

Abstract
Eukaryotic mRNA transcription by RNA polymerase II (RNAP II) is the first step in gene expression and a key determinant of cellular regulation. Elucidating the mechanism by which RNAP II synthesizes RNA is therefore vital to determining how genes are controlled under diverse biological conditions. Significant advances in understanding RNAP II transcription have been achieved using classical biochemical and structural techniques; however, aspects of the transcription mechanism cannot be assessed using these approaches. The application of single-molecule techniques to study RNAP II transcription has provided new insight only obtainable by studying molecules in this complex system one at a time.
Keywords: transcription, RNA polymerase II, single-molecule, optical tweezers, fluorescence, FRET, TIRF
Introduction
MRNA transcription by RNA polymerase II (RNAP II) is a highly regulated process that dictates levels of cellular gene expression. A small, but increasing number of studies has applied single-molecule techniques to studying different aspects of RNAP II transcription, providing new insight into this seminal process. A solid foundation and history of biochemical experiments have enabled much of the single-molecule work by elucidating important mechanistic details of the eukaryotic transcription reaction.1 This includes defining the general transcription factors (GTFs) that are necessary for promoter specific transcription: TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH. Assembly of these factors on promoter DNA is nucleated by the binding of TFIID; its TATA-binding protein (TBP) subunit binds to the TATA box region of the core promoter and the TAF (TBP associated factor) subunits make critical contacts with other core promoter DNA elements. The remaining GTFs and RNAP II assemble with TFIID at the core promoter to form a pre-initiation complex (PIC). Within the PIC, the TFIIH helicase facilitates DNA melting in an ATP-dependent process that marks the transition from a closed complex to an open complex. After transcription initiates, open complexes transform into elongation complexes via a series of structural changes that occur as the polymerase active site moves away from the start site of transcription and the RNA transcript grows. In cells, PIC assembly and the act of transcription occur on chromatin, in which DNA wrapping around nucleosomes provides a barrier that must be circumvented by the RNAP II machinery with the aid of regulatory proteins and nucleosome remodeling complexes.2
In addition to biochemical studies, crystallography and cryo-electron microscopy have elucidated important structural features of RNAP II, the GTFs, PICs, and elongation complexes. Crystal structures of RNAP II and elongation complexes containing RNAP II, promoter DNA and an RNA transcript have provided a framework for understanding the structural features of RNAP II that facilitate transcription and the transformations within RNAP II that occur as RNA is synthesized.3-5 Cryo-electron microscopy has revealed the overall architecture of closed and open complexes, and the positions of GTFs within these complexes.6,7 The body of structural work on assemblies of RNAP II and transcription factors has significantly advanced our understanding of transcription, but many mechanistic details about the transformation of PICs into elongation complexes, the dynamic process of nucleotide addition, and how RNAP II traverses nucleosomes remain to be resolved. Single-molecule techniques can complement the current knowledge by revealing aspects of the transcription mechanism that are unattainable from ensemble biochemical and structural approaches. Single molecule techniques allow the visualization of heterogeneous sub-populations that are masked by averaging in ensemble studies, and they can monitor dynamics in a way that structural approaches cannot. To date, most single-molecule studies of transcription have focused on viral and bacterial systems, which are reviewed elsewhere.8
A small but growing body of work has tailored single-molecule techniques to study key elements of the highly complex process of eukaryotic mRNA transcription, which is the focus of this review. These studies have provided unprecedented insight into complex assembly, initiation, and elongation in the presence and absence of nucleosomes, as well as regulatory factors that govern this process. The in vitro single-molecule techniques that have been applied to eukaryotic transcription can be divided into two broad, but distinct categories: fluorescence spectroscopy and force spectroscopy. Fluorescence spectroscopy involves labeling DNA, RNA, and/or protein subunits with fluorophores thereby allowing individual molecules to be observed using total internal reflection fluorescence (TIRF) microscopy. This approach is ideal for measuring distances between specific positions in macromolecular complexes, monitoring changes in conformation, and observing dynamics. Force spectroscopy often involves the use of optical traps to apply mechanical force to a system to monitor structural changes or observe effects on molecular movement as a function of that force. Hence, this technique is well suited to monitor motion and movement of individual molecules. Here we review advances in our understanding of eukaryotic mRNA transcription that have been obtained through the application of single-molecule fluorescence and force studies, and describe their unique contribution to building a mechanistic model for how RNAP II transcription occurs.
