

Abstract
Schizophrenia (SZ) and bipolar disorder (BPD) patients show a downregulation of GAD67, reelin (RELN), brain-derived neurotrophic factor (BDNF), and other genes expressed in telencephalic GABAergic and glutamatergic neurons. This downregulation is associated with the enrichment of 5-methylcytosine and 5-hydroxymethylcytosine proximally at gene regulatory domains at the respective genes. A pharmacological strategy to reduce promoter hypermethylation and to induce a more permissive chromatin conformation is to administer drugs, such as the histone deacetylase (HDAC) inhibitor valproate (VPA), that facilitate chromatin remodeling. Studies in mouse models of SZ indicate that clozapine induces DNA demethylation at relevant promoters, and that this action is potentiated by VPA. By activating DNA demethylation, clozapine or its derivatives with VPA or other more potent and selective HDAC inhibitors may be a promising treatment strategy to correct the gene expression deficits detected in postmortem brain of SZ and BPD patients.
Keywords: bipolar, chromatin remodeling, clozapine epigenetics, histone deacetylase inhibitor, neuroleptic, psychosis, schizophrenia
Abstract
Los pacientes con esquizofrenia (EQZ) y trastorno bipolar (TAB) muestran una regulación hacia abajo de GAD67, reelina (RELN), factor neurotrófico derivado del cerebro (BDNF) y otros genes expresados en neuronas glutamatérgicas y gabaérgicas del telencefálo. Esta regulación hacia abajo está asociada con un aumento de 5-metiIcitosina y 5-hidroximetilcitosina en la zona proximal de las regiones reguladoras de genes en los respectivos genes. Una estrategia farmacológica para reducir la hipermetilación del promotor e inducir una conformación de cromatina más permisiva es la administración de fármacos como el valproato (VPA), inhibidor de la histona deacetílasa (HDAC), que facilita la remodelación de cromatina. Estudios en modelos de ratones con EQZ indican que la clozapina induce desmetilación de ADN en promotores relevantes y que esta acción es potenciada por VPA. Al activar la desmetilación de ADN, la clozapina o sus derivados con VPA u otros inhibidores más potentes y selectivos de la HDAC pueden constituir una prometedora estrategia terapéutica para corregir los déficits en la expresión génica detectada en cerebros postmortem de pacientes con EQZ y con TAB.
Abstract
Les patients souffrant de schizophrénie (SZ) et de troubles bipolaires (BPD) présentent une régulation négative du GAD67, de la reelin (RELN), du facteur neurotrophique dérivé du cerveau (BDNF) et d'autres gènes exprimés dans les neurones télencéphaliques GABAergiques et glutamatergiques. Cette régulation négative est associée à l'enrichissement de la 5-méthylcytosine et de la 5-hydroxyméthylcytosine en position proximale au niveau des domaines régulateurs du gène dans les gènes respectifs. L'administration de médicaments, comme l'acide valproïque (VPA), inhibiteur de l'histone désacétylase (HDAC), qui facilite le remodelage de la chromatine est un moyen pharmacologique pour diminuer l'hyperméthylation du promoteur et pour induire une conformation plus souple de la chromatine. Des études sur des modèles murins de SZ indiquent que la clozapine induit la déméthylation de l'ADN au niveau des promoteurs essentiels, cette action étant potentialisée par le VPA. En activant la déméthylation de l'ADN, la clozapine ou ses dérivés avec le VPA ou d'autres inhibiteurs HDAC plus puissants et plus sélectifs peuvent être une stratégie thérapeutique prometteuse pour corriger les déficits de l'expression de gènes détectés dans les cerveaux postmortem des patients atteints de SZ et BPD.
Introduction
Progress in developing new, more effective, and less toxic antipsychotic drugs has been hampered by the lack of objective diagnostic tools to assess prodromes, progression severity, and therapeutic responses in schizophrenia (SZ) and bipolar disorder (BPD) patients. Additional fundamental barriers to the identification of new drugs to effectively treat SZ and BPD include the incomplete understanding of the etiopathogenetic mechanisms underlying the symptomatology of these diseases and the inability to reproduce the complex nature of these disorders in laboratory animals.
It is well established that SZ and BPD have a strong hereditary component. However, epigenetic studies indicate that altered DNA methylation may have an important role in the pathogenesis of these diseases and as a target mechanism for drug discoveries.1 The following topics will be addressed in this article: (i) altered expression of candidate genes in brain and blood cells of SZ and BPD patients treated with typical or atypical antipsychotics; (ii) dysregulated DNA methylation/demethylation processes as targets for antipsychotic drug action; (iii) effects of antipsychotics on epigenetic animal models of SZ. A greater understanding regarding the action of antipsychotic drugs on neuroepigenetic mechanisms will not only accelerate the development of novel pharmacological agents to treat SZ and BPD, but should also provide insights into their underlying neurobiological causes.
GABAergic and glutamatergic gene expression profiles altered in cortex and hippocampus in SZ or BPD: relation to antipsychotic treatment
In the last 20 years molecular biological studies have consistently detected a γ-aminobutyric acid (GABA) ergic and glutamatergic neuropathology in the hippocampus and cortex of SZ and BPD patients. (Table I).2-15
The GABAergic neuropathology is characterized by a decrease in the expression of glutamic acid decarboxylase 67 (GAD67, symbol=GAD1). GABAergic pathology is also characterized by decreased expression of nicotine acetylcholine receptor α4 (CHRNA4) and α7 (CHRNA7) subunits16 and by a decrease in other receptors abundantly expressed in GABAergic neurons, such as the N-methyl-D-aspartate (NMDA) receptor subunit NR2A (GRIN2A) and the kainate receptor subunit GluR5 (GRIK1).17-19 Further, there are decreases in somatostatin, tyrosine kinase B (TRKB) receptors, cholecystokinin (CCK), GABA transporter-1 (GAT1), and paralbumin (PVALB).17-19,20 Glutamatergic neuropathology is characterized by a decreased expression of brain-derived neurotrophic factor (BDNF), which potentially mediates a reduction in the neuropil detected in the prefrontal cortex (PFC) of SZ and BPD patients.9,13-15,21-24 It is important to mention that in SZ patients, reduced BDNF expression in pyramidal neurons is accompanied by a significant decrease in TRKB mRNA levels in GABAergic interneurons.9 Therefore, reduced levels of BDNF and TRKB as well as reelin (RELN) may be responsible for producing the decrease in spine density observed in the brains of SZ patients.
