

Abstract
The first total synthesis of the proposed structure of cycloinumakiol (1) and its C5-epimer (18) are achieved in a concise and efficient fashion: 9 and 5 steps from known 3-hydroxybenzocyclobutenone with overall yields of 15% and 33%, respectively. A key for the success of this approach is use of a catalytic C–C activation strategy for constructing the tetracyclic core of 1 through carboacylation of a sterically hindered trisubstituted olefin with benzocyclobutenone. In addition, the structure of the natural cycloinumakiol was reassigned to 19-hydroxyltotarol (7) through X-ray diffraction analysis. This work demonstrates the potential of C–C activation for streamlining complex natural-product synthesis.
Keywords: total synthesis, C–C activation, cycloinumakiol, structural revision, Rh catalysis
Podocarpaceae[1] is one of the most abundant evergreen trees distributed from Australia to the tropical and subtropical areas of Asia. Previous biological studies of the extracts from the leaves of this plant reveals high antibiotic as well as anti-cancer activities resulting from inumakiols,[1a] totarols[1b] and other norditerpenes (Figure 1).[1c,d] Recently, guided by an activator protein-1 (AP-1)-based bioassay, investigation on extracts of podocarpus latifolius at the National Cancer Institute (NCI) led to the isolation of three new inumakiol-family diterpenes, namely cycloinumakiol (1), inumakal (2) and inumakoic acid (3).[2] The proposed structure of cycloinumakiol (1) clearly distinguished itself from the rest tricyclic inumakiol-family members. First, its oxygen substitution on the aromatic ring is para instead of meta or ortho to the isopropyl group; second, oxidation and etherification of the angular C20-methyl group feature an unusual tetracyclic ring skeleton, along with an all-carbon quaternary stereocenter at the C10 position. Thus, the unique structure of 1 followed by perhaps a more intriguing question, that is how cycloinumakiol is produced from nature, makes cycloinumakiol an attractive target for synthesis. While a number of synthetic efforts has been carried out towards totarol-type diterpenes,[1b,3] to the best of our knowledge, no total synthesis was reported previously for cycloinumakiol (nor any other inumakiol family members). In this communication, we describe our development of a novel strategy for the first total synthesis of the proposed structure of cycloinumakiol and its C5-epimer, and disclose a structural revision of this natural product.
Figure 1.

Representative Natural Products from Podocarpaceae.
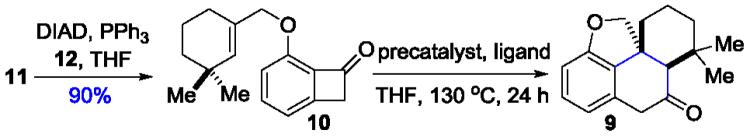
Our laboratory recently developed rhodium-catalyzed olefin and alkyne carboacylation methodologies through C–C activation of benzocyclobutenones (Scheme 1A).[4,5] This intramolecular “cut and sew” sequence allows for rapid access of fused-ring skeletons from relatively simple starting materials. We anticipate that the proposed cycloinumakiol (1) structure would provide an ideal platform to examine the applicability of this strategy for complex molecule synthesis. From a retrosynthetic viewpoint (Scheme 1B), we envisioned that cycloinumakiol (1) could be accessed through stereo-epimerization at the C5 position and ketone reduction from cis-terpenone 8. Compound 8 would be derived from a chemo- and site-selective arene-functionalization of ketone 9 that can potentially serve as a common intermediate for preparing cycloinumakiol analogues/derivatives. The tetracyclic core structure of 9 is expected to be rapidly constructed from benzocyclobutenone 10 through a catalytic intramolecular carboacylation of a hindered tri-substituted olefin. A Mitsunobu coupling of 3-hydroxylbenzocyclobutenone 11[6] and the known allyl alcohol 12[7] would afford compound 10.
Scheme 1.

Synthetic Strategy Design.
