

Summary
Mental illness is not due to mutations in a single gene, but rather involves molecular disturbances entailing multiple genes and numerous environmental factors that control their expression. Recent research has demonstrated that complex “epigenetic” mechanisms, which regulate gene activity without altering the DNA code, exert long-lasting effects within mature nerve cells. This review summarizes recent evidence for the existence of sustained epigenetic mechanisms of gene regulation in nerve cells that have been implicated in the long-lasting regulation of complex behavior, including behavioral abnormalities in several psychiatric disorders such drug addiction and depression. We also critique recent and still uncertain evidence that similar epigenetic changes, occurring in germ cells, might mediate the trans-generational transmission of behavioral experience and, possibly, mental illness.
Gene-environmental interactions in mental illness
I met Catherine when she was a 28 year old woman, the product of an upper middle class family in Massachusetts. Like most adolescents, she drank alcohol recreationally and occasionally smoked cigarettes. During college at an Ivy League university, she—like many students—was exposed to cocaine, however, unlike most students, she became severely addicted to the drug over the ensuing several years. By the time she presented for treatment at a community mental health center in Connecticut, she was homeless, prostituted herself to get money to support her addiction, and was disowned from her family for repeatedly stealing from them. Despite many years of treatment, she remained highly vulnerable to addiction, with periods of abstinence interspersed with repeated relapses. What made Catherine vulnerable to such a severe addiction? There was alcoholism in her family, but her several siblings were unaffected. Likewise, of all her classmates who experimented with cocaine, why did she alone become so severely affected? And what made Catherine’s addiction so persistent despite numerous efforts at treatment? How much of all this was genetic? What role did life experiences play? This age old debate between nature and nurture has recently taken on new meaning, as epigenetic mechanisms have been shown to regulate the interplay between genes and environment, often with very long-lived consequences.
Severe mental illness such as drug addiction or depression, like most common medical conditions, arises from a combination of genes and environment. Just the way an individual’s genetic constitution (e.g., variations in genes encoding enzymes that control cholesterol metabolism) combines with environmental exposures (e.g., a high fat diet) to determine whether that person has high blood levels of cholesterol and its many associated abnormalities such as heart disease, variations in neural genes and in environmental exposures combine to determine whether an individual develops a mental illness. All major forms of mental illness are highly heritable. For example, the risk for drug addiction and for depression is roughly 50% genetic, about the same heritability of high cholesterol. The specific genetic variations that comprise this risk are not yet known, but the mere presence of such variations is not sufficient to cause addiction or depression. Rather, individuals at such genetic risk must also be exposed to some environmental factors, which themselves remain poorly defined. Different forms of chronic stress are involved in some people, but apparently not in others, and the involvement of many other types of environmental exposures is not well understood. Moreover, it is likely that the development of addiction or depression involves the concerted actions of numerous genetic variations and of numerous environmental exposures.
Despite this relative lack of knowledge, scientists have gained increasing understanding of how genes and environment interact to control brain function under normal and pathological conditions. Genes encode messenger RNA’s (mRNA’s) which in turn encode proteins that determine all aspects of neuronal function, including an individual’s initial sensitivity to a host of environmental factors, for example, in the case of addiction and depression, drugs of abuse or stress. Likewise, environmental factors modify neuronal function and those behavioral responses by altering the expression of certain genes within the relevant brain circuits. The nature, magnitude, and rapidity of those environment-induced changes are also partly determined by an individual’s genetic variations: in other words, some genes do not influence an individual’s initial sensitivity to a drug of abuse or stress but can influence the ways in which the affected nerve cells (or neurons) adapt over time to repeated drug or stress exposure. These very complex, reciprocal interactions between genes and environment, which are mediated via tightly controlled alterations in gene expression, bring us to chromatin and epigenetic regulation.
What is chromatin?
Chromatin is the material within every cell’s nucleus and consists of that cell’s DNA and the many types of proteins that control the structure and function of the DNA. Each cell’s DNA would extend 2 meters long if stretched out completely. Yet, that DNA is miraculously packaged into a microscopic nucleus (Fig. 1). That extraordinary degree of condensation is achieved by interactions between DNA and many types of nuclear proteins, which together form chromatin. This packing is so tight, that most genes in a cell are not available for transcription into mRNA at any given point in time. Rather, just the right segments of DNA consisting of the needed genes are stretched out and available for expression into mRNA and protein according to the cell’s needs.
Figure 1.
