

Abstract
Electrochemical reactions are shown to be effective for the C—H functionalization of a number of heterocyclic substrates that are recalcitrant to conventional peroxide radical initiation conditions. Monitoring reaction progress under electrochemical conditions provides mechanistic insight into the C—H functionalization of a series of heterocycles of interest in medicinal chemistry.
Keywords: Electrochemistry, C—H activation, Radical Reactions, Reaction Mechanisms, Heterocycles
C—H functionalization by sulfinate-derived radicals has emerged as a convenient method for late stage functionalization of complex heterocycles (Scheme 1A)1 and has been shown to proceed cleanly in cases where other methodologies fail.2 Sulfinate radical sources enable the synthesis of complex alkyl and fluoroalkyl-substituted heterocycles that would be difficult or time-consuming to prepare de novo. The reaction exhibits high functional group tolerance with tunable and predictable regioselectivity.3 However, despite the rapidly expanding scope of this transformation and its potential as a general method for late-stage C—H functionalization, low yields persist for a number of substrates, and the factors which lead to a successful reaction with a given heterocycle are not yet well understood. Here we report that electrochemical initiation results in significantly enhanced yields in the C—H functionalization of a number of complex substrates of pharmaceutical interest (Scheme 1B). Initial studies implicate the controlled generation of the free radical as a major factor in improving yield.
Scheme 1.

Radical C—H Functionalization of Heteroarenes.
Our investigations of the functionalization of small heterocyclic pharmacophores as building blocks for fragment-based drug discovery showed that in the reaction initiated by peroxides (Scheme 1A, red pathway) some substrates, in particular 1,2 and 1,3-azoles, give low yields even with high sulfinate loading. We undertook investigations of the factors influencing radical generation and consumption with the goal of enabling the C—H functionalization of recalcitrant substrates of interest in drug discovery with a reduction in sulfinate loading and to develop predictive models of the reactivity of heterocycle and radical pairs.
Prior reaction calorimetric studies1a revealed a high, unproductive heat output upon contact between sodium trifluoromethane sulfinate (NaTFMS) and tert-butyl hydroperoxide (TBHP). Together with the high sulfinate and TBHP loadings required to achieve good yields with most substrates, this suggests that a significant proportion of CF3 radicals generated from the peroxide is lost to side reactions. Scattered reports on the electrochemical behavior of the [CF3SO2]− anion4 suggested that initiation of the reaction by an anode, rather than TBHP, might provide a more controlled environment for the generation and subsequent reactions of sulfinate-derived radicals. Bulk electrolysis5 allows either the driving force (potential), or rate of electron transfer (current) to be explicitly and separately tuned (Scheme 1A, green pathway).6 We reasoned that electrolysis at constant current could provide direct control over the rate of sulfinate oxidation and hence allow the radical flux to be precisely defined.
Cyclic voltammetry of a series of zinc and sodium fluoroalkylsulfinates in DMSO–NEt4ClO4 solution at a carbon working electrode showed irreversible oxidation waves at potentials between 1.0 and 1.4 V (vs Ag/AgCl)7 For each sulfinate, a distinct peak corresponding to loss of SO2 was observed at −0.7 V on the reverse scan, regardless of the oxidation potential observed in the forward scan. These data indicate, in accordance with previous reports,4a that rate limiting electron transfer gives a sulfinate radical followed by rapid cleavage to generate the fluoroalkyl radical.
The combination of the low reactivity of imidazoles such as 1 towards fluoroalkylation in the TBHP-initiated reaction and the prevalence of the imidazole moiety in drug development prompted our choice of 1 as a substrate for detailed investigation of the reaction under electrochemical conditions.8,9 Electrolysis of a solution of 1a and ZnTFMS was carried out in DMSO–NEt4ClO4 (0.1 M) at a carbon cloth anode at different constant current values.7 Reaction progress was monitored by rapid circulation of the reaction mixture through a transmission FTIR cell.7 The consumption of ZnTFMS and formation of product 2 were tracked by IR peaks at 1145 cm−1 and 1722 cm−1, respectively. Scheme 1C compares these processes under electrochemical and peroxide-initiated conditions. The TBHP-initiated reaction exhibits a faster initial production of 2. Rapid total consumption of sulfinate accounts for the stalling characteristic of these reactions. By contrast, the electrochemical initiation causes much slower consumption of the sulfinate concomitant with steady reaction proceeding to significantly higher conversion to product 2 (Scheme 1B, 25 vs 53% isolated yield).
