

Abstract
The transgenerational epigenetic programming involved in the passage of environmental exposures to stressful periods from one generation to the next has been examined in human populations, and mechanistically in animal models. Epidemiological studies suggest that gestational exposures to environmental factors including stress are strongly associated with an increased risk of neurodevelopmental disorders, including attention deficit-hyperactivity disorder, schizophrenia, and autism spectrum disorders. Both maternal and paternal life experiences with stress can be passed on to offspring directly during pregnancy or through epigenetic marks in the germ cell. Animal models of parental stress have examined relevant offspring phenotypes and transgenerational outcomes, and provided unique insight into the germ cell epigenetic changes associated with disruptions in neurodevelopment. Understanding germline susceptibility to exogenous signals during stress exposure and the identification of the types of epigenetic marks is critical for defining mechanisms underlying disease risk.
Keywords: development, fetal, germline, histone, maternal, methylation, microRNA, paternal, sperm
Abstract
Tanto en humanos como en modelos anímales se ha estudíado la programación epígenética transgeneracional involucrada en el paso de una generacíón a la otra de las exposicíones ambíentales a períodos de estrés. Los estudíos epídemiológicos sugíeren que las exposíciones a los factores ambientales durante la gestación, íncluyendo el estrés, están fuertemente asocíadas con un aumento del ríesgo para los trastornos del neurodesarrollo, como el trastorno por défícít de atencíón con híperactividad, la esquízofrenia y los trastornos del espectro autísta.
Las experiencias de vida con estrés, tanto maternas como paternas, pueden pasar a la descendencía directamente durante el embarazo o a través de marcas epigenéticas en las células germinales. Los modelos anímales de estrés parental han estudíado fenotípos relevantes en la descendencia y resultados transgeneracíonales, y han aportado una mírada oríginal a los cambíos epígenétícos de las células germinales asocíados con alteracíones en el neurodesarrollo. El conocer la susceptíbílídad de la línea germinal a las señales exógenas durante la exposición al estrés y el ídentíficar los típos de marcas epigenéticas resultan fundamentales para la definícíón de los mecanísmos que subyacen al ríesgo de enfermar.
Abstract
La programmation épigénétique transgénérationnelie impliquée dans la transmission d'une génération à la suivante de l'exposition environnementale à des périodes de stress, a été analysée chez les humains et de façon mécaniste dans des modèles animaux. Selon des études épidémiologiques, l'exposition gestationnelle à des facteurs environnementaux, dont le stress, s'associe fortement à un risque accru de troubles neurodéveloppementaux, comme le déficit de l'attention/hyperactivité, la schizophrénie et les troubles du spectre de l'autisme. Les expériences de stress paternelles et maternelles peuvent être transmises directement aux descendants pendant la grossesse ou par des marques épigénétiques dans les cellules germinales. Des modèles animaux de stress parental ont exploré des phénotypes de descendants et des résultats transgénérationnels pertinents et ont donné un aperçu unique des changements épigénétiques des cellules germinales associées aux perturbations du neurodéveloppement. La compréhension de la susceptibilité des lignées germinales aux signaux exogènes pendant l'exposition au stress et l'identification des types de marques épigénétiques sont déterminantes pour définir les mécanismes sous-tendant le risque de maladie.
