

Abstract
The 1H NMR spectroscopic analysis of the binding of ClO4− anion to the hydrophobic, concave binding site of a deep-cavity cavitand is presented. The strength of association between the host and ClO4− is controlled by both the nature and concentration of co-salts in a manner that follows the Hofmeister series. A model that partitions this trend into competitive binding of the co-salt anion to the hydrophobic pocket of the host, and counter-ion binding to its external carboxylates, successfully accounts for the observed changes in ClO4− affinity.
Keywords: Host-guest systems, Anions, Hofmeister Effect, Hydrophobic Effect
The growth in anion recognition during the last two decades has relied on many distinct functional groups,[1] but across all examples of receptors it is arguably those utilizing strong charge-charge attractions and hydrogen bond donation that have served the field best. Nevertheless, because attenuation of ion-ion interactions is significant in water, and because the high enthalpy of hydration of anions competes with ion recognition via hydrogen bonding,[2] the majority of anion complexations have been studied in organic solvent. This is unfortunate, for the chemistry of aqueous salt solutions is central to all aspects of life.[3] Furthermore, the chemistry of aqueous salt solutions is rich with ambiguities, the most general example of which is the Hofmeister Effect. Thus, for over 125 years it has been known that salts influence the solubility of molecules.[4] Salting-out salts such as NaF decrease solubility (apparently increasing the hydrophobic effect), whereas salting-in salts such as NaSCN increase solubility (apparently weakening it). Irrespective of the solute examined, studies repeatedly reveal the, “Hofmeister Series” of anions: F−, SO42−, AcO−, Cl−, Br−, NO3−, ClO3−, I−, ClO4− and SCN−. Furthermore, investigations examining salt effects on >38 macroscopic phenomena,[5] e.g., lower critical solution temperatures of polyamides,[6] surface tension changes,[7] and changes in water surface potentials[8] all reveal this Hofmeister Series.
Historically, the Hofmeister Effect has been attributed to salts modulating water structure. Hence salting-out salts are often referred to as water structure makers or “kosmotropes”,[5a] whilst salting-in salts have been termed water structure breakers or “chaotropes”.[9] However, there are also likely major contributions to the Hofmeister Effect from direct ion-solute interactions. Thus, there is increasing evidence of polarizable anions accumulating at the air-water interface[10],[5b] (without violating the thermodynamic theories arising from the observed negative Gibbs surface excess[11]), and powerful examples of surface-bulk ion partition models[7, 12] suggesting ion-solute interactions are important. Moreover, recently specific ion-solute interactions have been quantified; namely the interactions between anions and hydrogen bonding groups[6, 13] and anions with hydrophobic concavity.[14] Additionally, very recent studies of how salts interact with peptides have revealed a nuanced and complex interplay between ions and functional groups.[15] It is therefore evident that there is much to learn at the molecular level about the interactions between ions and solutes, and how this ‘folds’ into the Hofmeister Effect.
Recently, we demonstrated that salting-out salts enhanced the binding of a hydrophobic guest to the hydrophobic pocket of a host (octa-acid, OA, Figure 1),[16] whilst salting-in salts decreased this association.[14] Furthermore, it was ascertained that this latter effect was induced by strong (up to 2.7 kcal mol−1) anion affinity for the hydrophobic pocket of OA, which led to direct competitive binding between anion and hydrophobe. These results were intriguing because under the basic conditions used the host is ostensibly an octa-anion; Coulombic interactions did not play a role in the affinity between it and the salting-in anions.
Figure 1.
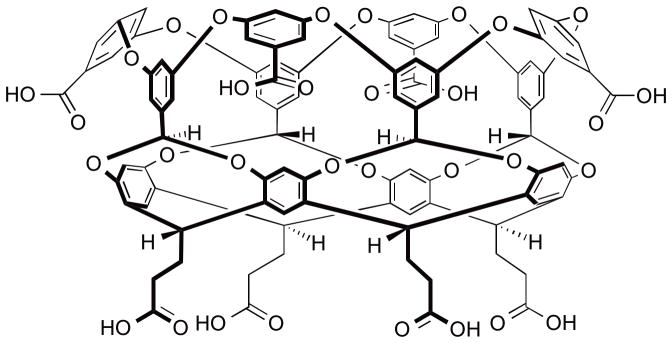
Structure of octa-acid host (OA).
