed in R 3.0, and fixed effects metaanalysis for these results was performed with the metafor package (27, 28). Local association plots were generated with LocusZoom software using the 1,000 Genomes EUR and AMR reference sample for linkage disequilibrium calculations for NHWs and AAs, respectively (29). Recombination rates were calculated from HapMap Phase 2 data.
For genome-wide significant loci, conditional genetic association analyses were performed in NHWs and AAs for all SNPs in a 500-kb window around the most significant SNP from metaanalysis. The threshold for significance of a conditional association was a false discovery rate less than 0.05, calculated from the tested SNPs using the QVALUE package (30, 31).
Enhancer and Promoter Enrichment Analysis in ENCODE and Roadmap Cell Lines
The lead SNPs from each genome-wide significant region were queried against the Haploreg database to identify overlap with epigenetic marks characteristic of enhancer and promoter regions as identified in ENCODE and Roadmap Epigenomics cell lines (32). In addition to the query SNPs, enhancer and promoter regions were identified for all SNPs in linkage disequilibrium (LD) with query SNPs at r2 greater than 0.8 in 1,000 Genomes Phase 1 data.
To test for global enhancer enrichment in top GWAS hits, an additional query of the Haploreg database was performed separately for each phenotype. For these phenotypes, all SNPs below a threshold of P less than 5 × 10−6 were queried against the Haploreg database for overlap with known enhancer regions using the same LD threshold of 0.8. The background set of SNPs used for comparison in the enhancer enrichment analysis consisted of all SNPs in 1,000 Genomes Phase 1 data. P values for enrichment were calculated using the binomial test as implemented by the Haploreg web application.
Results
Subject Characteristics
The characteristics of study subjects are shown in Table 1. In total, 9,743 subjects had complete LHE data available. The mild and moderate centrilobular emphysema patterns were frequently observed in the full study population, whereas the more severe LHE patterns (severe centrilobular and panlobular) were found almost exclusively among only moderate to very severe COPD cases.
Table 1.
Characteristics of Study Subjects
| NHW, All Subjects | NHW, Cases Only | AA, All Subjects | AA, Cases Only | |
|---|---|---|---|---|
| N | 6,533 | 2,760 | 3,210 | 799 |
| Age | 62.1 (8.8) | 64.7 (8.2) | 54.7 (7.2) | 59.0 (8.2) |
| Sex, % male | 52.5 | 55.9 | 55.4 | 54.9 |
| Pack-years | 42 (30–58.5) | 49.5 (37.8–70.5) | 34.5 (22.8–47.0) | 38.2 (25.8–51.9) |
| FEV1, % of predicted | 73.7 (25.9) | 49.8 (18.0) | 82.0 (23.8) | 52.4 (17.7) |
| Normal | 0.59 (0.30–0.77) | 0.30 (0.11–0.59) | 0.71 (0.50–0.81) | 0.36 (0.14–0.63) |
| Mild centrilobular | 0.24 (0.17–0.34) | 0.25 (0.18–0.34) | 0.22 (0.16–0.31) | 0.28 (0.19–0.36) |
| Moderate centrilobular | 0.07 (0.02–0.22) | 0.23 (0.07–0.41) | 0.03 (0.01–0.09) | 0.17 (0.04–0.37) |
| Severe centrilobular | 0.001 (0.0002–0.01) | 0.01 (0.001–0.07) | 0.0002 (0–0.002) | 0.004 (0.0004–0.04) |
| Panlobular | 0.0001 (0–0.005) | 0.005 (0.0001–0.03) | 0 (0–0.0002) | 0.0005 (0–0.01) |
| LAA-950 | 0.03 (0.008–0.09) | 0.09 (0.03–0.22) | 0.01 (0.004–0.03) | 0.05 (0.02–0.16) |
Definition of abbreviations: AA = African American; LAA-950 = proportion of low-attenuation area below −950 Hounsfield units; NHW = non-Hispanic white.
Values are mean (SD) or median (interquartile range).
Cases are defined as Global Initiative for Chronic Obstructive Lung Disease Spirometry Grade 2 or greater.
Emphysema values are proportions of total lung volume for local histogram-based emphysema patterns or proportion of total lung histogram for LAA-950.
GWAS Results
In total, the GWAS analyses included 9,614 COPDGene subjects with complete LHE and genetic data. Genome-wide association yielded significant associations for four of the five studied LHE patterns (Table 2). QQ plots did not show systematic inflation in the GWAS test statistics (see Figure E1 in the online supplement), and the lambda values after genetic ancestry adjustment ranged from 1.01 to 1.03. In total, seven distinct genomic loci achieved genome-wide significance with at least one of the LHE quantitative phenotypes. Of these, five are well-established COPD or spirometric loci identified in previous GWAS (the 15q25 region near IREB2/CHRNA3/CHRNA5 [6], the 4q31 region near HHIP [6, 33], the 19q13 region near CYP2A6/EGLN2/ADCK4 [34], the 11q22 region near MMP12 [5], and the 1q41 region near TGFB2 [5]), and two have not been previously reported. The two novel associations were identified on 13q14 (lead SNP rs9590614) and 17q11 (lead SNP rs379123) for the panlobular and severe centrilobular emphysema patterns, respectively. These novel GWAS peaks are present in the 3′ regions of the VWA8 and MYO1D genes, respectively. Of the loci achieving genome-wide significance, five of the seven loci were nominally significant in both the NHWs and AAs. The SNP genotypes with the strongest association signals at each locus were imputed, with the exception of rs379123, which was directly genotyped. At four of the seven loci, the most strongly associated directly genotyped SNP achieved genome-wide significance, with the strongest directly genotyped P values at the other three loci (1q41, 19q13, and 13q14) ranging from 5.6 × 10−8 to 7.5 × 10−6.