Introduction
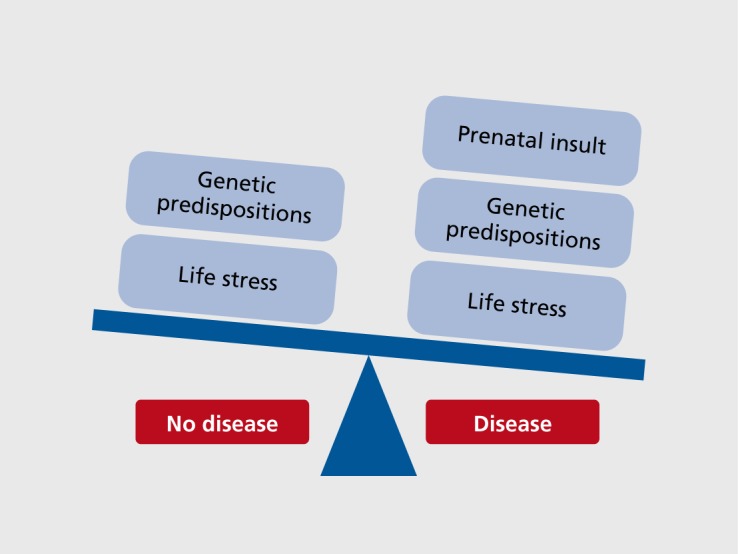
Stress pathway dysregulation, in which there can be either a heightened or a blunted sensitivity to stress, is the most common symptom across neuropsychiatry diseases, yet we understand little about the sensitive periods during which environmental perturbations may epigenetically program neurodevelopmental changes. Epidemiological studies suggest that gestational exposures to environmental factors such as stress are strongly associated with an increased incidence of neurodevelopmental disorders, including attention deficit-hyperactivity disorder (ADHD), schizophrenia, autism spectrum disorders (ASD), and depression1-4 Examination of birth cohorts has revealed strong associations between in utero exposure to stress, infections, hypoxia, and starvation, and an increased risk for schizophrenia, including disturbances of executive function, working memory, verbal memory, and structural brain abnormalities.5-7 Similarly, the maternal stress association with ASD has also been reported as exposure-timing sensitive where stress at 21 to 32 weeks of gestation and postnatal stressors such as the death of a relative in the first 6 months of life significantly increased ASD risk.8 A similar study using data from the Swedish registry found that the stress from the death of a close family member, specifically during the third trimester of pregnancy, increased the incidence of ADHD.9 Such epidemiological reports have repeatedly demonstrated the vulnerability of the prenatal and early life environment in influencing long-term disease risk. However, such factors are likely only one part of disease presentation. For example, when mapped on to a given genetic variation, the environmental exposure to prenatal stress may be a second hit in a two- or three-hit model Figure 1. In a study examining ADHD risk, a significant interaction was found for children with one of four latrophilin-3 (LPHN3) single nucleotide polymorphisms and exposure to maternal stress.10
Figure 1. Tipping the balance toward neurodevelopmental disease presentation likely depends on multiple interacting factors, ultimately combining a genetic predisposition with environmental insults experienced at key developmental/susceptible periods in life. For example, when mapped onto a given genetic variation that promotes susceptibility, an environmental exposure to prenatal stress may act as a second hit in a two- or three-hit model, where the next stress insult experienced during later points in life tips the scale in the direction of disease onset. In such a model, the prenatal exposure to stress is acting as a point of epigenetic programming due to an increased genetic vulnerability that when added together leads to sensitivity to later life perturbations.
When thinking about additional factors, genetic or otherwise, that may contribute to an increased disease risk, it is worth noting that most neurodevelopmental disorders exhibit a sex bias, where females present with affective disorders at 2 to 3 times the rate of males, ASD affect 4 times as many boys as girls, and schizophrenia and depression onset dramatically rises during the adolescent years, when major sex differences in brain maturation occur. Therefore, elucidation of the mechanisms by which sex-specific susceptibility arises is likely to provide critical insight into disease etiology, leading to the identification of novel targets for therapeutic development across disease areas within mental health, as well as on a broader scale across many diseases that are influenced via a gene x environment influence. While there are many factors that likely contribute to sex differences in disease predisposition, sex-specific responses to fetal antecedents occurring during sensitive windows of development may promote long-term programming outcomes that underlie disease bias.3,5
The mechanisms through which early life events contribute to disease development likely involve complex interactions between factors of genetic susceptibility, environmental exposure, and epigenetic programming. The idea of a G x E x D (or how our genes are affected by the environment during developmental periods) interaction is of critical importance in discerning potential sensitive periods and mechanisms in neurodevelopmental disorder risk. While we have gained a great appreciation for the importance of the epigenome, we know very little about the windows of vulnerability across the lifespan in which plasticity of this system exists. The identification of environmental programming effects on novel genes or functional gene sets during sensitive and dynamic periods in development would greatly push forward our understanding of where the greatest susceptibility lies. Such information can be more clearly examined in animal models of maternal and paternal stress experience.