Here we report an NMR study revealing that ClO4− binding to OA is modulated by the presence of other salts, and that this strength of association is controlled by the nature and concentration of co-salts in a manner following the Hofmeister Series. A model that partitions this trend into competitive binding of the co-salt anion to the pocket of the host, and counter-ion binding to its external carboxylates, successfully accounts for the observed changes in ClO4− affinity. To our knowledge the results represent the first link between anion binding to hydrophobicity and the Hofmeister series, and illustrate the complex relationship between the components of a salt and how they interact with a solute.
During NMR titration studies to quantify the affinity that anions such as ClO4− have for OA, we noted a strong dependence upon the overall ionic strength of the solution. Thus, using the benzal protons in the binding pocket of the host as reporter atoms, when NaCl was utilized to hold the ionic strength constant the Ka of ClO4− increased from 166 ± 3 M−1 in a solution of constant I = 120 mM, to 191 ± 3 M−1at I = 150 mM.
If the binding of anions to OA is dependent on I, does the nature of a co-salt also influence Ka? To address this question we opted for a more atom-economical and operationally straightforward titration procedure whereby I varied as NaClO4 was added to the host solution. We chose the sodium salts of six monovalent anions that cover the Hofmeister continuum and examined how the Ka of ClO4− to OA was affected by the concentration of these co-salts (phosphate buffer, pH 10.8). The results are presented in Figure 2.[17]
Figure 2.

The affect of [salt] on the Ka of ClO4− binding to OA host. Each point represents the average of two or three determinations. The lines represent fitting of the data using Eq. 2 (see text).
The figure shows that the addition of salting-out salts such as NaF enhance ClO4− binding, whilst those salts that reduce the apparent strength of the hydrophobic effect (e.g., NaSCN) weaken it. In between these extremes, NaClO3 (and to a lesser extent NaI) is observed to slightly enhance ClO4− binding at low concentrations but weaken it at higher concentrations. This suggests there are at least two factors leading to the observed data, which overall follows the Hofmeister series.
The major factor behind the reduction in the observed ClO4− binding constant (Kobs(ClO4−)) in the presence of salting-in anions is simple direct competition for the hydrophobic pocket of OA (Eq. 1).
| Eq. 1 |
where K(ClO4−) is the association constant of the ClO4− guest in the absence of any competing co-salt (95 M−1), K(salt) is the corresponding binding constant of the competing anion from the co-salt, and St is the total concentration of the co-salt. A plot of the difference between the data in Figure 2 and a model using Eq. 1 reveals that the equation slightly over-estimates the drop in Ka from competing ions (SI).
What is the cause of the increase in Kobs(ClO4−) as a function of I? At the most basic level this increase could be because the solution becomes less favorable to ClO4−, or because OA becomes a better host. This latter idea is primarily based on the notion that like polyelectrolyes such as DNA,[18] the −8 charge of the host means that cations condense strongly with it, and as I is increased so the host becomes less negatively charged and a stronger binder of anions.[19] Confirmation that this is the case came from ζ-potential measurements of OA as a function of added NaCl (SI). Thus, at concentrations from 0–280 mM added NaCl the ζ-potential of OA reduced in an approximately linear fashion from −39 to −21 mV. Two other lines of evidence support Na+ condensation. First, the increase in Kobs(ClO4−) in the presence of the more weakly condensing cesium ion[19] of CsCl (SI) was attenuated relative to NaCl. Second, half the concentration of Na2SO4 was required to bring about the same effect upon Ka as NaCl (SI).
Looking at cation condensation from the perspective of binding constants, to the author’s knowledge the Ka for Na+ complexation to carboxylate (–CO2−) in water has not been accurately determined. However, it has been estimated by the Cremer group to be 4.5 M−1 from changes in the lower critical solution temperature of elastin-like polypeptides.[20] To account for the observed enhanced binding by Na+ complexation, Eq. 1 was modified (SI) by defining K(salt) = (1 + αθ) K0,salt and K(ClO4−) = (1 + αθ) K0(ClO4−), where θ is the fraction of host-Na+ complex (θ = [HNa+]/Ht), K0(salt) and K0(ClO4−) are respectively the reference association constants for the co-salt and perchlorate at ‘zero’ added salt, and α is a unitless factor that relates the extent of cation condensation to the carboxylates and how this affects anion binding to the pocket. Substituting these definitions into Eq. 1 gives Eq. 2.