Table 2.
Genome-Wide Significant Associations for Local Histogram Emphysema Phenotypes, All Subjects
| LHE Pattern | Lead SNP | Nearest Gene | Locus | Position (BP) | Effect Allele | P Value Meta | NHW (n =
6,456) |
AA (n =
3,158) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | Effect (SE) | P Value | Frequency | Effect (SE) | P Value | |||||||
| Normal | rs17486278 | CHRNA5 | 15q25 | 78867482 | A | 8.3 × 10−13 | 0.63 | 0.03 (0.005) | 5.3 × 10−10 | 0.71 | 0.02 (0.006) | 2.0 × 10−4 |
| rs138641402 | HHIP | 4q31 | 145445779 | A | 1.7 × 10−9 | 0.64 | −0.03 (0.004) | 1.0 × 10−8 | 0.92 | −0.02 (0.01) | 0.06 | |
| rs1690789 | TGFB2 | 1q41 | 218698027 | C | 2.9 × 10−8 | 0.51 | 0.03 (0.004) | 6.6 × 10−9 | 0.89 | 0.006 (0.01) | 0.56 | |
| rs17368659 | MMP12 | 11q22 | 102742761 | G | 1.1 × 10−8 | 0.88 | −0.04 (0.007) | 1.8 × 10−7 | 0.97 | −0.04 (0.02) | 0.02 | |
| Moderate centrilobular | rs114205691 | CHRNA3 | 15q25 | 78901113 | C | 3.1 × 10−13 | 0.63 | −0.02 (0.003) | 9.4 × 10−11 | 0.82 | −0.01 (0.004) | 8.8 × 10−4 |
| rs56113850 | CYP2A6 | 19q13 | 41353107 | T | 1.3 × 10−9 | 0.40 | −0.02 (0.004) | 1.2 × 10−6 | 0.56 | −0.02 (0.004) | 2.1 × 10−4 | |
| rs17368582 | MMP12 | 11q22 | 102738075 | G | 2.7 × 10−9 | 0.88 | 0.02 (0.004) | 6.6 × 10−9 | 0.97 | 0.02 (0.01) | 0.13 | |
| rs1690789 | TGFB2 | 1q41 | 218698027 | C | 7.9 × 10−9 | 0.51 | −0.02 (0.002) | 3.3 × 10−9 | 0.89 | −0.005 (0.006) | 0.38 | |
| Severe centrilobular | rs9788721 | AGPHD1 | 15q25 | 78802869 | T | 1.8 × 10−13 | 0.62 | −0.007 (0.001) | 1.3 × 10−12 | 0.62 | −0.003 (0.0009) | 1.4 × 10−3 |
| rs379123 | MYO1D | 17q11 | 30891814 | T | 1.5 × 10−8 | 0.59 | −0.005 (0.001) | 1.5 × 10−6 | 0.34 | −0.003 (0.0009) | 1.1 × 10−3 | |
| Panlobular | rs11852372 | AGPHD1 | 15q25 | 78801394 | A | 1.5 × 10−10 | 0.65 | −0.003 (0.0006) | 1.1 × 10−7 | 0.83 | −0.003 (0.0008) | 2.9 × 10−3 |
| rs9590614 | VWA8 | 13q14 | 42175588 | G | 1.1 × 10−8 | 0.61 | −0.002 (0.0006) | 3.3 × 10−4 | 0.83 | −0.004 (0.0008) | 1.0 × 10−5 | |
Definition of abbreviations: AA = African American; BP = base pair location in hg19; Freq = effect allele frequency; LHE = local histogram-based emphysema; NHW = non-Hispanic white; SNP = single-nucleotide polymorphism.
SNPs associated with LHE phenotypes in all subjects at P less than 5 × 10−8 in metaanalysis in both NHW and AAs.
Effect: per allele effect. Phenotype ranges from 0 to 1, with a value of 1 indicating that 100% of computed tomography regions of interest were classified to the pattern of interest.
To determine whether these associations were driven by correlation between the presence of COPD affection status and emphysema, we examined only moderate-to-very severe cases (Table 3). In this analysis, the direction of effect was consistent with that seen in the total sample and the magnitude of effect was typically larger than the main analysis for the seven regions showing genome-wide significance. Consistent with resulting smaller sample sizes, the association P values were generally larger, although the P value did decrease for rs379123. Because some LHE patterns have nonnormal distributions, confirmatory analyses with ordinal logistic regression and logistic regression were also performed, and the results generally confirmed strong associations for these loci (see Tables E1–E4).
Table 3.