Reaction progress kinetics at different initial concentrations of 1 showed positive order in [substrate], and the productive reaction rate was not strongly influenced by current (Fig 1, green). The rate of sulfinate consumption followed zero order kinetics under electrochemical conditions and was controlled entirely by cell current (Fig. 1, red). This allows prediction of the time for total consumption of the reagent, which in effect dictates the window of time for productive reaction. The quantity of 2 formed per mole of sulfinate consumed is greater at lower current (Fig 1, blue), implying a shift in favor of productive vs. unproductive reactions when the radical is generated more slowly. Elevated temperature had no effect on the rate of sulfinate oxidation but resulted in slightly enhanced conversion. Good yields of 2 were obtained under electrochemical conditions using significantly lower [ZnTFMS] than in our previously reported conditions.7
Figure 1.

Initial rate of ZnTFMS consumption (blue, right axis), 2 production (green, right axis) and moles of 2 produced per mole of ZnTFMS consumed (red, left axis), as a function of current in the reaction of Scheme 1B.7
The magnitude of the observed redox potentials provides some insight into the privileged role of TBHP compared to other chemical oxidants that have been screened in previous work on these reactions. Reported reduction potentials for the tert-butoxy (tBuO) and tert-butylperoxy (tBuOO) radicals are –0.3410 and 0.75 V11 (vs Ag/AgCl) respectively, below the oxidation potential of the sulfinate, while weaker single-electron oxidants, e.g. ceric ammonium nitrate (CAN), possess much lower redox potentials.12 The redox behavior of TBHP indicates that Fenton-type cleavage of the hydroperoxide O—O bond by adventitious trace metal, as previously suggested,1a is unnecessary for initiation of the reaction.13
If the increased yield observed under electrochemical conditions could be rationalized solely by differences in the rate of radical generation, then slow addition of TBHP should result in a yield similar to that observed under electrochemical conditions. Interestingly, however, while addition of TBHP over 8 h (Figure 2, red curves) showed slow consumption of ZnTFMS, no significant increase in yield was observed compared to standard protocol (Figure 2, grey curves). This implies further differences between the two protocols, possibly in the second oxidation process, which may also be the cause of slight differences in regioselectivity between the two methods. The radical consumption/C—H functionalization step may also play an important role in the overall efficiency of these electrochemical reactions.
Figure 2.

Initial rate of ZnTFMS consumption (blue, right axis), 2 production (green, right axis) and moles of 2 produced per mole of ZnTFMS consumed (red, left axis), as a function of current in the reaction of Scheme 1B.7
A broad range of substrates show enhanced reactivity under electrochemical initiation of trifluoromethylation (Scheme 2). The rate of sulfinate oxidation was found to be invariant with substrate, which suggests that the reported yields reflect intrinsic differences in the second oxidation step (oxidation step b, Scheme 2). The reactions are typically highly selective. Pyrroles unexpectedly showed disubstitution under electrochemical initiation in some cases. Pyrazoles were among the least reactive substrates. Where necessary, regiochemistry was assigned using x-ray crystallography. X-ray structures of 5, obtained as an amorphous solid, 7, a highly volatile solid, and 12, a brown oil, were obtained from less than 2 mg of material using the recently reported metal-organic framework method of Fujita14 (Scheme 3). Difluoromethylation and trifluoroethylation were effective only for the most reactive substrates (Scheme 4). Though the sulfinate was oxidized in each case, products were obtained in low yield even at elevated temperature reflecting the poor reactivity of the CF2H and CH2CF3 radical–heterocycle pairs.
Scheme 2.