Animal models of parental stress
Maternal stress
Animal models of maternal stress have provided important opportunities to examine mechanisms underlying the programming of long-term offspring outcomes relevant to neurodevelopmental disorders. Offspring phenotypes vary depending upon the stressors utilized, outcomes examined, and timing of the stress event during pregnancy.11-14 In mice, rats, guinea pigs, and nonhuman primates, maternal stress has been clearly shown to alter the development and sensitivity of offspring hypothalamic-pituitary-adrenal (HPA) stress axis, behavioral stress reactivity, and cognitive deficits, all of which are endophenotypes relevant to neurodevelopmental disorders.13,15-21 However, the developing nervous system matures over the course of gestation and therefore is unlikely to show a uniform vulnerability to maternal stress experience during pregnancy. Our laboratory and others have reported findings from maternal stress models on a range of outcomes, including increased stress responsiveness, anhedonia, and cognitive deficits that were specific to the timing of stress exposure as well as the sex of the offspring.16,17,21-23 In comparing the effects of stress across early, mid, or late pregnancy on programming of offspring stress regulation, our lab utilized a chronic variable stress (CVS) paradigm in mice.17,18 In these studies, we found that CVS early in pregnancy (days 1 to 7) produced male offspring with increased stress sensitivity and reduced cognitive abilities.17,18 Mechanistically, this early stress exposure affected the programming of genes involved in offspring stress neurocircuitry, including elevated limbic corticotropin-releasing factor (CRF), and reduced hippocampal glucocorticoid receptors (GR).18 Surprisingly, these effects were specific to male offspring and only occurred with an early exposure to the dam's stress, as mid- and late-pregnancy stress did not program such effects. Interestingly, similarly to studies that have exposed rat dams to stress late in pregnancy, our early gestation CVS exposure appeared to dysmasculinize the males, where their physiological and behavioral responses to stress as adults were more like that of females than males.18,24 Similar findings have been reported in a guinea pig model by Kapoor et al, where the timing of maternal stress exposure predicted the offspring outcomes.13,25 A rat model of maternal social stress also produced adult offspring with enhanced HPA stress axis sensitivity and similar changes in gene expression patterns throughout the brain to those reported for maternal stress in mice and guinea pigs, including changes in hypothalamic CRF and hippocampal GR.12 This model of maternal social stress also appeared to change the adult offspring's maternal behavior, and as such perpetuated the stress dysregulation phenotype into the next generation.26
Postnatal maternal stress has also been modeled in animals, and has demonstrated an important role of maternal care in shaping of the developing brain. In a mouse model of maternal separation, in which dams were removed from their offspring and exposed to stress for 3 hours each day for the first 2 weeks postpartum, the male first-generation offspring showed significantly elevated stress reactivity in behavioral tests.27 Transgenerational programming of this phenotype was also examined where many of the behavioral effects persisted into the second and third generations. Interestingly, results from this study highlight the importance of offspring sex as a vulnerability or resiliency factor—while the first-generation males showed an effect of reduced maternal care, it was the second- and third-generation female offspring that subsequently presented with the phenotype, in contrast to their male littermates. Similar outcomes were programmed in a rat model of reduced maternal care, where maternal interactions and care of pups were reduced by placing the dam with her litter in an “impoverished” environment by limiting her bedding and nesting material for the first week postpartum.28 This fragmentation and unpredictability of maternal behavior models early life abuse and neglect, and produces adult offspring with reduced cognitive abilities and diminished dendritic arborization in the hippocampus.29
On the opposite side of fragmented care and neglect, work with rodent models has also examined effects on offspring resulting from augmented maternal care. Meaney et al have contributed a great deal of information to this field regarding the epigenetic processes that occur in early life as a function of the quality of maternal care.30,31 These studies examined maternal care—defined as high or low licking and grooming dams—and associated the dam's postnatal interactions with her litter with offspring adult behaviors, where high licking and grooming behaviors produced offspring with a hypoactive HPA stress axis and reduced levels of anxiety-like behaviors. The epigenetics examined in these outcomes, at least in part, reported changes in DNA methylation status of the GR in the hippocampus. High licking and grooming mothers produced enhanced tactile stimulation of their pups, resulting in increased expression of specific transcription factors during brain development that ultimately determined GR levels throughout life. Another maternal rat study modeled augmented maternal care by exposing dams to brief daily maternal separation from her pups over the first 6 postnatal days. This deprivation period resulted in dams that when returned to their litters showed enhanced maternal behaviors, including increased time spent licking and grooming and arched-back nursing.32 These changes in maternal care produced offspring with blunted hypothalamic reactivity, including fewer excitatory synapses on CRF neurons and reduced hypothalamic CRF expression. These results support an important temporal specificity for maternal stress, both during pregnancy and postpartum, to produce long-term changes in the stress pathway development of first-generation offspring that is reproducible across many different species and likely provides important and translatable relevance to human disease risk.
Paternal stress
It is clear from epidemiological studies in which risk factors for neurodevelopmental disorders and related outcomes have been examined that very little is known about paternal exposures, as most have focused on maternal experiences during pregnancy. Furthermore, we understand very little about germ cell epigenetic reprogramming, thus making it even more difficult to provide informed risk estimates without more clear insight into potential transgenerational influences. Epidemiological studies examining males exposed to the Swedish famine or the Dutch “Hunger Winter” famine have attempted to identify periods of development where germ cells may be more susceptible to perturbations in the environment.3,33,34 However, we still have much to learn about how nonimprinting-related epigenetic marks manifest in the germ cell and when in germ cell maturation this can occur.