Patients with SZ or BPD often receive antipsychotic medications. Since the control subjects never receive these medications, the question is whether the altered GABAergic and glutamatergic gene expression changes observed in the brains of these patients is the consequence of protracted antipsychotic treatment rather than the etiopathogenetic signature of SZ or BPD. Although, as shown in several postmortem studies, there is no correlation between the levels of GAD67, RELN, or BDNF mRNA and proteins and lifetime dosage of antipsychotic medications,7,21,25-26 the low statistical power of these postmortem studies may not be sufficient to draw reproducible conclusions.
Protracted haloperidol treatment of rats fails to change RELN mRNA content in cortex and cerebellum.7 In another study, it was shown that protracted haloperidol treatment fails to change the expression of GAD67 mRNA in the PFC of nonhuman primates.25 However, it was also reported27 that chronic (27 days) haloperidol and clozapine treatment increase, rather than decrease, the expression of GAD67 in cortico-limbic structures. Fatemi et al28 report that chronic olanzapine facilitates the expression of genes involved in signal transduction, cell communication, metabolism, immune responses, and an upregulation of RELN expression in frontal cortex of rats. Costa and his group reported previously that in rats the turnover rate of GABA fails to change with haloperidol but increases with clozapine treatment.29 Collectively, these data suggest that the downregulation of RELN and GAD67 in brains of SZ and BPD patients must be, at least in part, independent of haloperidol or clozapine treatment, although a more extensive study including additional typical or atypical antipsychotic treatment is needed.
Is an alteration in DNA methylation the molecular mechanism that mediates the GABAergic and glutamatergic dysfunction in SZ and BPD patients?
There are several epidemiological, clinical, and molecular peculiarities associated with SZ or BPD that are difficult to reconcile with Mendelian genetic disorders and, in contrast, correspond to features of an altered epigenetic homeostasis.30 Such features include: (i) incomplete phenotypic concordance between monozygotic twins; (ii) peaks of susceptibility to disease coinciding with major hormonal changes; and (iii) parent-of-origin effects. These observations have led to speculation regarding the importance of epigenetic factors in mediating changes in gene expression and psychosis susceptibility.
We previously reported that the downregulation of GAD67 or RELN expression in GABAergic neurons of SZ and BPD patients is associated with an overexpression of DNA methyltransferase 1 (DNMT1) and DNA methyltransferase 3a (DNMT3a, Table I).10-12,31-33
Table 1. Epigenetic signature in the prefrontal cortex of psychotic patients. APOBEC, apolipoprotein B mRNA-editing enzyme complex; BNDF, brain-derived neurotrophic factor; DNMT, DNA methyltransferase; GAD, glutamic acid decarboxylase; TET, tet methylcystosine dioxygenase.
DNMTs belong to a family of enzymes that catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to the 5 carbon of cytosines of many gene regulatory domains.1,34 Although increased DNA methylation induced by the overexpression of DNMTs in SZ and BPD patients may be the cause of GABAergic gene suppression, DNMTs also exert a negative action on gene expression through the formation of repressor complexes. Repressive chromatin complexes contain other specific proteins (eg, methyl CpG binding domain proteins, SIN3A, and histone deacetylases) that act to repress transcription via modifications of chromatin structure, shifting chromatin from a permissive open conformation to a repressive closed conformation.31,34-38
Support for the concept that an epigenetic pathology may be responsible for the GABAergic or glutamatergic gene transcriptional downregulation in SZ and BPD patients comes from the following experimental observations: (i) increased S-adenosyl-methionine (SAM) in the PFC39; (ii) hypermethylation or hyperhydroxymethylation of cytosines in CpG islands proximal to the RELN, GAD67, and BDNF promoters associated with reduced gene expression in the PFC21,26,37,40,41; and (iii) evidence of epigenetic dysregulation of several other GABAergic 42and glutamatergic genes in major psychosis (Table I).41
These data are consistent with the epigenetic theory of major psychosis31 and suggest that methylation/hydroxymethylation of GABAergic and glutamatergic gene promoters are important causal events in the pathogenesis of SZ and BPD. Furthermore, support for the hypothesis that an epigenetic chromatin remodeling pathology contributes to the downregulation of GABAergic or glutamatergic genes in psychotic patients comes from clinical studies conducted in the early 1970s (for review see ref 43). Methionine, the precursor of SAM (the methyl donor utilized by DNMTs to methylate cytosine in DNA), administered in large doses (10 to 20 g/day) for periods of 3 to 4 weeks to SZ patients, exacerbates psychotic symptomatology. In both mouse frontal cortex (FC) in vivo and rat neuronal cultures in vitro, the administration of large doses of methionine induces an increase in SAM and the hypermethylation of selective GABAergic promoters, including Gad67 and Reln, and facilitates the downregulation of their expression.35,44-47 Importantly, brain levels of GAD65 and various housekeeping genes were not affected by the treatment.46
DNA demethylation network process is altered in SZ and BPD patients
Inconsistent results with studies of RELN, GAD67, and BDNF promoter methylation in relation to mental illness41,48-50 suggest that the DNA methylation status of specific DNA regions in post-mitotic neurons is not stable but, in contrast, is rapidly fluid, maintained by the equilibrium between DNA methylation and demethylation.51-53 This theory is supported by several independent and particularly interesting findings showing that the 5-methylcytosine (5-mC) mark on promoter regions of specific genes can be oxidized to form 5-hydroxymethylcytosine (5-hmC) by members of the ten-eleven translocation (TET) protein family in mammalian brain.54-58
Further, it has been proposed that 5-hmC undergoes two subsequent, processing steps: (i) a deamination step catalyzed by the activation induced deaminase (AID)/apolipoproptein B RNA editing catalytic component (APOBEC) family of cytosine deaminases, that convert 5-hmC to 5-hydroxylmethyluracil (5-hmμ); and (ii) the base excision repair (BER) pathway, in which 5-hmμ is removed and replaced by cytosine by DNA glycosylases such as MBD4 and TDG (Figure 1).21,26,59 Growth Arrest and DNA Damage-inducible 45 (Gadd45)60-62 proteins are thought to coordinate this process in response to neuronal activity by recruiting deaminases and glycosylases to DNA enriched in 5-mC and 5-hmC.59,63
Figure 1. Schematic representation of putative DNA methylation/demethylation. DNMT, DNA methyltransferase; TET, ten-eleven translocation protein; APOBECs, activation induced deaminase/apolipoprotein B RNA editing catalytic components; TDG, thymine DNA glycosylase; C, cytosine; 5MC, 5 methylcytosine; 5HMC, 5-hydroxymethylcytosine; 5HMU, 5-hydroxymethyluridine.