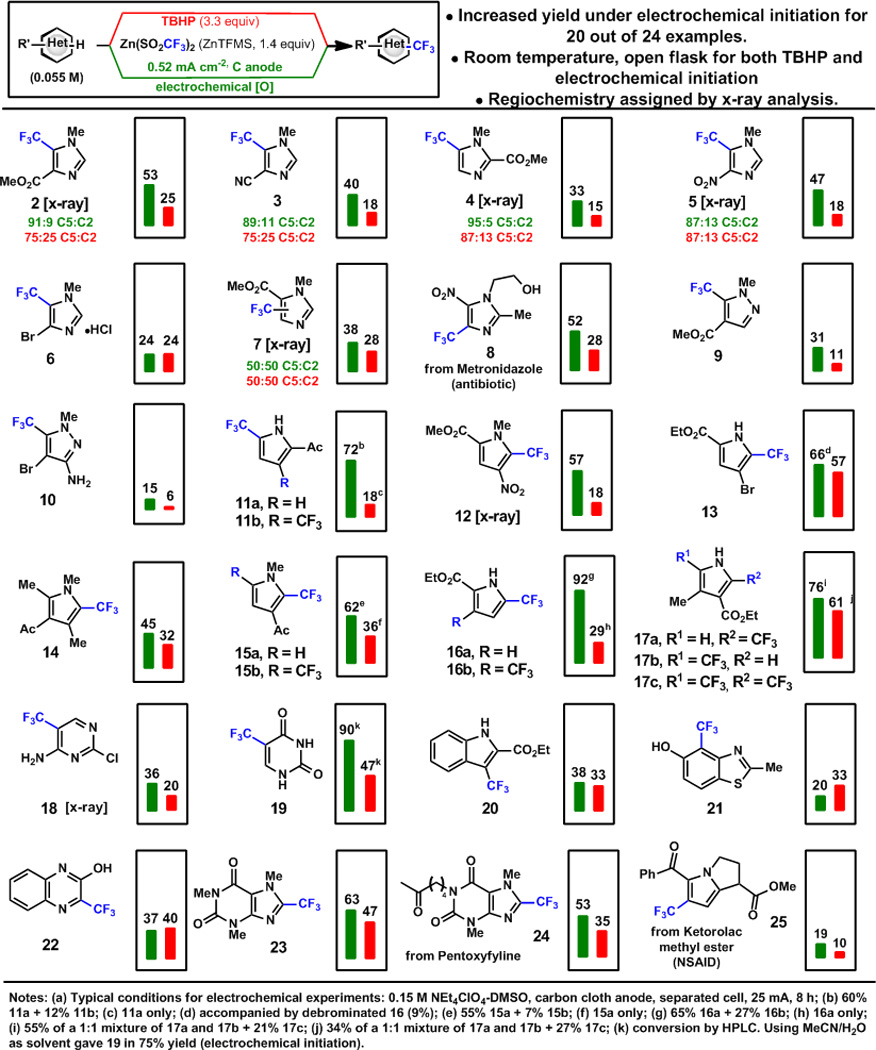
Comparison of Trifluoromethylation Under Electrochemical and TBHP Radical Initiation.7
Scheme 3.
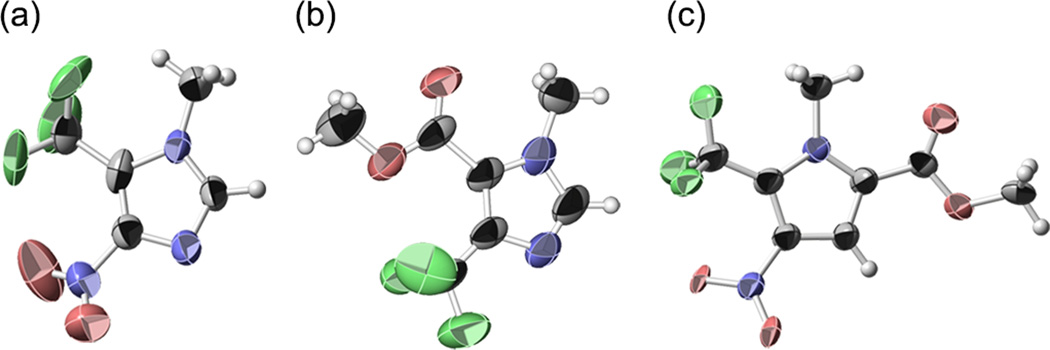
Structures of a) 5; b) 7; and c) 12; Determined by the Crystalline Sponge Method. Thermal ellipsoids are drawn at 50% probability.7,13
Scheme 4.