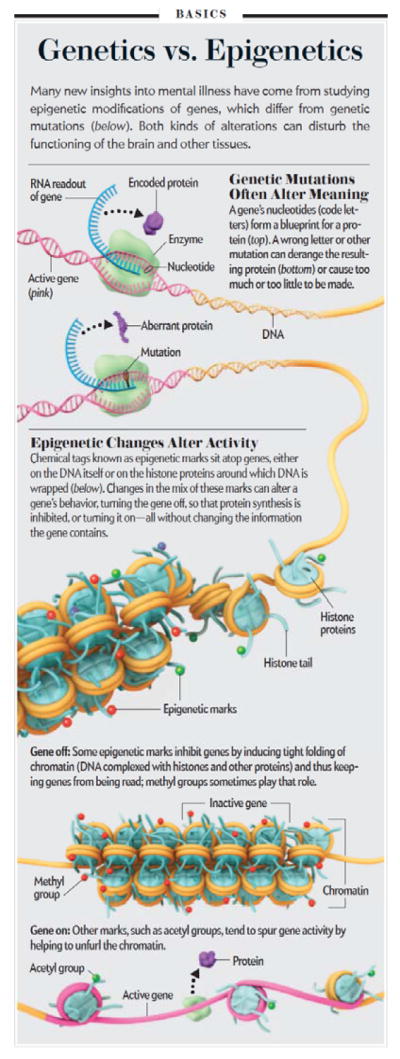
Epigenetic regulation. The figure compares genetics vs. epigenetics and describes several prominent features by which chromatin is regulated in the nervous system.
These functions are best understood during development, when identical stem cells differentiate into adult tissues. As stem cells give rise to neurons, liver cells (hepatocytes), or muscle cells (myocytes), for example, different segments of the DNA become condensed or open to allow for the expression of tissue-specific genes. Such large scale condensation and opening of chromatin, and the associated patterns of gene expression, remain constant for the life of the organism: neurons cannot be converted into hepatocytes, and vice versa. Nevertheless, more refined changes in gene expression are required throughout life to enable an individual to adapt to the environment. An example is provided by exercise. Using a muscle causes the muscle cells involved to express higher levels of muscle-specific genes such as myosin. This occurs through greater activation of the myosin gene, which in turn is mediated by the further opening of chromatin in the gene’s vicinity. This leads to more myosin protein, more myosin fibrils, larger muscle cells, and ultimately a larger and stronger muscle. In the same way, the ability of the brain to adapt to environmental challenges is mediated by the selective opening and closing of chromatin around neural genes, with the altered expression of those genes controlling changes in neural and brain function that seek to address the environmental challenges at hand. Such chromatin regulation mediates the ability of an individual to learn and remember, but these processes can go awry in extreme instances to mediate maladaptive changes to environmental exposures as seen in drug addiction and depression.
What is epigenetics?
Epigenetics describes processes by which lasting changes in gene expression can be accomplished without a change in DNA sequence. The best established example of epigenetic regulation in mammals concerns genetic imprinting, where a cell has only one active copy of a given gene through the inactivation of the paternally vs. maternally derived copy. In this way, genetically identical individuals may differ dramatically at the functional level: for example, one twin expresses the paternal version of a given neural gene, whereas the opposite might hold in the other twin. It is likely that more subtle epigenetic variations in the relative ability to activate or repress a given gene in response to some environmental stimulus is far more widespread, with numerous neural genes potentially affected. Such epigenetic modifications thereby underlie a dramatic third source of individual variability: not only do genes and environment matter, but stochastic epigenetic inactivation or activation of genes within distinct brain regions that occurs during development and adult life can dramatically influence an individual’s behavioral traits under normal and pathological conditions. In addition, epigenetics describes the changes in chromatin structure that mediates the ability of environmental stimuli, even in adult individuals, to cause lasting changes in gene expression. The term epigenetics was first introduced by Conrad Waddington in the 1940’s to describe such types of gene-environment interactions.
A still provocative idea is whether some of the epigenetic inactivation or activation of genes that occurs during the lifetime of an organism can be passed onto offspring. How such transmission would occur, and the extent to which it occurs, remain poorly understood, because in most models of cell division all epigenetic modifications of genes are erased during meiosis. Still, there is growing evidence that some epigenetic modifications are either not erased, or mechanisms exist to recreate them after meiosis, making such trans-generation transmission possible in at least some circumstances. This heritability of gene activation-inactivation, which today is established in plants only, led historically to the use of the term epigenetics to describe mechanisms of heritable transmission of traits that do not involve a change in DNA sequence. However, most people use the term far more broadly to refer to any lasting change in gene expression mediated at the level of DNA-protein interactions, that is, at the level of chromatin modifications. Whether epigenetic mechanisms drive heritable transmission of behavioral experiences remains an open question, as will be addressed toward the end of this review.