| Eq. 2 |
Examining different KNa+ values for θ, we fitted Eq. 2 to the obtained data by letting α float. All the other factors in this equation are knowns, although in the case of Br− and ClO3− we also opted to let K0(salt) float because their low affinities to the pocket of OA likely possess large errors. The results of the closest fit (KNa+ = 0.5 M−1) are given in Figure 2. These fits gave Ksalt for Br− and ClO3− very close to that anticipated (0.4 and 5.8 M−1 respectively), and α values from 21.8 to 40.8 (SI). The residuals from these fits are <21% of the empirical data, with the overwhelming majority within < 10% of the observed values (SI). With larger KNa+ values, e.g., 4.5 M−1,[20] the fits are poorer with slight curvatures to the lines for F−, and Cl−, and greater curvature for the Br− data; a reflection of greater saturation of the host with Na+ ions.
Overall these results reveal several key points: 1) anion binding to OA is dependent on both the ionic strength of the solution and the nature of co-salt used to alter the ionic strength; 2) the observed variation as a function of the nature of the co-salt follows the Hofmeister series; 3) relatively poorly hydrated and polarizable anions demonstrate strong salting-in properties via competitive binding; 4) in the absence of strong salting-in properties, the salts demonstrate typical salting-out binding enhancement that can be mostly accounted for by cation binding to the outside of OA reducing the net charge on the host; 5) salts such as NaClO3 demonstrate both (anion-induced) salting-in effects and (cation-induced) salting-out properties depending on its concentration.
The two binding sites of OA are, to a first approximation, operationally independent of each other. One, a hydrophobic pocket has strong affinity for salting-in anions but no affinity for either salting-out anions or metal cations. The other binding site of eight carboxylates has affinity for cations. For these reasons, OA differs considerably from other molecular probes,[15] used to illustrate the complex supramolecular relationships between anion, cation, and solute. We therefore anticipate that these cavitands offer a unique, molecular-level insight into the Hofmeister Effect.
It is also worth noting that the binding of large anions such as perchlorate to hydrophobic concavity suggests a new strategy for anion recognition; rather than counteract their relatively high enthalpy of hydration by attempting to remove all waters of solvation, an alternative approach is to take advantage of their thermodynamic preference to reside on the surface of water clusters.[21] In other words, partial dehydration may be sufficient to bring about their recognition in water. We will report progress on these notions at a future date.
Supplementary Material
Footnotes
The authors acknowledge the financial support of the National Institutes of Health (GM GM098141).
Supporting Information containing details of the experiments reported in this article is available on the WWW under http://...
References
- 1.a) Sessler JL, Gale PA, Cho W-S. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]; b) Gale PA, Busschaert N, Haynes CJ, Karagiannidis LE, Kirby IL. Chem Soc Rev. 2013 doi: 10.1039/c3cs60316d. [DOI] [PubMed] [Google Scholar]; c) Gale PA, Steed JW. John Wiley and Sons. 2012. [Google Scholar]; d) Caballero A, Zapata F, Beer PD. Coordination Chemistry Reviews. 2013;257:2434–2455. [Google Scholar]; e) Hirsch AKH, Fischer FR, Diederich F. Angew Chem Int Ed. 2007;46:338–352. doi: 10.1002/anie.200603420. [DOI] [PubMed] [Google Scholar]; f) Berryman O, Johnson DW. Chem Commun. 2009:3143–3153. doi: 10.1039/b823236a. [DOI] [PubMed] [Google Scholar]
- 2.Fernando IR, Surmann SA, Urech AA, Poulsen AM, Mezei G. Chem Commun (Camb) 2012;48:6860–6862. doi: 10.1039/c2cc32074f. [DOI] [PubMed] [Google Scholar]
- 3.Lynden-Bell RM, Morris SC, Barrow JD, Finney JL, Harper RLJ. CRC Press; Boca Raton: 2010. [Google Scholar]