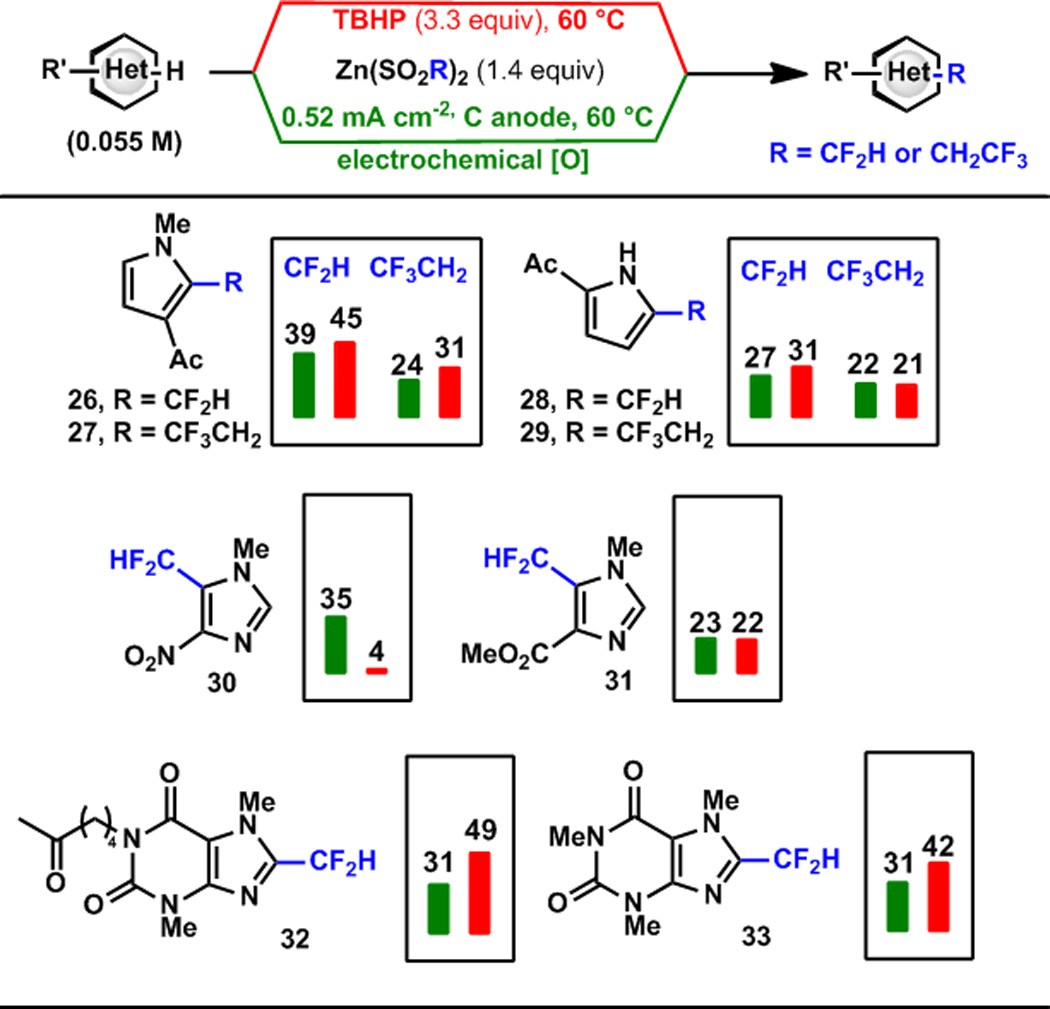
Comparison of Electrochemical and TBHP Radical Initiation with DFMS and TFES.7
In summary, monitoring of the reaction progress under electrochemical initiation allowed deconvolution of processes related to radical generation and radical consumption. Controlled radical formation mediates radical introduction into the system and increases the window of time for productive reaction, leading to enhanced yields for recalcitrant substrates of pharmaceutical interest. These results demonstrate the successful radical C—H functionalization of a wide variety of heterocycles up to gram scale under electrochemical initiation using significantly less sulfinate reagent than peroxide radical initiation methods.
Supplementary Material
Acknowledgements
DGB and PSB acknowledge funding from NIH/NIGMS (GM-106210) and Pfizer, Inc. AGO and AM acknowledge postdoctoral funding from Pfizer Inc. and Teijin Pharma Ltd., respectively. We thank Shota Yoshioka and Yuki Takahashi for x-ray analysis of 5, 7 and 12. We thank Prof. Arnold L. Rheingold and Dr. Curtis E. Moore for XRD of 2, 4 and 18.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Phil S. Baran, Email: pbaran@scripps.edu.
Donna G. Blackmond, Email: blackmond@scripps.edu.