The proposed synthetic approach is attractive for it would be the first time that a catalytic carboacylation of alkenes is employed as the key step in constructing natural-product skeletons,[8] allowing for investigation of whether a C–C activation strategy can streamline complex-molecule synthesis. However, the difficulty is obvious because success of the proposed approach relies on sterically hindered tri-substituted olefins can be employed as the coupling partner for the Rh-catalyzed carboacylation reaction. It is known that tri-substituted alkenes are highly challenging substrates for metal-catalyzed cycloaddition reactions,[9] due to that they generally have much lower binding affinity with transition metals compared to terminal olefins.[10] We recently discovered that, assisted by a Lewis-acid cocatalyst, 1-cyclohexenyl group can undergo intramolecular carboacylation to form poly-fused rings.[4a,11,12] However, the proposed total synthesis requires insertion of an even more sterically hindered tri-substituted olefin moiety that contains adjacent gem-dimethyl groups (i.e. compound 10). Thus, we hypothesized that a more active catalyst system is needed to furnish the proposed transformation.
In a forward direction, substrate 10 was prepared in 90% yield through a Mistunobu coupling between phenol 11 and allyl alcohol 12, both of which can be prepared in three steps from commercially available starting materials.[6,7] Under the previously optimized conditions ([Rh(cod)Cl]2/DPPB combination) for insertion of regular olefins,[4a] no desired product was observed (Table 1, entry 1), and addition of ZnCl2 led to undesired deallylation (entry 2). Use of common ligands, such as PPh3 and DPPF gave low conversions (entries 3 and 4). In contrast, use of more electron-rich phosphine ligands (e.g. PCy3) gave significant decomposition of the starting material and more deallylation products (entry 5). While cationic Rh species were not effective (entries 6 and 7), use of an electron-deficient Rh-precatalyst was discovered to be critical for this transformation: the desired tetracycle 9 was isolated in 22% yield when 5 mol% [Rh(CO)2Cl]2 was employed (entry 8). Excess of CO ligand was found detrimental (entry 9). It was interesting to observe that electron-deficient cobalt complexes, such as Co2(CO)8, can also afford the desired product, albeit in a lower yield (entry 10). Furthermore, we found use of π-acidic phosphine ligands was able to enhance the turnover of the Rh catalyst (entries 13-15). Finally, tetracycle 9 can be consistently isolated in 64-66% yield[13] by utilizing a 1:2 ratio of [Rh(CO)2Cl]2 and P(C6F5)3 (Rh:P = 1:1) as the catalyst, which was added in two portions to achieve optimal efficiency (entry 15). Delightfully, the [Rh(CO)2Cl]2/P(C6F5)3 catalyst combination can also be extended to other substrates containing a cyclic and acyclic tri-substituted olefin (Figure 2).[14] Under slightly modified conditions, tri- and tetracyclic compounds 9a-c were isolated in synthetically useful yields.
Table 1.
Selected Condition Optimization for the Key Step.[a]
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Precatalyst | Ligand | Conversion | Yield of 9[b] |
| 1[c] | [Rh(cod)2CI]2 | DPPB | 5% | NDP |
| 2 | [Rh(cod)2CI]2 | DppB[d] | 10% | NDP + 11 (10%) |
| 3[c] | [Rh(cod)2CI]2 | DPPF | <5% | NDP |
| 4[c] | Rh(PPh3)3CI[f] | PPh3 | <5% | NDP |
| 5[c] | [Rh(C2H4)2CI]2 | PCy3[e] | 100% | NDP + 11 (20%) |
| 6 | [Rh(acn)2(cod)]BF4 | None | <5% | NDP |
| 7 | [Rh(cod)2]BF4 | None | <5% | NDP |
| 8 | [Rh(CO)2CI]2 | None | 40% | 22% |
| 9 | [Rh(CO)2CI]2 | 5 atm CO | <5% | NDP |
| 10 | Co2(CO)8[g] | None | 48% | 6.4% |
| 11 | CpCo(PPh3)[f] | None | <5% | NDP |
| 12 | Rh(CO)2acac[f] | None | <5% | NDP |
| 13 | [Rh(CO)2CI]2 [f] | P(C6F5)3 | 100% | 66% |
| 14[h] | [Rh(CO)2CI]2 | P[3,5-(CF3)2C6H3]3 | 94% | 44% |
| 15[h] | [Rh(CO)2CI]2 | P(C6F5)3 | 100% | 64% |
Reaction conditions: 5 mol % Rh dimer precatalysts, 12 mol % bidentate phosphine or 20 mol % monodentate phosphine ligand, in THF at 130 °C for 24 h.
Isolated yield; conversion was determined based on recycled 10.
Toluene was used as solvent.
10 mol % ZnCl2 was added as an additive.