Association Evidence for Whole-Population Genome-Wide Significant Regions in COPD Cases (GOLD 2–4) Only
| LHE pattern | Lead SNP | Locus | Effect Allele | P Value Meta | Effect Size in NHW (SE) | P Value NHW | Effect Size in AA (SE) | P Value AA |
|---|---|---|---|---|---|---|---|---|
| Normal | rs17486278 | 15q25 | A | 5.5 × 10−9 | 0.04 (0.007) | 8.2 × 10−9 | 0.02 (0.01) | 0.15 |
| rs138641402 | 4q31 | A | 1.3 × 10−3 | −0.03 (0.008) | 1.6 × 10−9 | −0.02 (0.03) | 0.57 | |
| rs1690789 | 1q41 | C | 1.6 × 10−6 | 0.03 (0.007) | 1.2 × 10−6 | 0.01 (0.02) | 0.64 | |
| rs17368659 | 11q22 | G | 5.7 × 10−5 | −0.05 (0.01) | 1.2 × 10−5 | 0.06 (0.05) | 0.25 | |
| Moderate centrilobular | rs114205691 | 15q25 | C | 1.6 × 10−8 | −0.03 (0.005) | 9.4 × 10−9 | −0.01 (0.01) | 0.34 |
| rs56113850 | 19q13 | T | 3.9 × 10−7 | −0.03 (0.007) | 1.4 × 10−4 | −0.04 (0.01) | 6.6 × 10−4 | |
| rs17368582 | 11q22 | G | 6.3 × 10−5 | 0.03 (0.008) | 2.1 × 10−5 | −0.02 (0.03) | 0.53 | |
| rs1690789 | 1q41 | C | 1.1 × 10−6 | −0.02 (0.005) | 1.6 × 10−6 | −0.01 (0.02) | 0.40 | |
| Severe centrilobular | rs9788721 | 15q25 | T | 2.4 × 10−10 | −0.01 (0.002) | 1.8 × 10−9 | −0.007 (0.003) | 0.041 |
| rs379123 | 17q11 | T | 7.0 × 10−9 | −0.01 (0.002) | 3.1 × 10−7 | −0.01 (0.003) | 6.7 × 10−3 | |
| Panlobular | rs11852372 | 15q25 | A | 1.3 × 10−5 | −0.005 (0.001) | 9.6 × 10−5 | −0.007 (0.003) | 0.026 |
| rs9590614 | 13q14 | G | 5.2 × 10−6 | −0.004 (0.001) | 1.9 × 10−3 | −0.01 (0.003) | 1.8 × 10−5 |
Definition of abbreviations: AA = African American; COPD = chronic obstructive pulmonary disease; GOLD = Global Initiative for Chronic Obstructive Lung Disease; LHE = local histogram-based emphysema; Meta = meta-analysis P value of NHW and AA subjects; NHW = non-Hispanic white; SNP = single-nucleotide polymorphism.
NHW, n = 2,724; AA, n = 786.
LHE association in GOLD 2–4 subjects only for significant SNPs from whole cohort analysis.
Effect: Per allele effect. Phenotype ranges from 0 to 1, with a value of 1 indicating that 100% of computed tomography regions of interest were classified to the pattern of interest.
The mild centrilobular pattern is significantly associated with spirometric and functional measures in smoking control subjects, as previously reported (17). For this reason, we performed GWAS for this pattern in smoking control subjects to examine whether a genetic determinant for this pattern is evident in only this group, but no genome-wide significant associations were observed.
LHE phenotypes quantify distinct patterns of emphysema, but some patterns are highly correlated (17). To determine the extent to which GWAS of the five different LHE phenotypes provides distinct information, we calculated the Spearman rank correlation for the P values for all SNPs (n = 976) associated with any of the LHE phenotypes at P less than 5 × 10−6. This correlation for the top SNP results ranged from −0.40 to 0.79 (see Table E5), with the strongest correlation for top GWAS hits observed between the normal emphysema pattern and the moderate centrilobular pattern. The correlation in top results for the severe centrilobular and panlobular patterns was 0.50, indicating partial independence of the GWAS information from these two phenotypes. At a lower P value threshold of 5 × 10−7, the correlation between GWAS associations for these two LHE phenotypes was 0.57.
Fine Mapping Locus-Specific Association Signals in NHWs and AAs
For the seven loci exceeding genome-wide significance in the metaanalysis of NHW and AAs, we examined the plots within each racial group to assess the concordance of GWAS signals and to better localize candidate causal variants. Local association plots suggest a concordant pattern of association in NHWs and AAs for four of the seven regions (15q25, 19q13, 17q11, and 13q14) and discordant association patterns at the 4q31, 11q22, and 1q41 loci (see Figures E2–E4). For markers at 15q25, 19q13, and 13q14, the association peak was narrower in AA (Figures 1–3). The association peak at 17q11 was narrow in both races and bounded by two closely spaced recombination hotspots (Figures 4B and 4C). To test for allelic heterogeneity, conditional analysis was performed within each racial group, conditioning on the most significant SNP from the metaanalysis. A secondary signal was identified at the 19q13 and 1q41 loci. At 19q13 (Figure 2D), there was a clear secondary association peak in NHWs (but not AAs) approximately 100 kb from the top SNP (secondary lead SNP rs4560023; P = 2.3 × 10−5; q value = 0.02) (Figure 2). A similar pattern was observed at 1q41 (see Figure E4D), with significant primary and secondary signals separated by 57 kb in NHWs but not AAs (secondary lead SNP rs75011710; P = 6.2 × 10−5; q value = 0.04).