Single molecule fluorescence studies of RNAP II transcription
Fluorescence techniques that resolve individual molecules have been used to visualize the dynamics of binding events and map distances within nucleoprotein complexes important for RNAP II transcription. In the TIRF microscopy systems used, individual fluorescent molecules are tethered to a surface. Laser light is used to excite a sample at an angle greater than the critical angle, causing the light to be fully reflected and an evanescent field to be generated that extends ~100 nm into the sample (Fig. 1). Hence, only fluorescent molecules close to the surface are excited and ultimately visualized.
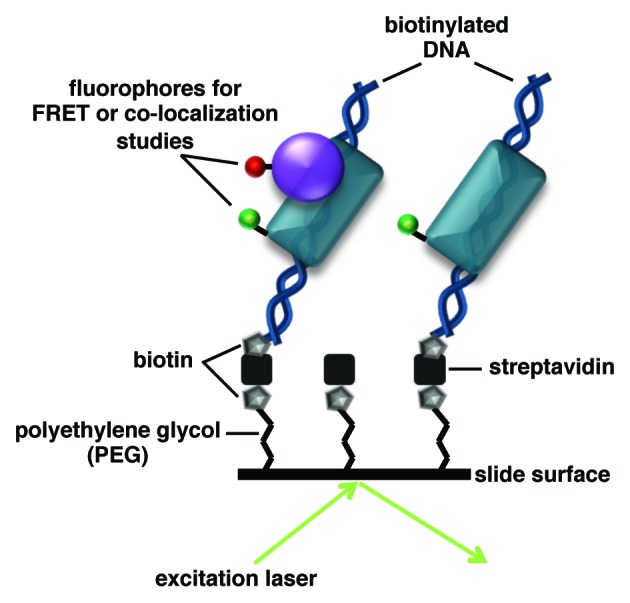
Figure 1. TIRF microscopy can be used study interactions between fluorescently labeled single molecules. In the example shown, the surface of a coverslip contains DNA immobilized via biotin-streptavidin linkages. The excitation laser intersects the slide at an angle greater than the critical angle, creating a ~100 nm evanescent field that allows only molecules on the surface of the sample to be visualized. Each protein is labeled with a chromatically unique fluorophore; this pair of fluorophores could be used to (1) co-localize the proteins on the DNA or (2) measure the distance between the proteins by FRET, both of which can be monitored over time. Single-molecule fluorescence microscopy will reveal heterogeneity within the sample by distinguishing which complexes contain both proteins or only one, and how this changes dynamically over time.
Two categories of single molecule fluorescence techniques have been used to study RNAP II transcription: 1) single-molecule fluorescence resonance energy transfer (smFRET) and 2) single molecule fluorescence co-localization. These techniques have revealed new kinetic and structural information unattainable from ensemble biochemical or structural studies. smFRET monitors distance changes between a donor fluorophore and an acceptor fluorophore that are attached to molecule(s) of interest (Fig. 1) on the millisecond timescale. Hence, smFRET can be used to obtain dynamic information about structural changes within individual molecules and/or macromolecular complexes. smFRET also allows distinct populations within a single sample that have unique FRET states or kinetic properties to be resolved. Single-molecule fluorescence co-localization, a tool for monitoring molecular interactions, involves labeling molecules of interest with chromatically unique fluorophores and mapping the overlap, or co-localization, between the different fluorescent signals over time (Fig. 1).
smFRET reveals new insight into how general transcription factors interact with DNA
Although TBP-induced DNA bending has been studied in detail using ensemble biochemical and structural approaches, more recent smFRET studies provided new insight into this phenomenon.9 Bending kinetics and the degree of DNA bending induced by human TBP was measured on DNA containing a TATA box flanked by donor and acceptor dyes. The FRET efficiencies observed for hundreds of molecules in the bent state were histogrammed and found to fit well to a single Gaussian; hence, as expected, TBP uniformly bent the consensus TATA box. Analysis of the kinetics of bending and unbending revealed two distinct subpopulations, one characterized by a short-lived bent state and a second that persisted in the bent state for considerably longer. Together the data led to a model in which the TBP/TATA complex exists in two kinetically distinct subpopulations, both of which contain DNA bent to the same extent. Interestingly, the smFRET studies also revealed that TBP bends a mutant sequence, TATA(A3), to the same extent as the consensus sequence, which contradicts previous ensemble studies showing that TBP bends TATA(A3) to a lesser extent.10,11 The discrepancy is likely attributable to difficulty saturating the TATA(A3) DNA with TBP in ensemble studies, due to the relatively low binding affinity of this interaction. Thus, the ensemble studies could have measured an average of bent and unbent populations.