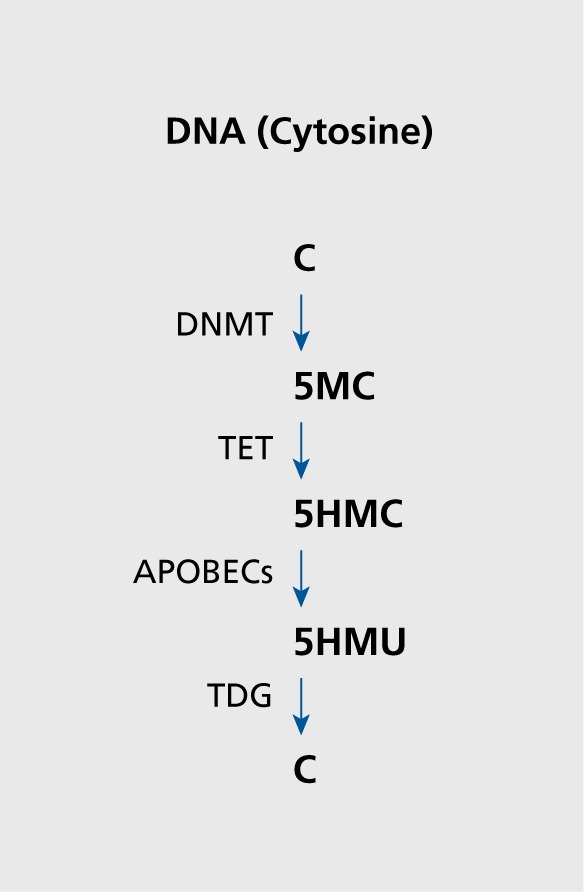
Although numerous studies of DNA methylation have been carried out in postmortem brains of psychotic patients,21,26,37,40,41 components of the DNA demethylation pathway, with the exceptions of Gadd45 and MBD4, remain largely unknown. Hence, we initiated an investigation into the expression of the TET gene family and AID/APOBEC deaminases in the inferior parietal lobule (IPL) and the cerebellum of a cohort of psychotic patients, which includes a group of BPD patients with psychosis and SZ patients. This cohort also includes a group of major depression (DEP) patients, and nonpsychiatric (control) subjects obtained from the Stanley Foundation Neuropathology Consortium Medical Research Institute (SFNC) (Bethesda, MD).
Our studies21,26 show that TET1 (mRNA and protein) is markedly increased (2- to 3-fold) in parietal cortex of psychotic patients and this increase is associated with an increase of 5-hmC level in genomic DNA containing the GAD67 and BDNF-IX promoters in proximity of their transcriptional start sites (TSSs, Table I). The increase may be specific to the cortex because the cerebellum of the same patients fails to show significant TET1 changes, even though the levels of TET1 are 3 to 4 times higher in cerebellum than in the cortex. The increase of TET1 in cortex of psychotic patients cannot be attributed to confounding demographic variables, nor to the type and dose of antipsychotic used during treatment. Furthermore, the increase of TET1 and of 5-hmC may not generalize to all major psychiatric disorders because it is absent in the depressed patient group.26
The role of TET1 may be to facilitate the removal of 5-mC from gene regions via formation of the intermediate 5-hmC so as to favor deamination catalyzed by the AID/APOBEC family of cytidine deaminases and the removal of 5-hmμ by the BER pathway (Figure 1). TET1-dependent active DNA demethylation and the concomitant increase in gene expression may occur in brain under physiological conditions.56 However, in the cortex of our psychotic patients we found an increase of TET1 that positively correlates with an increase of 5-hmC at genomic DNA containing promoters such as GAD67 and BDNF, which have been consistently associated with reduction in the expression of these target genes.14,21,26,42,64,65 A possible explanation for the unexpected finding is that increased TET1 is associated with a downregulation of some of the main APOBEC deaminating enzymes.26 An alternative explanation for the role of TET1 in transcriptional repression is that the repressive function of TET1 is independent of its catalytic activity. In fact it has been shown that TET1, which contains a CXXC domain,66-68 recruits polycomb repressive complex 2 and SIN3A corepressor proteins to target genes, suggesting that this repressive protein and SIN3A may play an important role in TET1-mediated gene silencing.67 Although our statistical analysis with medications as a covariant failed to show an influence of drug treatment on TET and 5-hmC expression, a role for medication in regulating gene expression in postmortem studies cannot be excluded. In fact, the medication history in the demographic records lacks detail and precision. In addition, there is evidence that different antipsychotics have different effects on epigenetic mechanisms.69
DNA methylation/demethylation as targets for antipsychotic medications
A limitation of postmortem studies on DNA methylation/demethylation in brain is the possibility that changes in the levels of the methylation/demethylation network proteins detected in SZ and BPD patients are induced, at least in part, by antipsychotic treatment and do not represent the pathophysiology of SZ and BPD. To investigate associations between epigenomics, SZ or BPD, and antipsychotic treatment in blood cells, Melas et al70 examined genome-wide DNA methylation levels in the leukocytes of SZ patients. They report a global DNA hypomethylation in SZ patients that was in part rescued by haloperidol treatment. This study, however, has a major limitation: the majority of patients showing global DNA hypomethylation were treated with antipsychotics different from haloperidol. Since several antipsychotic drugs, with the exception of haloperidol, display demethylating properties,69 the DNA hypomethylation observed in leukocytes of SZ patients treated with antipsychotics may be the consequence of chronic antipsychotic treatment that affects the transcription of genes by altering their epigenetic profiles. Aoyama et al71 reported that clozapine but not haloperidol ameliorates epigenetic and behavioral abnormalities induced by phencyclidine through activation of DA D1 receptors in mice. Melka et al72 examined the effect of olanzapine, an analog of clozapine, on DNA methylation status of genes of DA neurotransmission in rats. The results show that olanzapine causes methylation changes in genes associated with DA neurotransmission, not only in the hippocampus and cerebellum, but also in the liver. Bonsch et al73 studied methylation of genomic DNA and promoter methylation of RELN and SOX10 in peripheral blood of twins suffering from SZ. Global DNA methylation was significantly reduced in SZ twins but this reduction was more pronounced in nonmedicated rather than medicated SZ twins. In contrast, in discordant twins there was a relative hypermethylation of the SOX10 promoter.73 Taken together, these data underscore the need of further investigations into the action of antipsychotics on transcription of monoaminergic and other neurotransmitter genes in altering DNA methylation profiles in brain.