Figure 1.
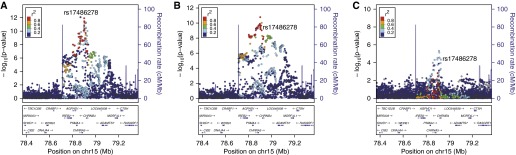
Normal local histogram-based emphysema pattern local association plots near rs17486278 in the 15q25 locus from (A) metaanalysis of non-Hispanic white and African American subjects, (B) non-Hispanic whites (n = 6,456), and (C) African Americans (n = 3,158).
Figure 3.
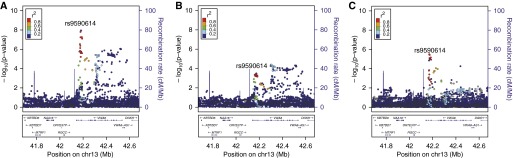
Panlobular local histogram-based emphysema pattern genome-wide association studies local association plots near rs9590614 in the 13q14 locus in (A) metaanalysis of non-Hispanic white and African American subjects, (B) non-Hispanic whites (n = 6,456), and (C) African Americans (n = 3,158).
Figure 4.
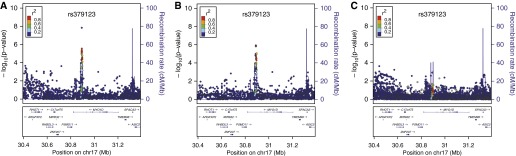
Severe centrilobular local histogram-based emphysema pattern genome-wide association studies local association plots near rs379123 in the 17q11 locus in (A) metaanalysis of non-Hispanic white and African American subjects, (B) non-Hispanic whites (n = 6,456), and (C) African Americans (n = 3,158).
Figure 2.

Moderate centrilobular local histogram-based emphysema pattern genome-wide association studies local association plots near rs56113850 in the 19q13 locus in (A) metaanalysis of non-Hispanic white and African American subjects, (B) non-Hispanic whites (n = 6,456), and (C) African Americans (n = 3,158). Conditional association in non-Hispanic whites conditioning on rs56113850 is shown in D.
Epigenetic Marks and DNase I Hypersensitive Regions in Top GWAS Loci
Most GWAS loci for complex diseases influence gene regulation (35–38). To determine the overlap of the seven genome-wide significant LHE loci found in this study with epigenetic marks and DNase I hypersensitive regions in cell lines from the ENCODE and the Epigenomics Roadmap Projects, we queried the Haploreg database for the seven lead GWAS SNPs, including SNPs in LD at an r2 threshold of 0.8 (32). Overlap of these SNPs with regulatory regions in lung-related cell lines is shown in Table 4, and the results in all cell lines are listed in Tables E6 and E7. Overlap between the most significant SNP (rs379123) for severe centrilobular emphysema with enhancer and DNase I hypersensitive regions is shown in Figure 5. This lead GWAS SNP lies within an annotated enhancer region that also encompasses a DNase I hypersensitive region in small airway epithelial cells.
Table 4.
Enhancer and DNase I Hypersensitive Regions in Lung-related Cell Types in Lead GWAS Loci
| Lead GWAS SNP | Variant | Chr | Position (hg19) | LD (r2) | Promoter Histone Marks | Enhancer Histone Marks | DNAse I Hypersenstivity |
|---|---|---|---|---|---|---|---|
| rs1690789 | rs10047116 | 1 | 218638291 | 0.85 | NHLF, IMR90 | ||
| rs623356 | 1 | 218647386 | 0.84 | NHLF | |||
| rs1764705 | 1 | 218648556 | 0.9 | NHLF, IMR90 | |||
| rs622912 | 1 | 218670357 | 0.96 | NHLF | |||
| rs143667728 | 1 | 218670655 | 0.94 | NHLF | |||
| rs550238 | 1 | 218690948 | 0.99 | LNG.FE | |||
| rs1690789 | 1 | 218698027 | 1 | NHLF, LNG.FE, IMR90 | NHLF | ||
| rs17368659 | rs17361668 | 11 | 102720344 | 0.91 | NHLF | NHLF | |
| rs72981675 | 11 | 102721251 | 0.94 | NHLF | |||
| rs72981680 | 11 | 102721859 | 0.94 | NHLF | |||
| rs9590614 | rs11840821 | 13 | 42162235 | 0.88 | NHLF | ||
| rs148558790 | 13 | 42163581 | 0.98 | NHLF, IMR90 | |||
| rs66700955 | 13 | 42169960 | 0.98 | NHLF, IMR90, LNG.FE | |||
| rs11840816 | 13 | 42171361 | 0.94 | NHLF, IMR90, LNG.FE | |||
| rs9594584 | 13 | 42171439 | 0.94 | NHLF, IMR90, LNG.FE | |||
| rs12584430 | 13 | 42171822 | 0.94 | NHLF, IMR90, LNG.FE | |||
| rs72224690 | 13 | 42171972 | 0.89 | NHLF, IMR90, LNG.FE | |||
| rs12584630 | 13 | 42172071 | 0.94 | NHLF, IMR90, LNG.FE | |||
| rs933010 | 13 | 42173406 | 0.94 | IMR90, LNG.FE | |||
| rs4299049 | 13 | 42173787 | 0.95 | IMR90, LNG.FE | |||
| rs7999090 | 13 | 42182493 | 0.94 | NHLF, LNG.FE, IMR90 | |||