The role of negative cofactor 2 (NC2) in inhibiting PIC formation has also been investigated using smFRET.12 NC2 binds DNA/TBP complexes and inhibits gene expression, but the underlying mechanism was unclear.13 The effect of NC2 on the distance between TBP (labeled with a donor fluorophore) and DNA (labeled with an acceptor fluorophore upstream of the TATA box) was monitored by smFRET, revealing dynamic changes. The smFRET fluctuated between three states after addition of NC2. Two of the populations corresponded with NC2-TBP-DNA complexes containing DNA in either a bent or unbent state. Additional ensemble biochemical experiments indicated that the third state arose from TBP-NC2 complexes moving away from the TATA box. Together these results support a model in which NC2 binds TBP-DNA complexes and unbends DNA to allow mobilization of TBP away from the TATA box. This suggests an interesting mechanism for NC2 regulated gene expression in which genes are not only repressed by removal of TBP from promoters, but NC2 may also fine-tune the position of TBP to optimize the position of initiation.12
smFRET elucidates structural changes in open complexes and elongation complexes
A nano-positioning system (NPS) was used to better understand the architecture of RNAP II open complexes and elongation complexes, and to monitor the path of RNA exit within RNAP II.14-16 NPS uses smFRET data in conjunction with X-ray crystallography models to map dynamic regions within complexes. The method involves labeling a protein or nucleic acid with a satellite dye molecule at a distinct location, and an antenna dye molecule at a position of interest that is not structurally established. The smFRET efficiency is measured between pairs of dye molecules consisting of the antennae dye molecule and individual satellite dye molecules placed at different positions. The data are analyzed using Bayesian parameter estimation to generate a probability density function for the unknown position (i.e., the position of the antennae dye molecule).16
Yeast RNAP II open complexes containing TBP, TFIIB, TFIIF, RNAP II, and heteroduplex DNA (i.e., contains an engineered bubble around the transcription start site) were studied using a NPS.15 Downstream DNA in open complexes was found to fluctuate between two locations: within and above the RNAP II cleft. The primary population had downstream DNA located within the cleft, which is also its location in elongation complexes. The dynamic fluctuation between the two positions occurred on the timescale of seconds. The comparison of the NPS data obtained with open complexes to structural models of the closed complex led the authors to propose a dramatic movement of upstream DNA to a position well above the cleft in the transition from closed to open complexes. The position of the upstream DNA in the open complex appeared to be stabilized by the presence of initiation factors, suggesting that this positional shift is a regulatory step in the structural rearrangements that occur to form open complexes. The repositioning of upstream DNA also caused DNA-bound TBP and the TFIIB core to move above the cleft and away from the surface of RNAP II. Because NPS is achieved by analyzing individual donor and acceptor dye pairs, structural subpopulations within the open complex were identified that would be indistinguishable due to averaging in ensemble techniques.
The RNAP II elongation complex has also been investigated using a NPS to monitor positional fluctuations of upstream DNA and the non-template DNA strand.14 The angle between the upstream and downstream duplexes was found to be ~80°, consistent with structural data from bacterial polymerases.17 Upstream DNA from -5 to -10 was exposed, while the template strand downstream of -5 was in the RNAP II cleft.14 The non-template strand of the melted DNA was shown to pass loop β10-β11 on RNAP II, and not between the lobe and the protrusion as was previously suspected. Moreover, the non-template strand was found to interact directly with the RNAP II rudder, which implicates it in the process of re-annealing the DNA behind the transcription bubble.