Epigenetic regulation: How is chromatin modified?
The fundamental unit of chromatin is the nucleosome, which consists of DNA wrapped around an octomer of histone proteins. The nucleosomal structure of chromatin allows DNA to be tightly packaged into the nucleus by organized folding (Fig. 1). As noted earlier, chromatin exists in an inactivated, condensed state, which is called heterochromatin, which does not allow transcription of genes, and in an activated, open state, called euchromatin, which allows individual genes to be transcribed (Fig. 1). In reality, chromatin can exist in many states in between these two extremes (Fig. 1). Portions of chromatin are highly repressed, and might never be accessible for transcription (e.g., liver genes in neurons). Other portions of chromatin are in repressed or permissive states; their basal activity may be low, but the genes are available for de-repression and activation in response to a given stimulus. Epigenetic modifications modulate gene expression with high temporal and spatial resolution by permitting small groups of nucleosomes to become more or less open, allowing or inhibiting access of the transcriptional machinery to specific genes.
The best characterized mechanism of chromatin regulation in brain is the post-translational, covalent modification of histones at distinct amino acid residues on their N-terminal tails. Such modifications include acetylation, ubiquitination, or SUMOylation at lysine (K) residues, methylation at lysine or arginine residues, phosphorylation at serine or threonine residues, and ADP-ribosylation at glutamate residues (Fig. 1). For example, hyperacetylation promotes decondensation of chromatin and increased gene activity, whereas hypoacetylation marks condensation and decreased activity. By contrast, histone methylation is more complex and can drive either gene activation or repression, depending on the Lys residue undergoing methylation. The diversity of histone modifications supports the “histone code” hypothesis, proposed by Dr. David Allis and colleagues at The Rockefeller University, which posits that the sum of modifications at a particular promoter region define a specific epigenetic state of gene activation or silencing. DNA methylation is another important mechanism of gene repression. The enzymes that control these covalent modifications are becoming increasingly understood as shown in Fig. 1. Dr. Allis has referred to these enzymes as the “writers” and “erasers” of epigenetic regulation.
These various histone and DNA modifications, called epigenetic “marks”, ultimately control the level of expression of a given gene through a set of chromatin remodeling proteins that bind to specific modifications. Such chromatin remodeling proteins can thereby be seen as “readers” of the epigenetic marks. For example, some readers bind to acetylated histones, or methylated histones (e.g., H3K4me3), to recruit other co-activators to the nearby gene, ultimately leading to the gene’s active transcription by the RNA polymerase II complex (Fig. 1). All told, it is estimated that a hundred or more proteins are recruited to such a gene to mediate its transcription or repression.
Mechanisms of chromatin regulation
The covalent modification of histones is controlled by enzymes referred to as “writers” and “erasers”. Histone acetyltransferases (HATs) and histone methytransferases (HMTs), which catalyze acetylation and methylation, respectively, can be seen as writers, whereas histone deacetylases (HDACs) and demethylases (HDMs) can be seen as erasers. Different types of HATs and HDACs expressed in cells likely govern the selective acetylation of many Lys residues on histone H3 and H4, and most of these reactions mediate gene activation. The situation is far more complicated for histone methylation, since distinct families of HMTs and HDMs differentially catalyze the mono-, di-, and trimethylation (or demethylation) of distinct Lys residues on H3, with each methylated state of each Lys residue mediating a distinct functional effect. Although the exact functions are not yet known with certainty, methylation of H3K4 is generally associated with gene activation, while di- and trimethylation of H3K9 and H3K27 is associated with gene repression, and di- and trimethylation of H3K36 is associated with elongation of gene transcription.
DNA methylation occurs by transfer of a methyl group from S-adenosyl methionine (SAM) to cytosine bases at the dinucleotide sequence CpG, and is catalysed by DNA methyltransferases (DNMTs). The amount of DNA methylation at a promoter correlates with the extent of gene inactivation. X chromosome inactivation (a form of genetic imprinting), is mediated through a combination of DNA and repressive histone methylation. While DNA methylation had been thought to be irreversible, it is likely that DNA demethylases exist to regulate DNA methylation in the adult brain.