| rs12585912 | 13 | 42183982 | 0.94 | NHLF, LNG.FE, IMR90 | |||
| rs9594585 | 13 | 42184824 | 0.92 | NHLF, LNG.FE, IMR90 | NHLF | ||
| rs17486278 | rs55853698 | 15 | 78857939 | 0.92 | NHLF, IMR90 | LNG.FE | |
| rs55781567 | 15 | 78857986 | 0.92 | NHLF, IMR90 | LNG.FE | ||
| rs8040868 | 15 | 78911181 | 0.81 | NHLF | IMR90 | A549 | |
| rs149959208 | 15 | 78912710 | 0.87 | IMR90 | A549 | ||
| rs379123 | rs225212 | 17 | 30896455 | 0.89 | SAEC |
Definition of abbreviations: A549 = lung cancer–derived lung epithelial cell line; Chr = chromosome; GWAS = genome-wide association studies; IMR90 = fetal lung fibroblast; LD (r2) = r2 between listed variant and lead GWAS SNP; LNG.FE = fetal lung; NHLF = normal lung fibroblast; SAEC = small airway epithelial cells; SNP = single-nucleotide polymorphism.
DNase I hypersensitivity and epigenetic marks associated with promoters and enhancer in lung-related cell types from the ENCODE and Roadmap projects.
Figure 5.
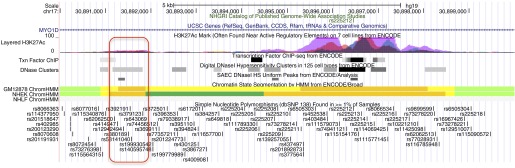
Enhancer and DNase I hypersensitive regions in selected ENCODE and Roadmap cell lines at the 17q11 locus. Red box indicates the lead genome-wide association studies single-nucleotide polymorphism, rs379123, and overlapping enhancer (orange bars) and DNase I hypersensitive regions in multiple cell lines, including small airway epithelial cells (SAEC). There are multiple transcriptionally active candidate regions in the 5′ end of the MYO1D. Image generated with UCSC Genome Browser.
To determine whether there is significant enrichment of enhancers for particular cell lines among the top LHE GWAS hits, we used the Haploreg web interface to perform enhancer enrichment analysis for all SNPs associated at P less than 5 × 10−6 for each of the five LHE phenotypes. Global enrichment for enhancer annotations was observed in at least one cell line for all phenotypes (enrichment P < 0.05; see Tables E8 and E9), and the strongest enrichments were observed with the panlobular (Table 5) and severe centrilobular LHE patterns. The strongest enrichment for top panlobular GWAS hits was observed in lung-related cell lines (i.e., pulmonary fibroblasts and fetal lung tissue), whereas the top hits for the severe centrilobular pattern showed the strongest enhancer enrichment in lymphoblastoid cell lines, CD4+ T cells, and breast myoepithelial cells.
Table 5.
Enhancer Enrichment among Top GWAS Signals for Panlobular LHE Pattern
| Source | Cell Line ID | Cell Line Description | All Enhancers |
Strongest Enhancers |
||||
|---|---|---|---|---|---|---|---|---|
| Observed | Expected | P Value | Observed | Expected | P Value | |||
| ENCODE | NHLF | Lung fibroblasts | 25 | 10.8 | 8.9 × 10−5 | 17 | 4.1 | 1.0 × 10−6 |
| Roadmap | ADI.NUC | Adipose nuclei | 25 | 13.4 | 2.1 × 10−3 | 19 | 6.4 | 2.6 × 10−5 |
| ADI.MSC | Adipose-derived mesenchymal stem cell cultured cells | 41 | 17.4 | <1 × 10−6 | 19 | 7 | 9.0 × 10−5 | |
| LNG.FE | Fetal lung | 29 | 11 | 2.0 × 10−6 | 12 | 3.3 | 1.5 × 10−4 | |
| IMR90 | IMR90 cell line | 25 | 13.1 | 1.5 × 10−3 | 16 | 7 | 2.0 × 10−3 | |
Definition of abbreviations: GWAS = genome-wide association studies; LHE = local histogram-based emphysema; SNP = single-nucleotide polymorphism.
Cell lines with enrichment P values for all enhancer and strong enhancer less than 0.005 considering all SNPs associated with the Panlobular emphysema pattern and P less than 5 × 10−6.
All enhancers: enrichment measures calculated for all chromosome segmentation states associated with enhancer activity.
Strongest enhancers: enrichment measures calculated only for those chromosome segmentation states annotated as strong enhancers.
P value from binomial test comparing the observed count of GWAS SNP-enhancer overlap to the expected count derived from all SNPs in 1,000 Genomes pilot phase.