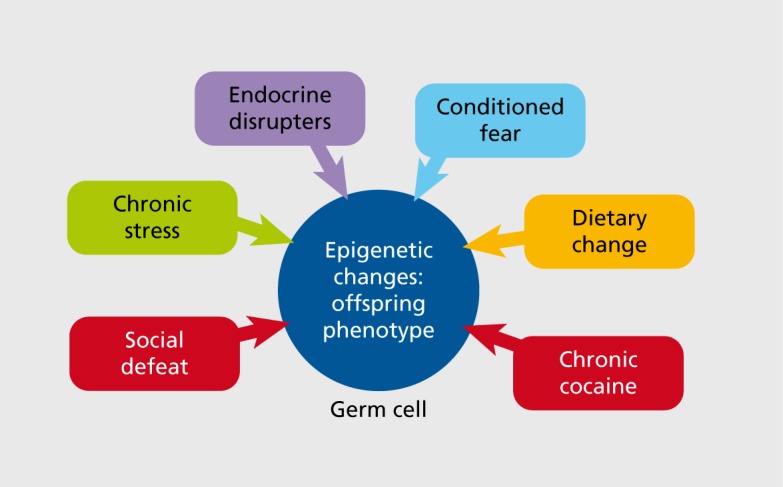
In animal studies, transgenerational outcomes following maternal exposures have been much more broadly examined than those of paternal modes of transmission in which the male was directly exposed prior to breeding. However, recent paternal studies have included models of social defeat, chronic stress, cocaine exposure, dietary manipulations, endocrine disrupters, and conditioned fear Figure 2. 35-42 While most maternal “exposures” are examined during pregnancy, and thus have the ability to affect the offspring more directly during somatic development, paternal studies have largely focused on stress experienced prior to mating and thus are thought to impact future offspring via germ cell reprogramming. One of the great advantages of studying transgenerational epigenetics in paternal rather than maternal transmission is that in most rodent studies males do not interact directly with their offspring and thus are only able to pass on information via germ cells, ie, if the sire's offspring are programmed differently as a result of his life experiences, then that information must be present in his sperm. This affords the unique ability to ascertain contributing epigenetic marks in a relatively homogenous and easily obtainable tissue source in which DNA methylation, histone modifications, or microRNAs (miRs) can be examined (as discussed below).
Figure 2. Animal models of paternal germ cell programming have demonstrated clear transgenerational epigenetic outcomes following exposure to various perturbations in which the male was exposed prior to breeding. Recent studies have included models of social defeat, chronic stress, cocaine exposure, dietary manipulations, endocrine disrupters, and conditioned fear.35-43 .
Recent stress models of paternal transmission have demonstrated a surprisingly broad timing of plasticity of the male germ cell across spermatogenesis. In the case of social defeat stress, offspring from defeat-subjected fathers showed changes in behavioral and physiological stress reactivity.35 Similarly, in a mouse model examining conditioned fear stress, males exposed to a shock paired with a specific odor passed on this conditioning effect to their first- and second-generation offspring.36 In an effort to more specifically define the timing of germ cell vulnerability to stress, we exposed male mice to 6 weeks of chronic variable stress either over the pubertal window or only as adults, and then bred these males to control females. We found, quite surprisingly, that stress during either time point, pubertal or adult, was able to reprogram the sperm and generate male and female offspring with a hypofunctioning HPA stress axis.37 The specificity of this effect was intriguing, as we found no change in any of the stress behavioral measures examined in these offspring, including lightdark box test, Barnes maze for spatial learning and memory, pre -pulse inhibition, or acoustic startle. The effect of stressing the father prior to breeding produced a very selective change in the offspring's physiological response to future stressors, suggesting that there may be an evolutionary advantage for the father to pass on to his offspring the ability to blunt their stress response in a stressful environment.