Similar in approach to NPS, triangulation smFRET was used to map the path the nascent RNA takes as it moves out of RNAP II.18,19 In these studies, smFRET was used to separately measure the distances between three donor/acceptor dye pairs placed on elongation complexes assembled with RNAP II and an RNA/DNA scaffold. One of the dyes in all three pairs labeled the RNA at a single nucleotide; the second dye in each pair was placed at a known location on the surface of RNAP II or on the downstream DNA. By analyzing the three distance measurements obtained from smFRET, the dye on the RNA could be localized on the RNAP II structure. Triangulation studies from two labs confirmed that RNA exits through the RNAP II exit tunnel, under the lid of the polymerase. Both smFRET analyses also found that once the growing RNA reaches 26 nucleotides in length its 5′ end can contact regions of RNAP II beyond the RNA exit tunnel; however, the specific regions of contact differed between the two studies. One study concluded that nascent RNA travels toward the Rpb4-Rpb7 subunits,19 while another found that the RNA contacts the RNAP II dock domain.18 This discrepancy could potentially arise from differing experimental conditions, such as ion concentrations.19 Interestingly, a NPS study found that in the absence of TFIIB exiting RNA was more likely to contact the dock domain of RNAP II, but upon addition of TFIIB, the contact shifted toward the Rpb4-Rpb7 subunits,16 suggesting that this switch could be regulated during early transcription.
Fluorescence co-localization can be used to investigate RNAP II initiation with single-molecule resolution
Transcript synthesis by human RNAP II has been directly visualized using single molecule fluorescence co-localization.20 Complete PICs containing all of the general transcription factors and RNAP II were assembled on fluorescently labeled, double stranded promoter DNA that was immobilized on specialized surfaces on a TIRF microscope. To detect RNA transcripts synthesized upon nucleotide addition, a fluorescently labeled oligonucleotide probe antisense to a region of the RNA transcript was included in the reactions. Upon transcription, the labeled probe annealed to the RNA transcript and the resulting fluorescent signals were analyzed for co-localization with individual fluorescent promoter DNA molecules, whose positions had been mapped on the surface.
Measuring transcription from single DNA molecules revealed that approximately 30% of the templates were transcribed when the strongest promoter was tested (an engineered “super core promoter”21). A mutant version of this promoter showed lower template usage and addition of the activator Sp1, which could bind GC boxes upstream of the TATA box, increased template usage. This system enabled experiments to ask whether promoters used in one round of initiation were used more frequently for subsequent rounds of transcription, which would be consistent with a reinitiation scaffold of proteins remaining at the core promoter.22,23 Single molecule data from templates exhibiting multiple round transcription fit a Poisson model; hence, these studies did not find evidence of reinitiation scaffolds under the conditions tested.
Force Spectroscopy Studies of RNAP II Transcription
Single-molecule force spectroscopy studies provide an ideal platform for studying molecular motion and movement. Hence, this technique is inherently useful for monitoring both the kinetics and mechanisms of transcript elongation, as well as pausing during elongation. Optical tweezers systems have been used to monitor RNAP II movement in response to a hindering or assisting force, to measure RNAP II force output during transcription, to study pausing by RNAP II during elongation, and to investigate the movement of RNAP II through nucleosomes. Atomic force microscopy (AFM) has been used to obtain snapshots of single RNAP II elongation complexes transcribing through a nucleosome.