10 mol % AgBF4 was added as an additive.
10 mol % catalyst was used.
1 equiv of Co2(CO)8 was used.
2.5 mol % [Rh(CO)2CI]2 and 5 mol % P(C6F5)3 were added initially; the reaction mixture was stirred at 140 °C for 12 h before another portion of the same catalyst was added.
DIAD= Diisopropyl azodicarboxylate, NDP= no desired product, DPPB= 1,1-bis(diphenyl-phosphino)butane, DPPF= 1,1-bis(diphenylphosphino)ferrocene, PCy3= tricyclohexylphosphine.
Figure 2.

Carboacylation with other substrates containing tri-substituted olefins. [a] 5 mol % [Rh(CO)2Cl]2, 40 mol % P(C6F5)3, in THF at 130 °C for 36 h.
With the cycloinumakiol core structure in hand (compound 9), we next set forth to introduce the isopropyl group at the C14 position through a site-selective arene functionalization (Scheme 2). To our delight, treatment of 9 with 2 equiv of NBS and a catalytic amount of NH4OAc[15] at room temperature led to a highly site-specific bromination with a near quantitative yield. Note that the activated C7 position remained intact under the oxidative conditions. A one-pot Suzuki cross-coupling with vinyl trifluoroborate salt 14[16] and hydrogenation sequence furnished the synthesis of cis-terpenone 8 in 86% overall yield from core compound 9.
Scheme 2.

Chemo and Site-selective Arene Functionalization.
We next attempted to directly invert the C5 stereocenter of compounds 9 and 8. However, under either strong basic or acidic conditions no epimization was observed, which is likely due to the rigid nature of the tetracyclic skeleton causing a high kinetic barrier (e.g. torsional strain by the angularly fused hydrofuran ring).[17] Nevertheless, we found a three-step sequence was effective to completely invert the C5 stereocenter (Scheme 3). The ketone was first converted to an olefin in high yields via LiAlH4 reduction and dehydration; ozonolysis/reduction followed by in situ treatment with DBU afforded dialdehyde 16 in the correct diastereomeric form. Subsequently, we took advantage of the McMurry coupling and restored the C6-C7 olefin.
Scheme 3.

Synthetic Sequence.
Finally, hydrogenation of alkene 17 provided the proposed cycloinumakiol (1) in 88% yield, the structure of which was unambiguously confirmed by 1H/13C-NMR, HRMS, IR and X-ray crystallography. While we are confident about our structural characterization of 1, unfortunately, the NMR spectra of synthetic cycloinumakiol do not match those reported in literature.[2] Thus, we anticipated there was a structural mis-assignment for cycloinumakiol.
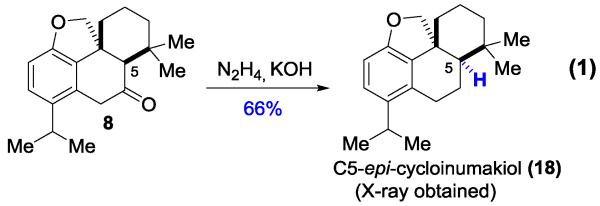
To examine whether the natural product is the other diastereomer of 1, we carried out Wolff-Kishner reduction of ketone 8 and obtained the C5-epi-cycloinumakiol 18 (Eq. 1). The structure of 18 was also unambiguously confirmed by 1H/13C-NMR, HRMS, IR and X-ray crystallography. Again, the NMR spectra of 18 do not match the reported data of cycloinumakiol.[2] After careful analysis and comparison between ours and reported 1H-NMR data, we found there is a significant difference in chemical shifts of the C20-methylene group: those of the natural sample are at 3.78 and 3.42 ppm while those of the synthetic samples (1 and 18) are around 4.6 and 4.1 ppm. Based on our previous experience on preparing benzohydrofuran compounds,[4] we suspected that the natural cycloinumakiol likely does not contain a benzohydrofuran motif.
Towards this end, thanks to a generous donation of the natural cycloinumakiol sample (ca 0.5 mg) from the NCI, we were able to further purify the sample, and gratifyingly, obtained a single crystal of this compound. X-ray crystallography analysis eventually revealed that the natural cycloinumakiol shares the same structure of 19-hydroxyltotarol (Figure 3).[18]
Figure 3.

Structure Elucidation of the Natural Cycloinumakiol.