Transgenerational programming: a mechanistic examination
An epigenetic mark must be present in the germ cell for a phenotypic outcome to be considered truly transgen erational. The ability for these epigenetic marks to be erased and re-established in the absence of re-exposure to the environmental stimuli determines whether future generations will be impacted. Of additional importance is the sex-specificity of transgenerational epigenetic programming, which requires examination at both the level of transmission of outcomes as well as the inheritance of them; whether both maternal and paternal lineages can pass on traits, and if both male and female offspring appear to inherit them. Epidemiological studies from Swedish famine periods have defined important and specific developmental windows during which germ cells may be more vulnerable to environmental influences. Males exposed to famine prior to or during the “slow growth” period prior to the start of puberty passed on health and disease risks to their children and grandchildren, suggesting that there is machinery present and able to reprogram epigenetic marks in the germ cell at this stage.44 Famine experienced outside this window diminished the transgenerational impact. Similar results have been reported in studies examining generational outcomes in exposures during the Dutch Hunger Winter at the end of World War II. Specific epigenetic changes were reported in individuals who were conceived during the stress of this food embargo where as adults they had reduced methylation of the imprinted gene IGF2 six decades after the initial exposure.34 This outcome was distinct from those who were prenatally exposed later in gestation and did not show a change in methylation at this locus as adults.
Both maternal and paternal exposures have been modeled in animals where tightly controlled environments can closely examine transgenerational outcomes and germ cell programming. As an example in mice, transgenerational effects were programmed in male offspring exposed to maternal stress in which these males presented with increased stress sensitivity themselves as adults, with increased HPA axis responsivity to acute restraint, and behavioral and cognitive deficits.18 When bred to control females, these males passed on this same phenotype to their male, but not female, offspring, demonstrating a germ cell involvement where epigenetic changes likely occurred during the initial gestational exposure to maternal stress.45
Germ cell reprogramming
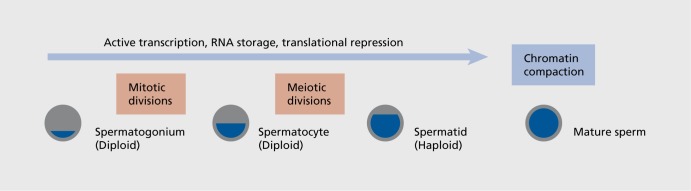
As described above, in order for the environment to influence the programming of future generations, epigenetic marks must be programmed in the germ cell. This poses important questions as to when and how this plasticity can occur Figure 3. 46 Epigenetic processes are important in regulation of germ cell development, determination of parental imprints, and as marks important in regulating gene expression, including DNA methylation and histone modifications. The methylation silencing of the maternal or paternal allele in genetic imprinting is critical for normal development, as dysregulation of this process can results in a number of disorders including Angelman, Prader-Willi, and Beckwith-Weidemann syndromes.47 During sexual differentiation, primordial germ cell imprints are largely erased and reset later in a sex-specific manner, either prior to (males) or following (females) birth. It is not entirely clear how the “memory” of these imprints following their erasure is maintained and then replaced; however, the epigenetic machinery involved may also function in reading and responding to changes in the environment, and is thus able to make new chromatin marks that will impact future generations. As an example, male mice fear-conditioned to a specific odor, acetophenone, which activates a specific odor receptor, Olfr151, not only passed on the fear of this odor to two future generations, but their sperm showed CpG hypomethylation of the Olfr151 gene, suggesting that the sire's germ cell was able to respond to a fear cue in the environment and epigenetically reprogram the locus such that future generations would know to avoid this odor.36 Similarly, in an early postnatal stress model in which mice continued to present with a stress-sensitive phenotype through three generations, comparable changes were detected in DNA methylation in the germline of these mice, suggesting an epigenetic mechanism by which the sire's stress experience was passed to his offspring.27 These studies showing interactions of the environment with the epigenome at the level of placing long-lasting marks on the chromatin that remain through cell divisions and across generations, support the plasticity of the germline to external influences that could increase disease susceptibility or build resilience of subsequent generations. It is also important to note that an environmental signal that influences the germline may not affect somatic tissue depending on the timing, and therefore phenotypes are effectively able to skip a generation, thus dissociating the exposure from its programmed outcome, making it more difficult to form clear risk analyses for pregnant women or other at-risk groups (ie, infants, adolescents). Once the primordial germ cells are sexually differentiated, the oocyte then enters into meiosis and quickly arrests until puberty, while male germ cells go into mitotic arrest until birth when they go through a proliferation phase, and completing meiosis at puberty.48
Figure 3. Points in spermatogenesis where environmental insults are most able to reprogram epigenetic marks. Prior to DNA compaction for the mature sperm, active transcription and storage of RNA is ongoing through the spermatogonium, spermatocyte, and spermatid stages.52 Once compaction of paternal DNA involving protamine swamping for most all histones has occurred, there is no clear means by which epigenetic marks can be altered in the mature sperm.