Optical tweezers studies reveal new aspects of the mechanism of transcript elongation by RNAP II
Dual-trap optical tweezers assays have been used to tether single molecules of DNA and RNAP II to different polystyrene beads that are trapped and manipulated by lasers (Fig. 2). As RNAP II transcribes, assisting force can be applied to a DNA tether upstream of RNAP II, or hindering force can be applied to a downstream tether.24 The change in distance between RNAP II and the DNA tether, and the force exerted on the DNA can be monitored as a function of time. This allows the rate of elongation, as well as the frequency and duration of RNAP II pausing events to be directly measured, providing important insight into mechanisms of elongation.24-26
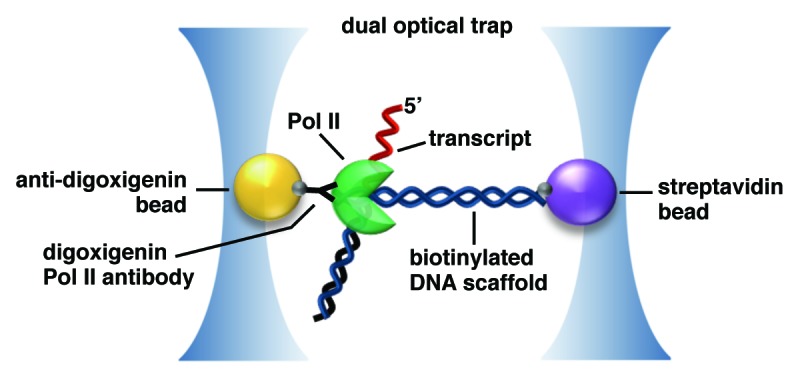
Figure 2. Dual-beam optical trapping assays can be used to monitor RNAP II transcription. In the example shown, an elongating RNAP II and a molecule of DNA are individually attached to beads, which are held in separate optical traps. The system can be used to impart hindering or assisting force on the polymerase during transcription to produce an RNA (in red). The position of RNAP II along the template can be monitored as a function of time to understand the duration and density of pausing events. The figure is adapted from Michaelis and Treutlein.35
RNAP II pausing and backtracking during elongation have been challenging to study with ensemble approaches that average pause duration and can mask kinetic subpopulations. Monitoring changes in the movement of single elongating RNAP II molecules as a function of hindering force provided key insight into the role of polymerase backtracking.25 Experiments showed that elongation was abrogated under 7.5 pN of hindering force. The force sensitive process was shown to be backtracking, which caused elongation to pause. Measuring the time required to exit such a pause supported a model in which RNAP II diffused back to a position in which the 3′ end of the nascent RNA was in the active site. Addition of hindering force inhibited this diffusive process and transcription ceased. Compellingly, in the presence of TFIIS, which cleaves the nascent RNA to generate a new 3′ end in the polymerase active site, elongation proceeded with up to 16.9 pN of hindering force, supporting the conclusion that the force-sensitive process was backtracking.
Single-molecule optical tweezers techniques were also used to investigate how the sequence and structure of the nascent RNA transcript affect RNAP II elongation.27 It was hypothesized that secondary structure in the nascent RNA would prevent RNAP II backtracking by presenting an energy barrier to backward movement of the polymerase. RNAP II pause duration and density was compared on GC-rich vs. AT-rich templates; transcripts made from the GC-rich templates would form stronger hairpins. Indeed, GC-rich templates exhibited shorter pauses and a lower density of pausing than AT-rich templates. Moreover, RNase A treatment eliminated the effect of sequence on pausing, supporting the conclusion that decreased pausing on the GC-rich templates was due to secondary structure formation in the RNA transcript.
Optical trapping assays were used to dissect the role of the highly conserved RNAP II trigger loop in elongation.26 These studies suggest the trigger loop affects both selection of the correct nucleotide as well as recognition of base mismatches in the RNA-DNA duplex after an incorrect nucleotide is incorporated. Transcriptional fidelity was measured by assessing the number of long RNAP II pauses per kilobase, or long pause density (LPD), which corresponds closely with the rate of nucleotide misincorporation. Nucleotide misincorporation was driven by adding saturating amounts of a single NTP and subsaturating amounts of the other NTPs, and the LPDs with wild type and trigger loop mutant RNAP II molecules were compared. The LPD for a trigger loop mutant RNAP II was much greater compared with wild type RNAP II, suggesting that the trigger loop mutant polymerase lacked the ability to discriminate between correct and incorrect nucleotides. To monitor mismatch recognition, ITP (inosine triphosphate) was included in elongation assays to facilitate increased misincorporation. As expected, the LPD for wild type polymerase increased upon addition of ITP. Surprisingly, the LPD for the trigger loop mutant decreased in the presence of ITP, which suggests that the RNAP II trigger loop plays a role in mismatch recognition.
Optical trapping assays reveal the impact of nucleosomes on transcript elongation by RNAP II
Nucleosomes are a prominent physical barrier that RNAP II must bypass during elongation. The mechanisms by which the polymerase traverses a nucleosome and mechanisms of nucleosomal repositioning are still being unraveled, and single-molecule optical trapping assays have provided unique insight into these processes because of the ability to analyze each pausing event associated with individual RNAP II molecules.28 The pause-free elongation velocity of tethered RNAP II on tethered naked DNA was compared with that on DNA containing a nucleosome. It was determined that when RNAP II encounters a nucleosome it does not actively unwrap the DNA from histones, but pauses and subsequently continues elongating once the equilibrium between wrapped and unwrapped DNA shifts to the unwrapped state.