These various epigenetic “marks” affect gene expression through the actions of effector proteins, which can be considered “readers”. Examples of readers are the SWI-SNF family of proteins, which bind to acetylated histones and use ATP-derived energy to disrupt nucleosomes and permit the sliding of the histone octamer along DNA during transcription. In contrast, other readers bind selectively to repressive methylated histone marks (e.g., H3K9me2/3 and H3K27me2/3), or to methylated DNA, to recruit many co-repressors to maintain gene repression. Examples of the latter are heterochromatin protein 1 (HP1) which binds to H3K9me3, and methyl binding domain proteins (MBDs) which bind to methylated DNA. Importantly, patterns of histone modifications and DNA methylation are intricately linked. For example, HDACs, repressive HMTs, and MBDs are associated with one another in large protein complexes to repress gene transcription.
Efforts to understand how environmental stimuli and communication among neurons lead to chromatin modifications have focused on a class of proteins termed transcription factors. Most transcription factors, such as CREB (cyclic AMP response element binding protein) or AP1 (activator protein 1; composed of Fos and Jun family proteins), once induced by synaptic activity, bind to specific short sequences of DNA, called response elements, present within target genes and recruit HATs, such as CBP (CREB binding protein), which despite its name is recruited by many such factors. This leads to the acetylation of nearby histones, to the recruitment of readers of acetylated histones (e.g., SWI/SNF factors) and many other co-activators, and to gene activation. Some transcription factors, like CLOCK (important in controlling circadian rhythms), even have intrinsic HAT activity. However, all of these transcription factors, in addition to activating many target genes, repress many others, even when binding to identical response elements in the various genes, and very little is known about what determines this differential activity. One possibility is that the effect of a transcription factor on activating vs. inhibiting a given target gene is determined by the chromatin state of that gene: in other words, genes to be activated may have very different chromatin regulatory proteins already bound to influence the consequences of transcription factor binding.
The best established mediator of gene repression is a transcription factor called REST (RE1-silencing transcription factor), also known as neuron-restrictive silencer factor or NRSF. REST binds to a DNA response element referred to as RE1, where it triggers the recruitment of several co-repressor proteins including HDACs and repressive HMTs, among others. In this way, REST keeps neural-specific genes turned off in non-neural cells, and has been shown to modulate the expression of many neural genes in response to synaptic activity. Whether other transcription factors repress genes via a similar cascade as REST remains unknown.
Epigenetic regulation of neural signaling
While chromatin remodeling is best understood for its influence on neural development, increasing evidence demonstrates a role throughout life, even in regulating mature, fully differentiated neurons. During synaptic transmission, neurons respond to neurotransmitters by receptor-mediated intracellular signal transduction events, which, among other actions, activate or inhibit transcription factors, proteins that bind to specific sites within gene promoters to increase or decrease gene expression. Transcription factors exert these effects on gene expression by recruiting co-activators or co-repressors to the gene and thereby triggering the many types of chromatin modifications described above. Epigenetic regulation is thus intimately linked to activation or repression of genes by synaptic activity and mediates the ability of synaptic activity to shape adaptive responses in neuronal signaling, survival, morphology, and ultimately the integrated regulation of complex behavior. A major challenge for the field is to understand the detailed mechanisms involved.
Epigenetic regulation in brain: Role in drug addiction
Drugs of abuse control human behavior by hijacking the brain’s natural reward centers, including the mesolimbic dopamine system. This circuit includes dopaminergic neurons of the ventral tegmental area (VTA), which project directly to the nucleus accumbens (NAc)–the ventral portion of the striatum. Over the past few decades much has been learned about the brain’s reward circuits and how drugs of abuse usurp its functions, but it is unclear why addictive behaviors persist long after drug abstinence, underlying high rates of relapse, as in the patient example provided at the outset. Many studies have identified drug-induced changes in mRNA levels in the VTA, NAc, and other brain reward regions. Some of these changes in gene expression persist even after months of abstinence. The longevity of these changes has driven research into epigenetic mechanisms as the molecular basis of sustained, even life-long, alterations in gene expression in brain reward regions.
Most studies to date have focused on cocaine and related psychostimulant drugs of abuse, such as amphetamine. Studies aimed at defining genome-wide changes in mRNA expression of genes have provided interesting insight into drug-induced epigenetic regulation in the NAc. An example is illustrated in Fig. 2. In this experiment, mice were given a single dose of cocaine and global changes in mRNA expression were studied by use of DNA microarrays. A bit less than 100 mRNA’s were shown to be induced 1 hr after cocaine administration. We then asked how prior chronic exposure to cocaine might alter these responses. We found that a small number of genes induced by acute cocaine show desensitization: they are not induced in mice previously treated with the drug. However, many more genes showed the opposite pattern of regulation: they were not affected by acute cocaine but were induced by a challenge dose of cocaine in animals previously exposed to the drug, and this increased sensitivity of gene induction lasted for at least one week after withdrawal from cocaine. This experiment suggests that chronic cocaine administration induces a state of increased permissiveness of gene expression.