In conclusion, we accomplished the first total synthesis of the proposed structure of cycloinumakiol (1) and its C5-epimer (18). This strategy features a Rh-catalyzed intramolecular coupling of a sterically hindered tri-substituted olefin with a benzocyclobutenone. Neither the synthesis of 1 nor 18 employed any protecting group. The conciseness of the total synthesis demonstrates that the “cut and sew” transformation can serve as a useful strategy to prepare structure motifs of high complexity, which is complementary to classical cycloaddition reactions. Furthermore, motivated by these synthetic endeavours, the structure of the natural cycloinumakiol was re-examined and revised as 19-hydroxyltotarol (7), suggesting chemical synthesis still plays important roles in validating the structures of natural products.[19] Investigation of the biological activity of the synthetic cycloinumakiol (1), the C5-epimer (18) and other intermediate compounds is ongoing through collaboration.
Supplementary Material
Footnotes
We thank UT Austin and CPRIT for a startup fund, NIGMS (R01GM109054-01) and the Welch Foundation (F 1781) for research grants. G. D. is a Searle Scholar. Dr. John Beutler from NCI is acknowledged for a donation of the natural-product sample. We thank Mr. Ren Zhi for the MM2 calculation. Dr. Vince Lynch is acknowledged for X-ray crystallography. We also thank Johnson Matthey for a generous donation of Rh salts.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- [1].Sato K, et al. Chem. Pharm. Bull. 2008;56:1691–1697. doi: 10.1248/cpb.56.1691. for a review of totarol, see: Bendall JG, Cambie RC. Aust. J. Chem. 1995;48:883–917. Park H-S, Takahashi Y, Fukaya H, Aoyagi Y, Takeya K. J. Nat. Prod. 2003;66:282–284. doi: 10.1021/np020334x. Park H-S, Yoda N, Fukaya H, Aoyagi Y, Takeya K. Tetrahedron. 2004;60:171–177.
- [2].Devkota KP, et al. J. Nat. Prod. 2011;74:374–377. doi: 10.1021/np100736y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For recent total synthesis of totarols, see: Barltrop JA, Rogers NAJ. J. Chem. Soc. 1958:2566–2572. Tada M, Kurabe J, Yasue H, Ikuta T. Chem. Pharm. Bull. 2008;56:287–291. doi: 10.1248/cpb.56.287. Kim MB, Shaw JT. Org. Lett. 2010;12:3324–3327. doi: 10.1021/ol100929z.
- [4].Xu T, Dong G. Angew. Chem. Int. Ed. 2012;51:7567–7571. doi: 10.1002/anie.201202771. Xu T, Ko HM, Savage NA, Dong G. J. Am. Chem. Soc. 2012;134:20005–20008. doi: 10.1021/ja309978c. for alkyne insertion, see: Chen P, Xu T, Dong G. Angew. Chem. Int. Ed. 2014;53:1674–1678. doi: 10.1002/anie.201310100.
- [5].For selected reviews on transition metal-mediated C-C bond activation, see: Jones WD. Nature. 1993;364:676–677. Murakami M, Ito Y. Top. Organomet. Chem. 1999;3:97–129. Rybtchiski B, Milstein D. Angew. Chem. Int. Ed. 1999;38:870–883. doi: 10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3. Perthuisot C, Edelbach BL, Zubris DL, Simhai N, Iverson CN, Müller C, Satoh T, Jones WD. J. Catal. Mol. A. 2002;189:157–168. van der Boom ME, Milstein D. Chem. Rev. 2003;103:1759–1792. doi: 10.1021/cr960118r. Jun C-H. Chem. Soc. Rev. 2004;33:610–618. doi: 10.1039/b308864m. Satoh T, Miura M. Top. Organomet. Chem. 2005;14:1–20. Jun C-H, Park JW. Top. Organomet. Chem. 2007;24:117–143. Necas D, Kotora M. Curr. Org. Chem. 2007;11:1566–1592. Crabtree RH. Chem. Rev. 1985;85:245–269. Kondo T, Mitsudo TA. Chem. Lett. 2005;34:1462–1467. Ruhland K. Eur. J. Org. Chem. 2012:2683–2706. Korotvicka A, Necas D, Kotora M. Curr. Org. Chem. 2012;16:1170–1214. Seiser T, Saget T, Tran DN, Cramer N. Angew. Chem. Int. Ed. 2011;50:7740–7752. doi: 10.1002/anie.201101053. Murakami M, Matsuda T. Chem. Commun. 2011;47:1100–1105. doi: 10.1039/c0cc02566f. Dermenci A, Coe PW, Dong G. Org. Chem. Front. 2014;1:567–581. doi: 10.1039/c4qo00053f. Xu T, Dermenci A, Dong G. Top. Curr. Chem. 2014 doi: 10.1007/128_2014_545. DOI: 10.1007/128_2014_545.