Epigenetic marks present and detected in sperm include noncoding RNAs, such as miRs and piRNAs, histone modifications (for the few retained histones in sperm), and DNA methylation.49 For instance, male germ cells have high levels of piRNAs that are required for normal spermatogenesis and function in gene silencing of transposons.50,51 The functioning of these piRNAs appears to be through interactions with DNA methyltransferases. Male and female germ cells also contain miRs. While their function in postfertilization programming is not clear, evidence points to a necessity of the oocyte RNA-induced silencing complex (RISC) in guiding early development.52 The RISC is a multi-protein complex where miRs and their target mRNAs are loaded and subsequently degraded by the endogenous RNase activity of one of the Argonaut proteins of the complex. For instance, in examination for germiine epigenetic marks in the sperm from male mice exposed to chronic stress, either over the pubertal window or as adults, nine specific miRNAs were significantly upregulated that were predictive of offspring stress axis dysregulation from both paternal time points of stress exposure.37 While it is becoming more clear as to how these noncoding RNAs may play important roles in programming the embryo, just how such epigenetic mechanisms can become transgenerational is less clear. Noncoding RNAs are transcribed from genes themselves, so if their expression levels were altered in the germ cell, it would suggest that an upstream epigenetic mark such as DNA methylation or histone modifications is the driving force. In the mature sperm, few histones are retained as most are exchanged for arginine-rich protamines during compaction. Interestingly, recent studies have pointed to an enrichment of retained nucleosomes at specific loci involved in early embryonic development where high levels of the histone marks H3K27me3 and H3K4me3 were found.53
Interestingly, environmental exposures such as drugs of abuse, odors, or high-fat diets can also produce germiine epigenetic marks transmitted to offspring capable of shaping their brain development and long-term behaviors.35-43 Such studies highlight how dynamic and plastic germ cells appear to be—far more than we thought. What is less clear, however, is how and when in germ cell development these epigenetic marks can be programmed. In the case of our paternal chronic stress study, stress exposure occurred over 6 weeks, which encompasses the start-to-end process of spermatogenesis and thus could impart effects at earlier points in which epigenetic machinery is still functional and present.37 However, in the odor fear-conditioned animals, the exposure to the shock-odor pairing in the males occurred over a much shorter window prior to breeding, thus suggesting that the epigenetic changes reported occurred in mature sperm.36 We are clearly just beginning to appreciate the mechanisms by which the environment can impart long-term epigenetic changes in germ cells, thus affecting future generations. Detailed studies are needed to determine the point in spermatogenesis where the germiine is susceptible to these exogenous signals and the types of epigenetic marks by which the signals are relayed. Studies are also needed to examine female prepregnancy life exposures and their effects on oocyte epigenetics. Such outcomes are exciting and have great translational appeal as potential biomarkers in predicting disease risk.
Summary
Epidemiological studies provide strong evidence for stressful early life experiences such as famine or war to program disease risk for future generations. The transgenerational passage of parental stress experiences involves changes in germ cell epigenetic marks. Animal models of maternal and paternal stress exposures have examined offspring phenotypes programmed either during pregnancy or prior to breeding, and have identified novel epigenetic mechanisms by which neurodevelopmental changes occurred. Future directions for this field will need to determine the windows and mechanisms during germ cell maturation where perturbations can have an influence. Further, there is a great need for understanding how these germ cell epigenetic changes direct postfertilization zygote development and eventually sculpt a different neuronal landscape. There are numerous black boxes here still to be determined—an exciting and promising era of transgenerational epigenetics that will no doubt provide new insight into mechanisms underlying neurodevelopmental disorders.
Acknowledgments
The work discussed in this review was funded in part by grants from the National Institutes of Health (NIH); MH091258, MH087597, and MH099910.