DNA promoter methylation patterns in animal models of SZ
To test whether antipsychotic drugs alter DNA methylation profiles in brain, we developed two animal models. A first model in which epigenetic modifications of GABAergic genes are induced by administering large doses of methionine,46,47 and a second model in which epigenetic modifications of GABAergic and glutamatergic genes were induced by prenatal stress.74,75
In the first model, mice were treated46-47,76 for protracted periods with methionine to induce Reln and Gad67 promoter hypermethylation as reflected by their reduced expression. The ratio of 5-mC to unmethylated cytosine (C) of the murine Reln promoter from -340 to + 160 bp or the murine Gad67 promoter from -760 to -31177 was quantified by measuring the fraction of Reln or Gad67 promoter immunoprecipitated by specific anti-5-mC, 5-hmC, or anti-methylcytosine binding protein-2 (MeCP2) antibodies with competitive RTPCR.21,26,78 Methionine administration induces: (i) an increase of SAM levels in brain; (ii) RELN and GAD67 promoter hypermethylation78,79; (iii) downregulation of RELN and GAD67 mRNA and protein expression46,47; and (iv) reprogramming of RELN and glucorticoid receptors (NR3C1) in hippocampal pyramidal neurons.80
GAD67 and RELN were not the only promoters hypermethylated by methionine treatment. ChlP-on-chip assays showed that in mice receiving methionine, about 5% of the promoters were hypermethylated in the FC of mice.79 Interestingly, the GAD65, neuron specific enolase (NSE), and glyceraldehyde-3 -phosphate dehydrogenase (G3PDH) promoters are not hypermethylated by the administration of methionine, suggesting that promoter hypermethylation is cell- and gene-specific.
Studies in cultured cortical neurons38,44 not only show that DNA hypermethylation induced by methionine is blocked by siRNA-mediated DNMT specific knockdown or by DNMT antagonists, but also that this blockade induces the overexpression of RELN, GAD67, or BDNF. Dong et al79 reported that if methionine is withdrawn from mice after 7 days of treatment, RELN hypermethylation decreases by ~50% after the seventh day and returns to control levels after 12 to 14 days of withdrawal.
In the second mouse model of SZ, we based our studies on the suggestion that a variety of prenatal stressors are related to high risk for cognitive and behavioral abnormalities associated with psychiatric illness.81 Using offspring of prenatal restraint stressed pregnant mice (PRS mice), we explored the long-term effect of PRS on behavior and on the expression of key chromatin remodeling factors including DNMT1 and TET and of GABAergic (GAD67, RELN) and glutamatergic (BDNF I-IX) target genes. Adult offspring of PRS-treated mothers demonstrate abnormalities in prepulse inhibition (PPI), locomotor activity, and social interaction. We found that there is a significant increase of DNMT1 and TET1 both in the FC and hippocampus of PRS offspring. Furthermore, there is a significant decrease in GAD67, RELN, and BDNF mRNA and protein levels.74-75 The results from methyl/hydroxymethyl-DNA immunoprecipitation (MeDIP/hMeDIP) studies show that 5-mC and 5-hmC levels are enriched at the GAD67, RELN, BDNF promoter regions. Thus, epigenetic changes in PRS mice are similar to changes observed in postmortem brains of psychiatric patients21,26,42 and represent, a reasonable model where the effect of drugs on altered DNA methylation may be studied.
DNA methylation/demethylation processes can be targets of antipsychotic drugs
Recent work demonstrates that methylation of a CpG island located ~30kb upstream of the gene encoding mitogen-activated protein kinase I (MEK1) is significantly correlated with lifetime antipsychotic use in postmortem BPD samples, with greater lifetime antipsychotic use associated with reduced levels of DNA methylation.41 This finding is interesting given the involvement of MAPK1 signaling pathways in mediating intra-neuronal signaling and the observation that clozapine activates this pathway by interacting with MEK1.82 As already discussed, leukocytes obtained from SZ patients receiving typical or atypical antipsychotics including clozapine but not haloperidol show global DNA hypomethylation.70 To address preclinically the issue of whether antipsychotic drugs acting on epigenetic mechanisms reduce methylation of hypermethylated DNA, correct the consequent downregulation of these genes in brain of SZ patients, and ameliorate SZ-like behavioral abnormalities, experiments were carried out in mice treated with methionine or in prenatally restraint mice. We found that clozapine and not haloperidol corrects the hyperlocomotor activity, the social interaction, and PPI deficit in methionine-treated and PRS mice. At the dose that corrects these abnormal behaviors, clozapine also induces chromatin remodeling (increase in the acetylation of histone 3 lysine 9) and facilitates demethylation of GABAergic promoters in the FC and striatum of methionine-treated mice. The effect of clozapine on GABAergic promoter methylation is shared by other dibenzepines including olanzapine and quetiapine, and by the benzamide sulpiride but not by haloperidol and risperidone (Table II).42,69,79
Table 2. Action of typical and atypical antipsychotics on DNA demethylation. a mg/kg: drugs were given s.c. twice a day for 3 days. b DNA demethylation refers to reelin and GAD67 promoter demethylation. Under basal conditions, 10% of methionine-induced hypermethylated reelin or GAD67 promoters is demethylated in 3 days after methionine withdrawal. (+) = 30-35% increase DNA demethylation over basal activity.(+++) = 75-90% increase DNA demethylation over basal activity. Data from ref 79.
| Drug | mg/kga | DNA demethylationb |
| Clozapine | 1.25 | +++ |
| Olanzapine | 10 | + |
| Quetiapine | 10 | + |
| Haloperidol | 1.5 | - |
| Risperidone | 10 | - |
| Sulpiride | 20 | ++ |
We found that the RELN and GAD67 promoters can be significantly demethylated in the FC of mice receiving 3 days of treatment with clinically relevant doses of clozapine and relatively high doses of quetiapine and olanzapine in the presence or absence of a threshold HDAC inhibitory dose of valproate (VPA).42,79 Because, in the same mice, RELN promoter methylation in the liver fails to change, we infer that clozapine and congeners modify methylation in the CNS and specifically in GABAergic and glutamatergic neurons, there-by increasing, for example, the expression of RELN, GAD67, and BDNF.