- [6].For an efficient and scalable 3-step synthesis of 3-hydroxybenzocyclo butenone 11, see: Chen P, Savage NA, Dong G. Tetrahedron. 2014;70:4135–4146. doi: 10.1016/j.tet.2014.03.080.
- [7].Allyl alcohol 12 can be synthesized from 2-methyl cyclohexenone in three steps, see: Bourdron J, Commeiras L, Audran G, Vanthuyne N, Hubaud JC, Parrain J-L. J. Org. Chem. 2007;72:3770–3775. doi: 10.1021/jo070045j.
- [8].To the best of our knowledge, the only example involving application of carboacylation in a total synthesis is reported with a stoichiometric cobalt-complex: South MS, Liebeskind LS. J. Am. Chem. Soc. 1984;106:4181–4185.
- [9].a) Trost BM, Jiang C. Synthesis. 2006:369–396. [Google Scholar]; b) Das JP, Marek I. Chem.Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; c) Mei T-S, Patel HH, Sigman MS. Nature. 2014;508:340–344. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schurig V. Inorg. Chem. 1986;25:945–949. [Google Scholar]
- [11].For decarbonylative insertion of cyclic (including trisubstituted) olefins to form spirocycles, see: Xu T, Savage NA, Dong G. Angew. Chem., Int. Ed. 2014;53:1891–1895. doi: 10.1002/anie.201310149.
- [12].For a single example of a carbocyanation of acyclic trisubstituted olefins, see: Nakao Y, Ebata S, Yada A, Hiyama T, Ikawa M, Ogoshi S. J. Am. Chem. Soc. 2008;130:12874–12875. doi: 10.1021/ja805088r. For the only other example (besides ref 4a) of carboacylation of a trisubstituted olefin, see: Souillart L, Parker E, Cramer N. Angew. Chem. Int. Ed. 2014;53:3001–3005. doi: 10.1002/anie.201311009.
-
[13].Around 5% yield of the decarbonylation product (9′) was isolated as the major byproduct.

- [14].Enantioselective transformation has been attempted using a chiral electron-deficient ligand, such as monophos; however, only 5 % yield and 4 % ee were obtained.
- [15].Tanemura K, Suzuki T, Nishida Y, Satsumabayashi K, Horaguchi T. Chem. Commun. 2004:470–471. doi: 10.1039/b315340a. [DOI] [PubMed] [Google Scholar]
- [16].a) Miyaura N. Top. Curr. Chem. 2002;219:11–59. [Google Scholar]; b) Suzuki A. J. Organomet. Chem. 1999;576:147–168. [Google Scholar]
- [17].A semi-empirical calculation (MM2 method) indicates that cis-terpenone 8 is about 1.5 kcal/mol less stable than its trans isomer.
- [18].For isolation and characterization of 19-hydroxyltotarol (7), see: Wenkert E, Beak P. Tetrahedron Lett. 1961;2:358–362. Crambie RC, Mander LN. Tetrahedron. 1962;18:456–475. Taylor DAH. J. Chem. Soc. 1963:1553–1560. Enzell CR, Wahlberg I. Acta. Chem. Scand. 1969;23:871–891. doi: 10.3891/acta.chem.scand.23-0871. Crambie EC, Cox RE, Croft KD, Sidwell D. Phytochem. 1983;22:1163–1166. Ying B-P, Kubo I. Phytochem. 1991;30:1951–1955. For semisynthesis of 19-hydroxyltotarol (7), see: Day AC. J. Chem. Soc. 1964:3001–3005.
- [19].Nicolaou KC, Snyder SA. Angew. Chem. Int. Ed. 2005;44:1012–1044. doi: 10.1002/anie.200460864. [DOI] [PubMed] [Google Scholar]
- [20].CCDC 999131 (1), 99729 (8) 997288 (18) and 997289 (7) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.