The individual effects of histone tails and histone-DNA contacts on RNAP II elongation kinetics were dissected by reconstituting nucleosomes with mutant histones in an optical trapping system.29 RNAP II pause density and duration were monitored as a function of distance from the nucleosome. Eliminating or acetylating histone tails decreased RNAP II pauses just prior to the nucleosome, indicating that histone tails impede the dynamics of RNAP II entry into the nucleosomal region of the DNA, which can be attenuated by acetylation. The effect of histone-DNA contacts on elongation was tested by using histones H3 and H4 containing point mutations in their core regions. Nucleosomes containing mutant H3 and H4 showed lower RNAP II pausing within the central regions of the nucleosome corresponding to where RNAP II directly contacts the nucleosome dyad. Moreover, the H3 and H4 mutations shifted the equilibrium for the outer region of the DNA in the nucleosome toward the unwrapped state. These data suggest the inherent rate of DNA wrapping and unwrapping around histones can directly affect the amount of RNAP II pausing.
Investigating nucleosomal repositioning using atomic force microscopy
AFM, a scanning probe microscopy technique, can elucidate structural and mechanistic features of a system by immobilizing a sample on a surface and probing the sample-surface interface. AFM subjects molecules adhered to a surface to a tapping force from a probe and measures fluctuations of the probe to record structural features of single molecules.30 Specifically, laser light is deflected by a cantilever that is linked to the surface probe; fluctuations in contact between the probe and the sample are monitored by changes in deflection of the laser.
AFM was used to characterize mechanisms of nucleosomal repositioning during transcription by reconstituting transcription elongation complexes upstream of a nucleosome and fixing/imaging either before or after addition of NTPs.31 Because RNAP II is much larger than a nucleosome, the two complexes could be readily distinguished in the images. This provided snapshots of how the DNA length and protein positions changed during the elongation process and a means to evaluate how histones reposition as RNAP II transcribes. The total length of DNA not occupied by RNAP II or histones was measured per molecule and fit with Gaussians.31 The data from populations in which RNAP II was actively transcribing through the nucleosome revealed a collection of intermediate states, some of which showed nucleosome contacts with upstream and downstream DNA. Moreover, the average unbound DNA length in these complexes was ~30 nm shorter than the unbound DNA length from populations in which RNAP II was not actively transcribing through the nucleosome, supporting a model in which DNA loops form to facilitate transfer of histones. The AFM data were consistent with a model in which elongating RNAP II encounters a nucleosome, and DNA loops as histones are transferred to a position on the DNA behind the polymerase. To achieve this, the nucleosome downstream of transcribing RNAP II partially unwraps and its histones also contact DNA upstream of RNAP II to form a loop, thus facilitating the transfer of the nucleosome behind RNAP II. This corroborates data obtained from an optical tweezers study that showed that when RNAP II moved through a nucleosome, histones were not displaced from the DNA and, moreover, nucleosomal transfer did not occur when the DNA was subjected to force.28 This would suggest that histones are transferred through a looping mechanism that is unable to form under tension.
AFM also revealed that when RNAP II elongated through a nucleosome, there was a decrease in the height of the histone structures, suggesting disassembly of octamers.31 It was determined that octamers can decay into hexamers and this transformation is dependent upon the elongation rate; slower elongation favors less disassembly due to less histone unwrapping. At higher transcription rates, unwrapping occurs more quickly, which favors disassembly of the histone octamers. Hence, this study revealed a competing kinetic interplay between RNAP II transcription elongation rate, histone transfer via a looping mechanism, and histone dissociation.
Future Directions
Single-molecule studies of eukaryotic mRNA transcription have provided novel insight into the mechanism of this reaction and have built a comprehensive framework that will undoubtedly be expanded by future studies. The established approaches will be invaluable for further dissecting the dynamics and heterogeneity of the RNAP II transcription machinery during all stages of the transcription reaction. In addition, other single-molecule techniques that have been used to study bacterial RNA transcription will likely also be applied to the eukaryotic system. For example, magnetic tweezers studies have been used to analyze DNA scrunching by E. coli RNA polymerase during abortive initiation32; this technique has the potential to provide insight into the process of initiation by RNAP II as well. Though cell-based studies have not been discussed in this review, advances in live-cell imaging at the single-molecule level provide a means to test the biological relevance of mechanisms identified in vitro, and to further explore the dynamics and activities of single molecules in the nuclear environment.33,34
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by grant MCB-1244518 from the National Science Foundation. A.E.H. was supported in part by NIH Training Grant T32 GM-065103.