Effect of clozapine on DNA demethylation
The precise mechanism whereby clozapine and its congeners or VPA and other HDAC inhibitors induce DNA demethylation of select promoters remains to be elucidated. Recently, it was reported that electroconvulsive treatment (i) induces Gadd45 β expression; (ii) increases Gadd 45 β binding to cytosine deaminase or G/T mismatch glycosylase; and (iii) induces DNA demethylation at specific promoters (ie, Bdnf, Fgf-1). The observed DNA demethylation is abolished in Gadd45 KO mice.63 Hence, it is thought that Gadd45 proteins exert a regulatory role on DNA-methylation/demethylation processes. We examined the possibility that antipsychotic drugs elicit functionally relevant DNA-demethylation changes altering the expression or activity of Gadd45 α, β, or γ. Clozapine, in doses that induce promoter demethylation, increases Gadd45 β expression.42 Taken together, the data suggest that in addition to DNMTs, neuronal promoter methylation can be regulated by the activity of additional proteins that participate in removing the methyl group operating under constraints imposed by Gadd45. Hence, evidence suggests that in neurons, promoter methylation is a dynamic process that can be altered in response to environmental factors, such as stress, drugs, and various psychopathologies.
Histone deacethylase inhibitors and DNA demethylation
VPA (a mood stabilizer and anticonvulsant, drug) has been coadministered for over a decade with atypical antipsychotics to medicate BPD and in some cases SZ patients.83-84 VPA or other histone deacethylase (HDAC) inhibitors (ie, the benzamide MS275) induce promoter demethylation by activating DNA-demethylation mechanisms.42,79,85 VPA and MS275, like clozapine, correct the RELN and GAD67 promoter hypermethylation, the decrease in GAD67 and RELN expression and the PPI and social interaction deficits induced by protracted methionine treatment.47,79 Furthermore, VPA corrects the hyperactivity, social interaction, and PPI deficits in offspring of prenatally stressed mothers at doses that have no major effect in control mice.74 The coadministration of VPA and clozapine but not that of VPA and haloperidol, in parallel with a synergistic increase of DNA demethylation, induces histone 3 (H3) hyperacetylation at RELN or GAD67 promoters in the mouse FC.47,79 It is possible that VPA elicits functionally relevant DNA demethylation changes by altering the expression or activity of Gadd45 α or β. In fact, we found that in FC of mice that had been given 70 mg/kg of VPA (three days/twice a day), there is an increase of Gadd45 β mRNA compared to vehicle-treated controls.42
The data presented in this review strongly support the provocative concept that the coadministration of VPA with clozapine, by activating DNA demethylation, reverses a repressed nuclear epigenetic function expressed in postmitotic cortical GABAergic neurons of SZ or BPD patients. Although VPA per se fails to elicit antipsychotic activity, when administered in combination with antipsychotics such as haloperidol, it restores the capacity of aged mice to respond to haloperidol in the condition avoidance test.86
Concluding remarks
Recent breakthroughs in the study of aberrant molecular mechanisms operative in SZ and BPD point to a decreased expression of several genes in GABAergic interneurons, most likely caused by promoter hypermethylation mediated by overexpression of DNMT and TET in these cells.1,10-12,26,33,38
An epigenetic modulation of telencephalic GABAergic function may be responsible for disinhibiting pyramidal neurons that provide an excitatory input to dopamine cells in the ventral tegmental area (VTA) or serotonin cells in the raphe nucleus. The resulting hyper-dopaminergic or -serotoninergic state further increases pyramidal neuron excitability and presumably induces psychotic symptoms in SZ and BPD patients87,88. Taken together, these data suggest that to produce a significant symptomatic improvement of SZ or BPD morbidity, it may be desirable to pharmacologically reverse promoter hypermethylation in vulnerable GABAergic neurons. We have attempted to establish a preclinical strategy for evaluating drugs that facilitate DNA demethylation by either (i) reducing DNMT activity (ie, administering DNMT inhibitors); or (ii) promoting recruitment of DNA demethylation complexes to facilitate changes in chromatin remodeling.
The ability of clozapine, in clinically relevant doses on the one hand, and the inability of haloperidol or risperidone on the other, to influence chromatin remodeling and induce DNA demethylation in experimental animals may explain why clozapine is considered the “gold standard” among medications used for the treatment of cognitive and negative symptoms in antipsychotic-resistant SZ patients.89 In fact, in addition to its well-known effect on monoamine receptors, clozapine is unique in that it also has an effect on chromatin remodeling in clinically relevant, doses. An analysis of the data of Table II suggests that the action of clozapine and its derivatives on chromatin remodeling is independent of its action on catecholamine or serotonin receptors. To validate this concept, the effect of clozapine and congeners on chromatin remodeling should be studied in mice with a genetic ablation of dopamine or 5-HT receptor subtypes. It must be stressed that the current preclinical work in experimental animals has limitations because the animal models fail to reach the complex pathophysiology underpinning of SZ morbidity.
In postmortem brain studies, we and others reported that in the cortex of psychotic patients the DNA promoter methylation/hydroxymethylation of GABAergic and glutamatergic genes is increased while the expression of the respective mRNAs is decreased (Table I). These changes appear to be independent of antipsychotic or VPA treatment. Hence, we cannot exclude the possibility that antipsychotic drugs only partially correct the epigenetic alterations of GABAergic and glutamatergic genes, because the cause-effect relationship between GABAergic or glutamatergic promoter methylation and gene expression may be altered in psychotic patients. For example, methylation of specific promoters can be increased in the absence of changes in the levels of DNA-methylating enzymes by increasing brain SAM levels, possibly by administering methionine.46,47 An example of a nonlinear cause-effect relationship between BER (base excision repair) protein levels and promoter methylation is that increased expression of Gadd45 β in psychosis is not followed by a decrease of BDNF promoter methylation.21 One explanation for these unexpected results is suggested by chromatin immune precipitation experiments. In these experiments, despite high levels of GADD45 β, psychotic patients show reduced Gadd45 β binding to BDNF-IX compared with nonpsychiatric controls. It has been suggested that a restrictive chromatin state (ie, histone tail methylation) in psychosis90 could obstruct or fail to recruit Gadd 45β binding to specific promoters.21 Furthermore, the relationship between TET1 expression and enrichment of 5-hmC at specific promoters may not be linear. In fact, a bidirectional biological role of TET1 has been proposed: one in which TET1 removes aberrant DNA methylation and another that proposes a nonenzymatic role of TET1 in transcriptional repression.