- 4.a) Hofmeister F. Arch Exp Pathol Pharmakol. 1888;24:247–260. [Google Scholar]; b) Kunz W, Lo N, Ninham BW. Curr Opin Colloid In. 2004;9:1–18. [Google Scholar]
- 5.a) Collins KD, Washabaugh MW. Quarterly Review of Biophysics. 1985;18:323–422. doi: 10.1017/s0033583500005369. [DOI] [PubMed] [Google Scholar]; b) Jungwirth P, Tobias JW. Chem Rev. 2006;106:1259–1281. doi: 10.1021/cr0403741. [DOI] [PubMed] [Google Scholar]; c) Ball P. Chem Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]; d) Lo Nostro P, Ninham BW, Milani S, Lo Nostro A, Pesavento G, Baglioni P. Biophys Chem. 2006;124:208–213. doi: 10.1016/j.bpc.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 6.a) Zhang Y, Furyk S, Bergbreiter DE, Cremer PS. J Am Chem Soc. 2005;127:14505–14510. doi: 10.1021/ja0546424. [DOI] [PubMed] [Google Scholar]; b) Zhang YJ, Cremer PS. Annu Rev Phys Chem. 2010;61:63–83. doi: 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]
- 7.Pegram LM, Record MTJ. J Phys Chem B. 2007;111:5411–5417. doi: 10.1021/jp070245z. [DOI] [PubMed] [Google Scholar]
- 8.Randles JEB. Physics and Chemistry of Liquids. 1977;7:107. [Google Scholar]
- 9.Hamaguchi K, Geiduschek EP. J Am Chem Soc. 1962;84:1329–1338. [Google Scholar]
- 10.a) Petersen PB, Saykally RJ. Annu Rev Phys Chem. 2006;57:333–364. doi: 10.1146/annurev.physchem.57.032905.104609. [DOI] [PubMed] [Google Scholar]; b) Verreault D, Allen HC. Chemical Physics Letters. 2013;586:1–9. [Google Scholar]
- 11.Onsager L, Samaras NNT. The Journal of Chemical Physics. 1934;2:528. [Google Scholar]
- 12.a) Pegram LM, Wendorffa T, Erdmanna R, Shkela I, Bellissimoa D, Felitskya DJ, Record MTJ. Proc Natl Acad Sci U S A. 2010;107:7716–7721. doi: 10.1073/pnas.0913376107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pegram LM, Record MTJ. Chemical Physics Letters. 2008;467:1–8. doi: 10.1016/j.cplett.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Baldwin RL. Biophys J. 1996;71:2056–2063. doi: 10.1016/S0006-3495(96)79404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zangi R, Hagen M, Berne BJ. J Am Chem Soc. 2007;129:4678–4686. doi: 10.1021/ja068305m. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Cremer PS. Curr Opin Chem Biol. 2006;10:658–663. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Gibb CLD, Gibb BC. J Am Chem Soc. 2011;133:7344–7347. doi: 10.1021/ja202308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Xie WJ, Gao YQ. The Journal of Physical Chemistry Letters. 2013;4:4247–4252. doi: 10.1021/jz402072g. [DOI] [PubMed] [Google Scholar]; b) Gao YQ. Journal of Physcial Chemistry B. 2012;116:9934–9943. doi: 10.1021/jp305532h. [DOI] [PubMed] [Google Scholar]; c) Paterova J, Rembert KB, Heyda J, Kurra Y, Okur HI, Liu WR, Hilty C, Cremer PS, Jungwirth P. J Phys Chem B. 2013;117:8150–8158. doi: 10.1021/jp405683s. [DOI] [PubMed] [Google Scholar]
- 16.a) Gibb CLD, Gibb BC. J Am Chem Soc. 2004;126:11408–11409. doi: 10.1021/ja0475611. [DOI] [PubMed] [Google Scholar]; b) Liu S, Whisenhunt-Ioup SE, Gibb CLD, Gibb BC. Supramolecular Chemistry. 2011;24:480–485. doi: 10.1080/10610278.2010.550290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.SCN− and ClO4− induced similar shifts of the reporter signal, such that above 50 mM SCN− the observed overall Δδ was < 0.05 ppm. This resulted in unacceptable errors in the Ka values and hence prevented determinations above 50 mM.
- 18.Manning GS. The Journal of Chemical Physics. 1969;51:924. [Google Scholar]
- 19.Hunter RJ. Zeta Potential in Colloid Science. Academic Press; Osney Mead: 1981. [Google Scholar]
- 20.Kherb J, Flores SC, Cremer PS. J Phys Chem B. 2012;116:7389–7397. doi: 10.1021/jp212243c. [DOI] [PubMed] [Google Scholar]
- 21.a) Stuart SJ, Berne BJ. Journal of Physical Chemistry A. 1999;103:10300–10307. [Google Scholar]; b) Stuart SJ, Berne BJ. J Phys Chem. 1996;100:11934–11943. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.