REFERENCES
- 1.Meyer U., Feldon J., Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation?. Pediatr Res. 2011;69(5 Pt 2):26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howerton CL., Bale TL. Prenatal programing: at the intersection of maternal stress and immune activation. Horm Behav. 2012;62:237–242. doi: 10.1016/j.yhbeh.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bale TL., Baram TZ., Brown AS., et al Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72:1272–1276. doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stress. 2011;14:348–356. doi: 10.3109/10253890.2011.586447. [DOI] [PubMed] [Google Scholar]
- 6.Susser E., St Clair D., He L. Latent effects of prenatal malnutrition on adult health: the example of schizophrenia. Ann N Y Acad Sci. 2008;1136:185–192. doi: 10.1196/annals.1425.024. [DOI] [PubMed] [Google Scholar]
- 7.Brown AS., Vinogradov S., Kremen WS., et al Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am J Psychiatry. 2009;166:683–690. doi: 10.1176/appi.ajp.2008.08010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angelidou A., Asadi S., Alysandratos KD., Karagkouni A., Kourembanas S., Theoharides TC. Perinatal stress, brain inflammation and risk of autismReview and proposal. BMC Pediatr. 2012;12:89. doi: 10.1186/1471-2431-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Class QA., Abel KM., Khashan AS., et al Offspring psychopathology following preconception, prenatal and postnatal maternal bereavement stress. Psychol Med. 2014;44:71–84. doi: 10.1017/S0033291713000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhry Z., Sengupta SM., Grizenko N., et al LPHN3 and attentiondeficit/hyperactivity disorder: interaction with maternal stress during pregnancy. J Child Psychol Psychiatry. 2012;53:892–902. doi: 10.1111/j.1469-7610.2012.02551.x. [DOI] [PubMed] [Google Scholar]
- 11.Clarke AS., Schneider ML. Prenatal stress has long-term effects on behavioral responses to stress in juvenile rhesus monkeys. Dev Psychobiol. 1993;26:293–304. doi: 10.1002/dev.420260506. [DOI] [PubMed] [Google Scholar]
- 12.Brunton PJ., Russell JA. Prenatal social stress in the rat programmes neuroendocrine and behavioural responses to stress in the adult offspring: sex-specific effects. J Neuroendocrinoi. 2010;22:258–271. doi: 10.1111/j.1365-2826.2010.01969.x. [DOI] [PubMed] [Google Scholar]
- 13.Kapoor A., Matthews SG. Short periods of prenatal stress affect growth, behaviour and hypothalamo-pituitary-adrenal axis activity in male guinea pig offspring. J Physiol. 2005;566(Pt 3):967–977. doi: 10.1113/jphysiol.2005.090191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cottrell EC., Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 2009;3:19. doi: 10.3389/neuro.08.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider ML., Moore CF., Kraemer GW., Roberts AD., DeJesus OT. The impact of prenatal stress, fetal alcohol exposure, or both on development: perspectives from a primate model. Psychoneuroendocrinology. 2002;27:285–298. doi: 10.1016/s0306-4530(01)00050-6. [DOI] [PubMed] [Google Scholar]
- 16.Kapoor A., Kostaki A., Janus C., Matthews SG. The effects of prenatal stress on learning in adult offspring is dependent on the timing of the stressor. Behav Brain Res. 2009;197:144–149. doi: 10.1016/j.bbr.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Mueller BR., Bale TL. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol Behav. 2007;91:55–65. doi: 10.1016/j.physbeh.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Mueller BR., Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemaire V., Koehl M., Le Moal M., Abrous DN. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc Natl Acad Sci U S A. 2000;97:11032–11037. doi: 10.1073/pnas.97.20.11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darnaudery M., Maccari S. Epigenetic programming of the stress response in male and female rats by prenatal restraint stress. Brain Res Rev. 2008;57:571–585. doi: 10.1016/j.brainresrev.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Weinstock M. Alterations induced by gestational stress in brain morphology and behaviour of the offspring. Prog Neurobiol. 2001;65:427–451. doi: 10.1016/s0301-0082(01)00018-1. [DOI] [PubMed] [Google Scholar]
- 22.Mueller BR., Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. 2006;88:605–614. doi: 10.1016/j.physbeh.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 23.Richardson HN., Zorrilla EP., Mandyam CD., Rivier CL. Exposure to repetitive versus varied stress during prenatal development generates two distinct anxiogenic and neuroendocrine profiles in adulthood. Endocrinology. 2006;147:2506–2517. doi: 10.1210/en.2005-1054. [DOI] [PubMed] [Google Scholar]
- 24.Ward IL. Prenatal stress feminizes and demasculinizesthe behavior of males. Science. 1972;175:82–84. doi: 10.1126/science.175.4017.82. [DOI] [PubMed] [Google Scholar]
- 25.Kapoor A., Leen J., Matthews SG. Molecular regulation of the hypothalamic-pituitary-adrenal axis in adult male guinea pigs after prenatal stress at different stages of gestation. J Physiol. 2008;586 (Pt 17):4317–4326. doi: 10.1113/jphysiol.2008.153684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunton PJ. Effects of maternal exposure to social stress during pregnancy: consequences for mother and offspring. Reproduction. 2013;146:R175–R189. doi: 10.1530/REP-13-0258. [DOI] [PubMed] [Google Scholar]