Considering the complex nature of the epigenetic mechanisms underlying the symptomatology of SZ and BPDs, our ability to correct altered promoter methylation with typical or atypical antipsychotic drugs appears inadequate. However, interestingly, we have shown that dibenzepine derivatives (clozapine, quetiapine, and olanzapine), and not the butyrophenone derivative haloperidol and the piperidyl-benzisoxazole derivative risperidone, induce chromatin remodeling changes and activate DNA-demethylation of GABAergic and glutamatergic promoters (Table II). This observation suggests that dibenzepines by reducing promoter hypermethylation may contribute to correcting the dysregulation of GABAergic and glutamatergic transmission present in the brain of animal models of SZ and perhaps in the brain of SZ and BPD patients. In the future, the identification of factors contributing to DNA demethylation and a better understanding of how drugs activate or inhibit DNA demethylation pathways in brain may pave the way towards a better understanding of the disease and improve pharmacotherapeutic strategies.
Acknowledgments
Supported by IROIMH093348 and IROIMH101043 to AG.
Selected abbreviations and acronyms
- APOBEC
activation induced deaminase/apolipoprotein BRNA editing catalytic component
- BPD
bipolar disorder
- DNMT
DNA methyltransferase
- GABA
γ-aminobutyric acid
- GAD
glutamic acid decarboxylase
- RELN
reelin
- SZ
schizophrenia
- TET
ten-eleven translocation protein
- VPA
valproate
Contributor Information
Alessandro Guidotti, Psychiatric Institute, Department of Psychiatry, College of Medicine, University of Illinois at Chicago, Illinois, USA.
Dennis R. Grayson, Psychiatric Institute, Department of Psychiatry, College of Medicine, University of Illinois at Chicago, Illinois, USA.
REFERENCES
- 1.Grayson DR., Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38:138–166. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akbarian S., Kim JJ., Potkin SG., et al Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 3.Benes FM., Beretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 4.Benes FM., Lim B., Matzilevich D., Walsh JP., Subburaju S., Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104:10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benes FM., Vincent SL., Alsterberg G., Bird ED., SanGiovanni JP. Increased GABAA receptor binding in superficial layers of cingulate cortex in schizophrenics. J Neurosci. 1992;12: 924–929. doi: 10.1523/JNEUROSCI.12-03-00924.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fatemi SH., Earle JA., McMenomy T. Reduction in reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry. 2000;5:654–665. doi: 10.1038/sj.mp.4000783. [DOI] [PubMed] [Google Scholar]
- 7.Guidotti A., Auta J., Davis JM., et al Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 8.Impagnatiello F., Guidotti A., Pesold C., et al A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci USA. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis DA., Hashimoto T., Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 10.Veldic M., Caruncho JH., Liu WS., et al DNA methyltransferase-1 (DNMT1) is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc Natl Acad Sci USA. 2004;101:348–353. doi: 10.1073/pnas.2637013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veldic M., Guidotti A., Maloku E., Davis JM., Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc Natl Acad Sci U S A. 2005;102:2152–2157. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veldic M., Kadriu B., Maloku E., et al Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr Res. 2007;91:51–61. doi: 10.1016/j.schres.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong J., Hyde TM., Cassano HL., Deep-Soboslay A., Kleinman JE., Weickert CS. Promoter specific alterations of brain-derived neurotrophic factor mRNA in schizophrenia. Neuroscience. 2010;169:1071–1084. doi: 10.1016/j.neuroscience.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weickert CS., Hyde TM., Lipska BK., Herman MM., Weinberger DR., Kleinman JE. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2003;8:592–610. doi: 10.1038/sj.mp.4001308. [DOI] [PubMed] [Google Scholar]
- 15.Ikegame T., Bundo M., Sunaga F., et al DNA methylation analysis of BDNF gene promoters in peripheral blood cells of schizophrenia patients. Neurosci Res. 2013;77:208–214. doi: 10.1016/j.neures.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Breese CR., Lee MJ., Adams CE., et al Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23:351–364. doi: 10.1016/S0893-133X(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 17.Bitanihirwe BKY., Lim MP., Kelley JF., Kaneko T., Woo TUW. Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry. 2009;9:71. doi: 10.1186/1471-244X-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo T-U., Shrestha K., Lamb D., Minns MM., Benes FM. N-methyl-d-aspartate receptor and calbindin-containing neurons in the anterior cingulate cortex in schizophrenia and bipolar disorder. Biol Psychiatry. 2008;64:803–809. doi: 10.1016/j.biopsych.2008.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woo T-U., Walsh JP., Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express n-methyl-d-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2004;61:649–657. doi: 10.1001/archpsyc.61.7.649. [DOI] [PubMed] [Google Scholar]
- 20.Guidotti A., Auta J., Davis JM., et al GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology. 2005;180:191–205. doi: 10.1007/s00213-005-2212-8. [DOI] [PubMed] [Google Scholar]
- 21.Gavin DEP., Sharma RP., Chase KA., Matrisciano F., Dong E., Guidotti A. Growth arrest and DNA-damage-inducible, beta (GADD45b)-Mediated DNA demethylation in major psychosis. Neuropsychopharmacology. 2012;2:531–542. doi: 10.1038/npp.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldman-Rakic PS., Selemon LD. Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr Bull. 1997;23:437–58. doi: 10.1093/schbul/23.3.437. [DOI] [PubMed] [Google Scholar]