Glossary
Abbreviations:
- AFM
atomic force microscopy
- GTF
general transcription factor
- ITP
inosine triphosphate
- LPD
long pause density
- NC2
negative cofactor 2
- NPS
nano-positioning system
- PIC
preinitiation complex
- RNAP II
RNA polymerase II
- smFRET
single-molecule fluorescence resonance energy transfer
- TAF
TBP associated factor
- TBP
TATA binding protein
- TIRF
total internal reflection fluorescence
References
- 1.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–78. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 2.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–76. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 4.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–82. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 5.Kostrewa D, Zeller ME, Armache KJ, Seizl M, Leike K, Thomm M, Cramer P. RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature. 2009;462:323–30. doi: 10.1038/nature08548. [DOI] [PubMed] [Google Scholar]
- 6.He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495:481–6. doi: 10.1038/nature11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murakami K, Elmlund H, Kalisman N, Bushnell DA, Adams CM, Azubel M, Elmlund D, Levi-Kalisman Y, Liu X, Gibbons BJ, et al. Architecture of an RNA polymerase II transcription pre-initiation complex. Science. 2013;342:1238724. doi: 10.1126/science.1238724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbert KM, Greenleaf WJ, Block SM. Single-molecule studies of RNA polymerase: motoring along. Annu Rev Biochem. 2008;77:149–76. doi: 10.1146/annurev.biochem.77.073106.100741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blair RH, Goodrich JA, Kugel JF. Single-molecule fluorescence resonance energy transfer shows uniformity in TATA binding protein-induced DNA bending and heterogeneity in bending kinetics. Biochemistry. 2012;51:7444–55. doi: 10.1021/bi300491j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hieb AR, Halsey WA, Betterton MD, Perkins TT, Kugel JF, Goodrich JA. TFIIA changes the conformation of the DNA in TBP/TATA complexes and increases their kinetic stability. J Mol Biol. 2007;372:619–32. doi: 10.1016/j.jmb.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 11.Wu J, Parkhurst KM, Powell RM, Brenowitz M, Parkhurst LJ. DNA bends in TATA-binding protein-TATA complexes in solution are DNA sequence-dependent. J Biol Chem. 2001;276:14614–22. doi: 10.1074/jbc.M004402200. [DOI] [PubMed] [Google Scholar]
- 12.Schluesche P, Stelzer G, Piaia E, Lamb DC, Meisterernst M. NC2 mobilizes TBP on core promoter TATA boxes. Nat Struct Mol Biol. 2007;14:1196–201. doi: 10.1038/nsmb1328. [DOI] [PubMed] [Google Scholar]
- 13.Goppelt A, Stelzer G, Lottspeich F, Meisterernst M. A mechanism for repression of class II gene transcription through specific binding of NC2 to TBP-promoter complexes via heterodimeric histone fold domains. EMBO J. 1996;15:3105–16. [PMC free article] [PubMed] [Google Scholar]
- 14.Andrecka J, Treutlein B, Arcusa MA, Muschielok A, Lewis R, Cheung AC, Cramer P, Michaelis J. Nano positioning system reveals the course of upstream and nontemplate DNA within the RNA polymerase II elongation complex. Nucleic Acids Res. 2009;37:5803–9. doi: 10.1093/nar/gkp601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treutlein B, Muschielok A, Andrecka J, Jawhari A, Buchen C, Kostrewa D, Hög F, Cramer P, Michaelis J. Dynamic architecture of a minimal RNA polymerase II open promoter complex. Mol Cell. 2012;46:136–46. doi: 10.1016/j.molcel.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Muschielok A, Andrecka J, Jawhari A, Brückner F, Cramer P, Michaelis J. A nano-positioning system for macromolecular structural analysis. Nat Methods. 2008;5:965–71. doi: 10.1038/nmeth.1259. [DOI] [PubMed] [Google Scholar]
- 17.Rees WA, Keller RW, Vesenka JP, Yang G, Bustamante C. Evidence of DNA bending in transcription complexes imaged by scanning force microscopy. Science. 1993;260:1646–9. doi: 10.1126/science.8503010. [DOI] [PubMed] [Google Scholar]