- 27.Franklin TB., Russig H., Weiss IC., et al Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68:408–415. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 28.Ivy AS., Brunson KL., Sandman C., Baram TZ. Dysfunctional nurturing behavior in rat dams with limited access to nesting material: a clinically relevant model for early-life stress. Neuroscience. 2008;154:1132–1142. doi: 10.1016/j.neuroscience.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunson KL., Kramar E., Lin B., et al Mechanisms of late-onset cognitive decline after early-life stress. J Neurosci. 2005;25:9328–9338. doi: 10.1523/JNEUROSCI.2281-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hackman DA., Farah MJ., Meaney MJ. Socioeconomic status and the brain: mechanistic insights from human and animal research. Nat Rev Neurosci. 2010;11:651–659. doi: 10.1038/nrn2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weaver IC., Diorio J., Seckl JR., Szyf M., Meaney MJ. Early environmental regulation of hippocampal glucocorticoid receptor gene expression: characterization of intracellular mediators and potential genomic target sites. Ann N Y Acad Sci. 2004;1024:182–212. doi: 10.1196/annals.1321.099. [DOI] [PubMed] [Google Scholar]
- 32.Korosi A., Shanabrough M., McClelland S., et al Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J Neurosci. 2010;30:703–713. doi: 10.1523/JNEUROSCI.4214-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown AS., Susser ES. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophrenia Bull. 2008;34:1054–1063. doi: 10.1093/schbul/sbn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heijmans BT., Tobi EW., Stein AD., et al Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dietz DM., Nestler EJ. From father to offspring: paternal transmission of depressive-like behaviors. Neuropsychopharmacology. 2012;37:311–312. doi: 10.1038/npp.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dias BG., Ressler KJ. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci. 2014;17:89–96. doi: 10.1038/nn.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodgers AB., Morgan CP., Branson SL., Revello S., Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neuroscience. 2013;33:9003–9012. doi: 10.1523/JNEUROSCI.0914-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng SF., Lin RC., Laybutt DR., Barres R., Owens JA., Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467:963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- 39.Vassoler FM., White SL., Schmidt HD., Sadri-Vakili G., Pierce RC. Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci. 2013;16:42–47. doi: 10.1038/nn.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carone BR., Fauquier L., Habib N., et al Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anway MD., Cupp AS., Uzumcu M., Skinner MK. Epigenetic transgenerational actions of endocrine disrupters and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson LM., Riffle L., Wilson R., Travlos GS., Lubomirski MS., Alvord WG. Preconceptional fasting of fathers alters serum glucose in offspring of mice. Nutrition. 2006;22:327–331. doi: 10.1016/j.nut.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Gapp K., Jawaid A., Sarkies P., et al Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Wat. Neurosci. 2014;17:667–669. doi: 10.1038/nn.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaati G., Bygren LO., Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents' and grandparents' slow growth period. Eur J Hum Genet. 2002;10:682–688. doi: 10.1038/sj.ejhg.5200859. [DOI] [PubMed] [Google Scholar]
- 45.Morgan CP., Bale TL. Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J. Neurosci. 2011;31:11748–11755. doi: 10.1523/JNEUROSCI.1887-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kotaja N., Sassone-Corsi P. The chromatoid body: a germ-cell-specific RNA-processing centre. Nat Rev Mol Cell Biol. 2007;8:85–90. doi: 10.1038/nrm2081. [DOI] [PubMed] [Google Scholar]
- 47.Lawson HA., Cheverud JM., Wolf JB. Genomic imprinting and parentof-origin effects on complex traits. Nat Rev Genet. 2013;14:609–617. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Apostolou E., Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature. 2013;502:462–471. doi: 10.1038/nature12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jenkins TG., Carrell DT. The sperm epigenome and potential implications for the developing embryo. Reproduction. 2012;143:727–734. doi: 10.1530/REP-11-0450. [DOI] [PubMed] [Google Scholar]
- 50.Wang G., Reinke V. AC. elegans Piwi, PRG-1, regulates 21 U-RNAs during spermatogenesis. Curr Biol. 2008;18:861–867. doi: 10.1016/j.cub.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siomi MC., Sato K., Pezic D., Aravin AA. PlWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 2011;12:246–258. doi: 10.1038/nrm3089. [DOI] [PubMed] [Google Scholar]
- 52.Lykke-Andersen K., Gilchrist MJ., Grabarek JB., Das P., Miska E., Zernicka-Goetz M. Maternal Argonaute 2 is essential for early mouse development at the maternal-zygotic transition. Mol Biol Cell. 2008;19:4383–4392. doi: 10.1091/mbc.E08-02-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brykczynska U., Hisano M., Erkek S., et al Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol. 2010;17:679–687. doi: 10.1038/nsmb.1821. [DOI] [PubMed] [Google Scholar]