- 23.McAllister AK., Katz LC., Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 24.Hill JJ., Kolluri N., Hashimoto T., Wu Q., Sampson AR., Monteggia LM. Analysis of pyramidal neuron morphology in an inducible knockout of brain derived neurotrophic factors. Biol Psychiatry. 2005;57:923–934. doi: 10.1016/j.biopsych.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Volk DW., Austin MC., Pierri JN., Sampson AR., Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- 26.Dong E., Gavin DP., Chen Y., Davis J. Upregulation of TET1 and down-regulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry. 2012;2:e159. doi: 10.1038/tp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipska BK., Lerman DN., Khaing ZZ., Weickert CS., Weinberger DR. Gene expression in dopamine and GABA systems in an animal model of schizophrenia:effects of antipsychotic drugs. Eur J Neurosci. 2003;18:391–402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- 28.Fatemi SH., Folsom TD., Reutiman TJ., Novak J., Engel RH. Comparative gene expression study of the chronic exposure to clozapine and haloperidol in rat frontal cortex. Schizophr Res. 2011;134:211–208. doi: 10.1016/j.schres.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 29.Marco E., Mao CC., Cheney DL., Revuelta A., Costa E. The effects of antipsychotics on the turnover rate of GABA and acetylcholine in rat brain nuclei. Nature. 1976; 264:363–365. doi: 10.1038/264363a0. [DOI] [PubMed] [Google Scholar]
- 30.Ptak C., Petronis A. Epigenetics and complex disease: from etiology to new therapeutics. Ann Rev Pharmacol Toxicol. 2008;48:257–276. doi: 10.1146/annurev.pharmtox.48.113006.094731. [DOI] [PubMed] [Google Scholar]
- 31.Costa E., Chen Y., Davis J., et al Reelin and schizophrenia: a disease at the interface of the genome and the epigenome. Mol Interv. 2002;2:47–57. doi: 10.1124/mi.2.1.47. [DOI] [PubMed] [Google Scholar]
- 32.Ruzicka WB., Zhubi A., Veldic M., Grayson DR., Costa E., Guidotti A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol Psychiatry. 2007;12:385–397. doi: 10.1038/sj.mp.4001954. [DOI] [PubMed] [Google Scholar]
- 33.Zhubi A., Veldic M., Puri NV., et al An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schiz Res. 2009;111:115–122. doi: 10.1016/j.schres.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanEmburgh BO., Robertson KD. DNA methyltransferases and methyl CpG binding proteins as multifunctional regulators of chromatin structure and development in mammalian cells. In: Tost J, ed. Norwich, UK: Caister Academic Press; Epigenetics. 2008:23–62. [Google Scholar]
- 35.Chen Y., Dong E., Grayson DR. Analysis of the GAD1 promoter: Transacting factors and DNA methylation converge on the 5' untranslated region. Neuropharmacology. 2011;60:1075–1087. doi: 10.1016/j.neuropharm.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y., Kundakovic M., Agis-Balboa RC., Pinna G., Grayson DR. Induction of the reelin promoter by retinoic acid is mediated by Sp1. J Neurochem. 2007;183:650–665. doi: 10.1111/j.1471-4159.2007.04797.x. [DOI] [PubMed] [Google Scholar]
- 37.Grayson DR., Jia X., Chen Y., Sharma RP., Mitchell CO., Guidotti A., Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kundakovic M., Chen Y., Guidotti A., Grayson DR. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol. 2009;75:342–354. doi: 10.1124/mol.108.051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guidotti A., Ruzicka W., Grayson DR., Veldic M., Pinna G., Davis JM., Costa E. S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. Neuroreport. 2007;18:57–60. doi: 10.1097/WNR.0b013e32800fefd7. [DOI] [PubMed] [Google Scholar]
- 40.Abdolmaleky HM., Cheng KH., Russo A., et al Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Genet. 2005;134B:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- 41.Mill J., Tang T., Kaminsky Z., et al Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guidotti A., Auta J., Chen Y., et al Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology. 2011;60:1007–1016. doi: 10.1016/j.neuropharm.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wyatt RJ., Termini BA., Davis J. Biochemical and sleep studies of schizophrenia. A review of the literature 1960-1970. Schiz Bull. 1971;4:10–44. [Google Scholar]
- 44.Noh JS., Sharma RP., Veldic M., et al DNA methyltransferase 1 regulates reelin mRNA expression in mouse primary cortical cultures. Proc Natl Acad Sci USA. 2005;102:1749–1754. doi: 10.1073/pnas.0409648102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell CP., Chen Y., Kundakovic M., Costa E., Grayson DR. Histone deacetylase inhibitors decrease reelin promoter methylation in vitro. J Neurochem. 2005;93:483–492. doi: 10.1111/j.1471-4159.2005.03040.x. [DOI] [PubMed] [Google Scholar]
- 46.Tremolizzo L., Carboni G., Ruzicka WB., et al An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci U S A. 2002;99:17095–17100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tremolizzo L., Doueiri M-S., Dong E., et al Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 48.Dempster EL., Mill J., Craig IW., Collier DA. The quantification of COMTmRNA in post mortem cerebellum tissue: diagnosis, genotype, methylation and expression. BMC Med Genet. 2006;7:10. doi: 10.1186/1471-2350-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lintas C., Persico AM. Neocortical RELN promoter methylation increases significantly after puberty. Neuro Report. 2010;2:114–118. doi: 10.1097/WNR.0b013e328334b343. [DOI] [PubMed] [Google Scholar]
- 50.Tochigi M., Iwamoto K., Bundo M., Komori A., Sasaki T., Kato N., Kato T. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry. 2008;63:530–533. doi: 10.1016/j.biopsych.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Szyf M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry (Moscow). 2005;70:533–544. doi: 10.1007/s10541-005-0147-7. [DOI] [PubMed] [Google Scholar]
- 52.Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009;49:243–263. doi: 10.1146/annurev-pharmtox-061008-103102. [DOI] [PubMed] [Google Scholar]
- 53.Dong E., Chen Y., Gavin DP., Grayson DR., Guidotti A. Valproate induces DNA demethylation in nuclear extracts from adult mouse brain. Epigenetics. 2010;5:730–735. doi: 10.4161/epi.5.8.13053. [DOI] [PubMed] [Google Scholar]