Genetic Associations with Previously Identified COPD and Emphysema SNPs
We observed strong, but not genome-wide significant, associations for some SNPs previously associated with emphysema or COPD susceptibility in GWAS in other studies (see Tables E10 and E11) including SNPs near AGER and RIN3. Details are included in the online supplement.
Discussion
This study demonstrates for the first time genetic associations with distinct CT emphysema patterns based on established pathologic categories of centrilobular and panlobular emphysema. Five of the genome-wide significant loci identified here have been previously established in COPD case-control or lung function GWAS studies, and two markers near the VMA8 and MYO1D genes represent novel associations. There is strong enrichment of cell-type–specific enhancer regions in these top GWAS results, particularly for lung fibroblasts, indicating that regulation of gene expression in specific lung cell types may be a key functional mechanism for genetic factors influencing emphysema.
Two previous studies have performed GWAS for emphysema phenotypes. Kong and coworkers (10) performed GWAS in 2,383 subjects for visually assessed emphysema and quantitative emphysema (%LAA-950), identifying a genome-wide significant association for visual emphysema in a secondary analysis of severe emphysema (defined as >25% emphysematous involvement). The lead SNP (rs161976) in their analysis was not significantly associated with any of the five LHE phenotypes in our study. Manichaikul and coworkers (11) performed GWAS for %LAA-950 in 7,914 subjects from the MESA Lung/SHARe study, a multiethnic, general population cohort consisting primarily of non-COPD subjects with a median %LAA-950 between 2 and 4%. This study identified genome-wide significant associations between %LAA-950 and variants near SNRPF and PPT2/AGER that were previously reported to be associated with lung function in the general population. Our findings replicated the association at the PPT2/AGER locus and provided suggestive evidence of association near SNRPF.
The association with markers in 13q14 with the panlobular LHE pattern is located at the 3′ end of the VWA8 gene. This gene has two validated RefSeq isoforms, and the region of strongest signal is located in the longer isoform in a region containing a von Willebrand factor type A conserved protein domain. von Willebrand factor type A domains, originally described in the von Willebrand factor protein, are present in many other proteins, including integrins and collagens, and have been implicated in multiple cellular functions including cell-cell signaling and cell migration (39). However, VWA8 has not previously been implicated in COPD pathogenesis. VWA8 is expressed in many tissues, including homogenized lung tissue, fetal lung, and bronchial and tracheal epithelium (40).
The novel association between the severe centrilobular LHE pattern and markers at 17q11 locus encompasses the 3′ end of MYO1D, a class I atypical myosin gene. Class I myosins have been implicated in membrane trafficking and cell motility (41). The GWAS peak in MYO1D is narrow and lies within two closely spaced recombination peaks, and the top GWAS SNP (rs379123) lies within an enhancer region that also includes a DNase I hypersensitive region in small airway epithelial cells. MYO1D has not been previously related to COPD or emphysema.
Genome-wide significant associations were observed for five genomic regions (15q25 near IREB2 and CHRNA3/5, 4q31 near HHIP, 11q22 near MMP12, 19q13 near CYP2A6/ADCK4, and 1q41 near TGFBR2) previously associated with risk of COPD. The presence of these loci among the top signals for LHE phenotypes supports the validity of quantitative LHE phenotypes, and leads to questions about whether these markers are associated with COPD only, emphysema only, or both phenotypes. The 15q25 locus, for example, has shown a complex pattern of phenotypic association with smoking behavior (42, 43), COPD status (6), CT emphysema measures (44), and lung cancer (45, 46). To determine whether these associations (and the two novel associations) were driven solely by correlation of emphysema with COPD status, we performed GWAS in the subset of smokers with Global Initiative for Chronic Obstructive Lung Disease Stage 2–4 COPD, and these results were consistent with their genome-wide significance in the overall analysis of emphysema patterns.
The COPDGene Study enrolled large numbers of NHW and AA smokers, providing the opportunity for replication across racial groups within the same study. The different LD structure between populations is potentially useful for localization of causal variants, and the GWAS peak in AAs was narrower than in NHWs for three of seven genome-wide significant loci. Interestingly, the associations at 11q22, 19q13, and 1q41 showed different patterns of association across racial groups. For 11q22, the association was very strong in NHWs and weak in AAs, and at the 19q13 and 1q41 regions, NHWs showed a clear secondary association signal, whereas AAs did not. These findings may be caused by lower power in the smaller AA sample, but they may also be caused by allelic heterogeneity both within and across ethnic groups. It is also possible that synthetic association caused by rare genetic variation could play a role in apparent allelic heterogeneity; however, the role of synthetic association as an overall explanation for GWAS associations is likely to be small (47).
Previous studies have implicated genetic control of gene expression as a key functional mechanism for most GWAS associations, and publicly available data from the ENCODE and Roadmap Epigenomics projects provide an unprecedented opportunity to link genetic variation with experimental regulatory data from a wide array of cell lines (32, 35, 37, 48, 49). Integration of LHE GWAS results with ENCODE and Roadmap regulatory data confirms strong enrichment of enhancer regions among our top LHE GWAS loci and points to a role for multiple cell types in the pathogenesis of emphysema, particularly lung fibroblasts. However, this integrative analysis has some important limitations. First, publicly available cell line data are extensive but not comprehensive, and important emphysema-related cell types may not be present in ENCODE and Roadmap data resources. Second, available data on variability of regulatory annotation in specific cell types in various conditions are limited.