References
- 1.a) Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc. Natl. Acad. Sci. USA. 2011;108:14411. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fujiwara Y, Dixon JA, Rodriguez RA, Baxter RD, Dixon DD, Collins MR, Blackmond DG, Baran PS. J. Am. Chem. Soc. 2012;134:1494. doi: 10.1021/ja211422g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fujiwara Y, Dixon JA, O’Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herlé B, Sach N, Collins MR, Ishihara Y, Baran PS. Nature. 2012;492:95. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) O’Hara F, Baxter RD, O’Brien AG, Collins MR, Dixon JA, Fujiwara Y, Ishihara Y, Baran PS. Nat. Protoc. 2013;8:1042. doi: 10.1038/nprot.2013.059. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhou Q, Gui J, Pan C-M, Albone E, Cheng X, Suh EM, Grasso L, Ishihara Y, Baran PS. J. Am. Chem. Soc. 2013;135:12994. doi: 10.1021/ja407739y. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Baxter RD, Blackmond DG. Tetrahedron. 2013;69:5604. [Google Scholar]
- 2.Stout EP, Choi MY, Castro JE, Molinski TF. J. Med. Chem. 2014 doi: 10.1021/jm4016922. ASAP, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Hara F, Blackmond DG, Baran PS. J. Am. Chem. Soc. 2013;135:12122. doi: 10.1021/ja406223k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smertenko EA, Datsenko SD, Ignat’ev NV. Russ. J. Electrochem. 1998;34:46. Tommasino J-B, Brondex A, Médebielle M, Thomalla M, Langlois BR. B. R. Synlett. 2002:1697.for a related photochemical example, see Wilger DJ, Gesmundo NJ, Nicewicz DA. Chem. Sci. 2013;4:3160. d) for a conceptually related example of anodic CF3 generation from trifluoroacetate, see: Utley JHP, Holman RJ. Electrochim. Acta. 1976;21:987.
- 5.For elegant recent examples, see: Morofuji T, Shimizu A, Yoshida J. J. Am. Chem. Soc. 2014;136:4496. doi: 10.1021/ja501093m. Ashikari Y, Shimizu A, Nokami T, Yoshida J. J. Am. Chem. Soc. 2013;135:16070. doi: 10.1021/ja4092648. Morofuji T, Shimizu A, Yoshida J. J. Am. Chem. Soc. 2013;135:5000. doi: 10.1021/ja402083e. Nguyen BH, Redden A, Moeller KD. Green Chem. 2014;16:69. Redden A, Perkins RJ. K. D. Angew. Chem. Int. Ed. 2013;52:49. doi: 10.1002/anie.201308739. Zeng C, Zhang N, Lam CM, Little DR. Org. Lett. 2012;14:1314. doi: 10.1021/ol300195c. Kirste A, Elser B, Schnakenburg G, Waldvogel SR. J. Am. Chem. Soc. 2012;134:3571. doi: 10.1021/ja211005g.
- 6.a) Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. Wiley; 2000. [Google Scholar]; b) Moeller KD. Tetrahedron. 2000;56:9527. [Google Scholar]
- 7.See Supporting Information for experimental details.
- 8.For a previous report of electrochemical functionalization of imidazoles, see: Médibielle M, Oturan MA, Pinson J, Savéant J-M. J. Org. Chem. 1996;61:1331.
- 9.An example of current-controlled reactivity: Fedkiw PS, Chao JC. AIChE. J. 1985;31:1578.
- 10.Bietti M, DiLabio GA, Lanzalunga O, Salamone M. J. Org. Chem. 2010;75:5875. doi: 10.1021/jo100931a. [DOI] [PubMed] [Google Scholar]
- 11.Nath Das T, Dhanasekaran T, Alfassi ZB, Neta P. J. Phys. Chem. A. 1998;102:280. [Google Scholar]
- 12.Connelly NG, Geiger WE. Chem. Rev. 1996;96:877. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 13.The active oxidant may be neutral TBHP. For an example, see: Jones CM, Burkitt MJ. J. Am. Chem. Soc. 2003;125:6946. doi: 10.1021/ja034416z.
- 14.Inokuma Y, Yoshioka S, Ariyoshi J, Arai T, Hitora Y, Takada K, Matsunaga S, Rissanen K, Fujita M. Nature. 2013;495:4. doi: 10.1038/nature11990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.