- 54.Ito S., D'Alessio AC., Taranova OV., Hong K., Sowers LC., Zhang Y. Role of Tet proteins in 5-mC to 5-hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tahiliani M., Koh KP., Shen Y., et al Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo JU., Su Y., Zhong C., Ming GL., Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kriaucionis S., Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009; 324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhutani N., Burns DM., Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Ann Rev Genetics. 2009;43:143–166. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morgan HD., Dean W., Coker HA., Reik W., Petersen-Mahrt SK. Activation induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 61.Rai K., Huggins IJ., James SR., Karpf AR., Jones DA., Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barreto G., Schafer A., Marhold J., et al Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 63.Ma DK., Jang MH., Guo JU., et al Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buckley PF., Pillai A., Howell KR. Brain-derived neurotrophic factor: findings inschizophrenia. Curr Opin Psychiatry. 2011;24:122–127. doi: 10.1097/YCO.0b013e3283436eb7. [DOI] [PubMed] [Google Scholar]
- 65.Roth TL., Lubin FD., Sodhi M., Kleinman JE. Epigenetic mechanisms in schizophrenia. Biochem Biophys Acta. 2009;1790:869–877. doi: 10.1016/j.bbagen.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lyer LM., Tahiliani M., Rao A., Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams K., Christensen J., Pedersen MT., et al TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H., Zhang X., Clark E., Mulcahey M., Huang S., Shi YG. TET1 is a DNA binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390–1393. doi: 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- 69.Guidotti A., Dong E., Kundakovic M., Satta R., Grayson DR., Costa E. Characterization of the action of antipsychotic subtypes on valproateinduced chromatin remodeling. Trends Pharmacol Sci. 2009;30:55–60. doi: 10.1016/j.tips.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 70.Melas PA., Rogdaki M., Ösby U., Schalling M., Lavebratt C., Ekström TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012;26:2712–2718. doi: 10.1096/fj.11-202069. [DOI] [PubMed] [Google Scholar]
- 71.Aoyama Y., Mouri A., Toriumi K., et al Clozapine ameliorates epigenetic and behavioral abnormalities induced by phencyclidine through activation of dopamine D1 receptor. Int J Neuropsychopharmacol. 2013;17:1–15. doi: 10.1017/S1461145713001466. [DOI] [PubMed] [Google Scholar]
- 72.Melka MG., Laufer Bl., McDonald P., et al The effects of olanzapine on genome-wide DNA methylation in the hippocampus and cerebellum. Clin Epigenetics. 2014;6:1. doi: 10.1186/1868-7083-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bönsch D., Wunschel M., Lenz B., Janssen G., Weisbrod M., Sauer H. Methylation matters? Decreased methylation status of genomic DNA in the blood of schizophrenic twins. Psychiatry Res. 2012;198:533–537. doi: 10.1016/j.psychres.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 74.Matrisciano F., Tueting P., Dalai I., et al Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–194. doi: 10.1016/j.neuropharm.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matrisciano F1., Tueting P., Maccari S., Nicoletti F., Guidotti A. Pharmacological activation of group-ll metabotropic glutamate receptors corrects a schizophrenia-like phenotype induced by prenatal stress in mice. Neuropsychopharmacology. 2012;37:929–938. doi: 10.1038/npp.2011.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tueting P., Davis JM., Veldic M., Pibiri F., Kadriu B., Guidotti A., Costa E. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Neuroreport. 2010;21:543–548. doi: 10.1097/WNR.0b013e3283373126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satta R., Maloku E., Zhubi A., Pibiri F., Hajos M., Costa E., Guidotti A. Nicotine targets the epigenetic mechanisms in selected populations of mouse telencephalic GABAergic neurons. Proc Natl Acad Sci U S A. 2008;105:16356–16361. doi: 10.1073/pnas.0808699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dong E., Guidotti A., Grayson DR., Costa E. Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proc Natl Acad Sci U S A. 2007;104:4676–4681. doi: 10.1073/pnas.0700529104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dong E., Nelson M., Grayson DR., Costa E., Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate nuclear DNAdemethylation in the brain. Proc Natl Acad Sci U S A. 2008;105:13614–13619. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weaver IC., Meaney MJ., Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety mediated behaviors in the offspring that are reversible in the adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Markham JA1., Koenig JL. Prenatal stress: role in psychotic and depressive diseases. Psychopharmacology (Berl). 2011;214:89–106. doi: 10.1007/s00213-010-2035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Browning JL., Patel T., Brandt PC., Young KA., Holcomb LA., Hicks PB. Clozapine and the mitogen-activated protein kinase signal transduction pathway: implications for antipsychotic actions. Biol Psychiatry. 2005;57:617–623. doi: 10.1016/j.biopsych.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Kelly DL., Conley RR., Feldman S., Yu Y., McMahon RP., Richardson CM. Adjunct divalproex or lithium to clozapine in treatment-resistant schizophrenia. Psychiatr Q. 2006;77:81–95. doi: 10.1007/s11126-006-7963-9. [DOI] [PubMed] [Google Scholar]
- 84.Wassef AA., Dott SG., Harris A., Brown A., O'Boyle M., 3rd Meyer WJ., Rose RM. Randomized, placebo-controlled pilot study of divalproex sodium in the treatment of acute exacerbations of chronic schizophrenia. J Clin Psychopharmacol. 2000;20:357–361. doi: 10.1097/00004714-200006000-00011. [DOI] [PubMed] [Google Scholar]
- 85.Detich N., Bovenzi V., Szyf M. Valproate induces replication-independent active DNA demethylation. J Biol Chem. 2003;278: 27586–27592. doi: 10.1074/jbc.M303740200. [DOI] [PubMed] [Google Scholar]
- 86.Montalvo-Ortiz JL., Keegan J., Gallardo C., et al HDAC Inhibitors restore the capacity of aged mice to respond to haloperidol through modulation of histone acetylation. epub ahead of print. Neuropsychopharmacology. doi: 10.1038/npp.2013.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewis DA., Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- 88.Lisman JE., Coyle JT., Green RW., Javitt DC., Benes FM., Heckers S., Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gray JA., Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12:904–922. doi: 10.1038/sj.mp.4002062. [DOI] [PubMed] [Google Scholar]
- 90.Houston I., Peter CJ., Mitchell A., Straubhaar J., Rogaev E., Akbarian S. Epigenetics in the human brain. Neuropsychopharmacology. 2013;38:183–197. doi: 10.1038/npp.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]