- 18.Andrecka J, Lewis R, Brückner F, Lehmann E, Cramer P, Michaelis J. Single-molecule tracking of mRNA exiting from RNA polymerase II. Proc Natl Acad Sci U S A. 2008;105:135–40. doi: 10.1073/pnas.0703815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen CY, Chang CC, Yen CF, Chiu MT, Chang WH. Mapping RNA exit channel on transcribing RNA polymerase II by FRET analysis. Proc Natl Acad Sci U S A. 2009;106:127–32. doi: 10.1073/pnas.0811689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Revyakin A, Zhang Z, Coleman RA, Li Y, Inouye C, Lucas JK, Park SR, Chu S, Tjian R. Transcription initiation by human RNA polymerase II visualized at single-molecule resolution. Genes Dev. 2012;26:1691–702. doi: 10.1101/gad.194936.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juven-Gershon T, Cheng S, Kadonaga JT. Rational design of a super core promoter that enhances gene expression. Nat Methods. 2006;3:917–22. doi: 10.1038/nmeth937. [DOI] [PubMed] [Google Scholar]
- 22.Zawel L, Kumar KP, Reinberg D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev. 1995;9:1479–90. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- 23.Yudkovsky N, Ranish JA, Hahn S. A transcription reinitiation intermediate that is stabilized by activator. Nature. 2000;408:225–9. doi: 10.1038/35041603. [DOI] [PubMed] [Google Scholar]
- 24.Palangat M, Larson MH, Hu X, Gnatt A, Block SM, Landick R. Efficient reconstitution of transcription elongation complexes for single-molecule studies of eukaryotic RNA polymerase II. Transcription. 2012;3:146–53. doi: 10.4161/trns.20269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galburt EA, Grill SW, Wiedmann A, Lubkowska L, Choy J, Nogales E, Kashlev M, Bustamante C. Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature. 2007;446:820–3. doi: 10.1038/nature05701. [DOI] [PubMed] [Google Scholar]
- 26.Larson MH, Zhou J, Kaplan CD, Palangat M, Kornberg RD, Landick R, Block SM. Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II. Proc Natl Acad Sci U S A. 2012;109:6555–60. doi: 10.1073/pnas.1200939109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zamft B, Bintu L, Ishibashi T, Bustamante C. Nascent RNA structure modulates the transcriptional dynamics of RNA polymerases. Proc Natl Acad Sci U S A. 2012;109:8948–53. doi: 10.1073/pnas.1205063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science. 2009;325:626–8. doi: 10.1126/science.1172926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bintu L, Ishibashi T, Dangkulwanich M, Wu YY, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal elements that control the topography of the barrier to transcription. Cell. 2012;151:738–49. doi: 10.1016/j.cell.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Billingsley DJ, Bonass WA, Crampton N, Kirkham J, Thomson NH. Single-molecule studies of DNA transcription using atomic force microscopy. Phys Biol. 2012;9:021001. doi: 10.1088/1478-3975/9/2/021001. [DOI] [PubMed] [Google Scholar]
- 31.Bintu L, Kopaczynska M, Hodges C, Lubkowska L, Kashlev M, Bustamante C. The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes. Nat Struct Mol Biol. 2011;18:1394–9. doi: 10.1038/nsmb.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Revyakin A, Liu C, Ebright RH, Strick TR. Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching. Science. 2006;314:1139–43. doi: 10.1126/science.1131398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, Maniatis T, Xie XS. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods. 2013;10:421–6. doi: 10.1038/nmeth.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hocine S, Raymond P, Zenklusen D, Chao JA, Singer RH. Single-molecule analysis of gene expression using two-color RNA labeling in live yeast. Nat Methods. 2013;10:119–21. doi: 10.1038/nmeth.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michaelis J, Treutlein B. Single-molecule studies of RNA polymerases. Chem Rev. 2013;113:8377–99. doi: 10.1021/cr400207r. [DOI] [PubMed] [Google Scholar]