In previous work we have shown that LHE patterns are more strongly associated with COPD-related measures of physiology and function than %LAA-950 (17), despite correlation between LHE patterns and %LAA-950 (Spearman rank correlation ranging from 0.30 to 0.96). We now present data indicating that LHE patterns are also strongly associated with both novel and previously established COPD risk variants, providing further evidence that LHE patterns capture information from CT that is physiologically and biologically relevant.
This study has the following strengths and limitations. We analyzed quantitative phenotypes that capture distinct emphysema patterns based on established pathologic categories in a large, biracial study population. Although these LHE patterns are distinct, they are also correlated and some represent different gradations of a single pathologic type of emphysema (i.e., mild, moderate, and severe centrilobular emphysema). Some patterns shared top GWAS loci extensively (i.e., the normal and moderate centrilobular pattern), and other LHE patterns provided distinct GWAS signals. These results support the hypothesis that genetic determinants of distinct emphysema patterns are, to an extent, nonoverlapping, and they confirm the value of developing novel methods for analyzing quantitative measures from CT that provide more precise phenotypic characterization.
LHE measures are an improvement over standard, threshold-based quantitative emphysema measures (17); however, emphysema quantification methods are likely to continue to improve. In particular, paraseptal emphysema was not examined in this study because of limitations of the LHE method in detecting this emphysema pattern. The LHE phenotypes did not include information about distribution of each pattern within the lung. In the future such an analysis may provide additional information about the genetic determinants of the distribution of specific LHE patterns. The severe centrilobular and panlobular LHE patterns have heavily skewed distributions, complicating the analysis of these phenotypes by linear regression, and a degree of caution is required in interpreting the significant associations with these phenotypes. However, the genome-wide significant associations with these phenotypes remain highly significant in ordinal and logistic regression analyses.
In summary, GWAS of distinct, quantitative emphysema patterns in NHW and AA smokers in COPDGene identifies five established COPD loci and two novel associations. Both novel associations are located within genes controlling cell-cell signaling and cell motility, and enhancer enrichment analysis suggests control of gene expression is a key functional mechanism for genes associated with emphysema. There is significant enrichment of enhancer and DNase I regions from many cell types among these top emphysema GWAS associations, with pulmonary fibroblasts prominent among the cell types showing the strongest enrichment.
Footnotes
The COPDGene Study (NCT00608764) was supported by award numbers R01HL089897 (J.D.C.) and R01HL089856 (E.K.S.) from the National Heart, Lung, and Blood Institute. This work was also supported by National Institutes of Health grants P01HL105339 and R01 HL075478 (E.K.S.), K08HL102265 (P.J.C.), K25HL104085 and R01HL116931 (R.S.E.), K08 HL097029, R01 HL113264, and the Alpha-1 Foundation (to M.H.C.), K23HL089353 and HL107246-03 (G.W.). The COPDGene project is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion.
Author Contributions: Conception and design, P.J.C., R.S.J.E., E.K.S., and M.H.C. Acquisition, analysis, and/or interpretation, P.J.C., R.S.J.E., E.K.S., M.H.C., M.-L.N.M., T.H.B., N.L., J.D.C., and G.W. Drafting the manuscript for important intellectual content, P.J.C., R.S.J.E., E.K.S., M.H.C., M.-L.N.M., T.H.B., N.L., J.D.C., and G.W.
Originally Published in Press as DOI: 10.1164/rccm.201403-0569OC on July 9, 2014
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 2.Rennard SI, Vestbo J. The many “small COPDs”: COPD should be an orphan disease. Chest. 2008;134:623–627. doi: 10.1378/chest.07-3059. [DOI] [PubMed] [Google Scholar]
- 3.Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am J Respir Crit Care Med. 2013;188:941–947. doi: 10.1164/rccm.201302-0263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, DeMeo DL, Hunninghake GM, Litonjua AA, Sparrow D, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho MH, McDonald ML, Zhou X, Mattheisen M, Castaldi PJ, Hersh CP, Demeo DL, Sylvia JS, Ziniti J, Laird NM, et al. NETT Genetics, ICGN, ECLIPSE and COPDGene Investigators. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2:214–225. doi: 10.1016/S2213-2600(14)70002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, et al. ICGN Investigators. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YM, Chen T-H, Barr RG, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soler Artigas M, Loth DW, Wain LV, Gharib SA, Obeidat M, Tang W, Zhai G, Zhao JH, Smith AV, Huffman JE, et al. International Lung Cancer Consortium; GIANT consortium. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011;43:1082–1090. doi: 10.1038/ng.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, Zhao JH, Ramasamy A, Zhai G, Vitart V, et al. Wellcome Trust Case Control Consortium; NSHD Respiratory Study Team. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong X, Cho MH, Anderson W, Coxson HO, Muller N, Washko G, Hoffman EA, Bakke P, Gulsvik A, Lomas DA, et al. ECLIPSE Study NETT Investigators. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. Am J Respir Crit Care Med. 2011;183:43–49. doi: 10.1164/rccm.201004-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manichaikul A, Hoffman EA, Smolonska J, Gao W, Cho MH, Baumhauer H, Budoff M, Austin JH, Washko GR, Carr JJ, et al. Genome-wide study of percent emphysema on computed tomography in the general population: the Multi-Ethnic Study of Atherosclerosis Lung/SNP Health Association Resource Study. Am J Respir Crit Care Med. 2014;189:408–418. doi: 10.1164/rccm.201306-1061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müller NL, Staples CA, Miller RR, Abboud RT. “Density mask”: an objective method to quantitate emphysema using computed tomography. Chest. 1988;94:782–787. doi: 10.1378/chest.94.4.782. [DOI] [PubMed] [Google Scholar]
- 13.Hogg JC, Senior RM. Chronic obstructive pulmonary disease - part 2: pathology and biochemistry of emphysema. Thorax. 2002;57:830–834. doi: 10.1136/thorax.57.9.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246:697–722. doi: 10.1148/radiol.2462070712. [DOI] [PubMed] [Google Scholar]
- 15.Sørensen L, Shaker SB, de Bruijne M. Quantitative analysis of pulmonary emphysema using local binary patterns. IEEE Trans Med Imaging. 2010;29:559–569. doi: 10.1109/TMI.2009.2038575. [DOI] [PubMed] [Google Scholar]
- 16.Mendoza CS, Washko GR, Crapo JD, Ross JC, Diaz AA, Lynch DA, Silverman E, Acha B, Serrano C, Estépar RSJ. Emphysema quantification in a multi-scanner HRCT cohort using local intensity distributions; Proc IEEE Int Symp Biomed Imaging; 2012. pp. 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castaldi PJ, San José Estépar R, Mendoza CS, Laird N, Hersh CP, Crapo JD, Lynch DA, Silverman E, Washko GR. Distinct quantitative computed tomography emphysema patterns are associated with physiology and function in smokers. Am J Respir Crit Care Med. 2013;188:1083–1090. doi: 10.1164/rccm.201305-0873OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castaldi P, Estepar RSJ, Mendoza CS, Cho MH, Crapo JD, Lynch DA, Beaty TH, Washko GR, Silverman E. Genome-wide association study for local histogram emphysema patterns identifies loci near CHRNA3/5 and MMP12/MMP3 [abstract] Proc Am Thorac Soc. 2012;185:A3808. [Google Scholar]
- 19.Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, Curran-Everett D, Silverman EK, Crapo JD. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7:32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.American Thoracic Society. Standardization of spirometry, 1994 update. Am J Respir Crit Care Med. 1995;152:1107–1136. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 26.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. Available from: http://www.R-project.org/
- 28.Viechtbauer W.Conducting meta-analyses in R with the metafor package J Stat Softw 2010361–48 [Google Scholar]
- 29.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storey JD.A direct approach to false discovery rates J R Stat Soc Ser B 200264479–498 [Google Scholar]
- 31.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilk JB, Chen T-H, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, Myers RH, Borecki IB, Silverman EK, Weiss ST, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, Demeo DL, Himes BE, Sylvia JS, Klanderman BJ, Ziniti JP, et al. ICGN Investigators; ECLIPSE Investigators; COPDGene Investigators. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21:947–957. doi: 10.1093/hmg/ddr524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy A, Chu JH, Xu M, Carey VJ, Lazarus R, Liu A, Szefler SJ, Strunk R, Demuth K, Castro M, et al. Mapping of numerous disease-associated expression polymorphisms in primary peripheral blood CD4+ lymphocytes. Hum Mol Genet. 2010;19:4745–4757. doi: 10.1093/hmg/ddq392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becker A-KA, Mikolajek H, Paulsson M, Wagener R, Werner JM. A structure of a collagen VI VWA domain displays N and C termini at opposite sides of the protein. Structure. 2014;22:199–208. doi: 10.1016/j.str.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 1997;13:163. doi: 10.1016/s0168-9525(97)01103-7. [DOI] [PubMed] [Google Scholar]
- 41.Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:527–533. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- 42.Caporaso N, Gu F, Chatterjee N, Sheng-Chih J, Yu K, Yeager M, Chen C, Jacobs K, Wheeler W, Landi MT, et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS ONE. 2009;4:e4653. doi: 10.1371/journal.pone.0004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siedlinski M, Cho MH, Bakke P, Gulsvik A, Lomas DA, Anderson W, Kong X, Rennard SI, Beaty TH, Hokanson JE, et al. COPDGene Investigators; ECLIPSE Investigators. Genome-wide association study of smoking behaviours in patients with COPD. Thorax. 2011;66:894–902. doi: 10.1136/thoraxjnl-2011-200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pillai SG, Kong X, Edwards LD, Cho MH, Anderson WH, Coxson HO, Lomas DA, Silverman EK ECLIPSE and ICGN Investigators. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:1498–1505. doi: 10.1164/rccm.201002-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 47.Anderson CA, Soranzo N, Zeggini E, Barrett JC. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol. 2011;9:e1000580. doi: 10.1371/journal.pbio.1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]