Figure 2.

Epigenetic mechanisms of addiction. The figure illustrates some of the ways in which drugs of abuse—through changes in chromatin mechanisms—produce a lasting, addicted state.
Indeed, we now know that this permissive state is mediated via several global changes in histone and DNA modifications that render the genome generally more activatable in response to subsequent drug exposures. Chronic cocaine administration decreases the activity of certain HDACs, repressive HMTs, and DNMTs selectively in the NAc. Moreover, decreasing levels of each of these enzyme in the NAc, by use of inducible and localized genetic knockouts or direct infusion of small molecule inhibitors of the enzymes, mimics the permissive state of gene regulation and increases an animal’s sensitivity to the rewarding effects of cocaine.
By combining such DNA microarray experiments with methods that enable assessment of chromatin modifications at specific genes, it is possible to understand, for the first time, the detailed transcriptional mechanisms by which cocaine or other drugs of abuse regulate individual genes within the NAc and other brain reward regions. Thus, while studies to date have focused on drug regulation of steady-state levels of mRNA’s, any investigation of transcriptional mechanisms has had to rely on cell culture experiments, even though we know that what happens in cultured cells—even cultured neurons—is not an accurate reflection of what happens in brain. Epigenetic research makes it possible to push the frontier of this work one important step further. This is achieved by use of an experimental procedure called chromatin immunoprecitation (ChIP), where chromatin fragments are immunoprecipitated with an antibody directed against some chromatin modification of interest (e.g., an antibody to acetylated histones) and the immunoprecipitated DNA is analyzed: a) for a specific gene promoter of interest by PCR, b) on promoter chips (ChIP-chip), or c) by high throughput sequencing (ChIP-seq) (. The latter two methods provide a genome-wide assessment of epigenetic regulation. These methods have made it possible to identify the hundreds of genes that show histone or DNA modifications, selectively within the NAc, in response to chronic cocaine administration and to define with increasingly completeness the complexes of co-activators and co-repressors (such as those shown in Fig. 2) that are recruited to specific genes, in concert with their activation or repression, during a course of cocaine exposure.
The unprecedented understanding of genes that are regulated by cocaine is revealing the many molecular pathways and cellular processes that are altered in the brain’s reward regions to underlie distinct features of drug addiction. The next key challenge is use this information, as a template or key, to develop improved treatments of cocaine addiction by designing small molecules that interfere with these drug-induced maladaptive changes in gene expression and epigenetic regulation in brain.
Epigenetic regulation in brain: Role in depression
Depression is a common, chronic, and debilitating disease; it is the number one cause worldwide of disease burden. Although many patients benefit from antidepressant medications, electroconvulsive seizures, or psychotherapy, fewer than half of depressed patients show a complete remission, which underscores the need for more effective agents. The mechanisms that precipitate depression, such as stress, are incompletely understood. One mystery of the disease is its long-lasting nature and delayed response to antidepressant treatment. This persistence is thought to be mediated by slowly developing but stable adaptations, thus implicating epigenetic regulation.
Chronic social defeat stress is one animal model of depression. Mice are subjected to larger, more aggressive mice over a period of 10 days and thereafter display many symptoms of human depression, including anhedonia (reduced ability to experience pleasure), social withdrawal, and obesity and related metabolic derangements. Some of these abnormalities are long-lasting—they persist for months—and can be reversed by chronic (but not acute) administration of standard antidepressant medications, such as fluoxetine or imipramine. Another interesting feature of the social defeat model is that roughly one-third of mice do not develop this behavioral syndrome when subjected to chronic defeat stress, rather, they appear “resilient,” and this coincides with the knowledge that most people can cope with periods of chronic stress without developing depression or a related syndrome.
Our group has studied the transcriptional mechanisms responsible for the persisting behavioral abnormalities that characterize social defeat and has demonstrated a role for epigenetic mechanisms. To date, this work has focused on two brain regions implicated in depression, the NAc and hippocampus. We have used ChIP assays to demonstrate profound and long-lasting regulation of numerous genes within these brain regions that occur in susceptible mice. Stress regulation of large subsets of these genes can be reversed by antidepressant medications, and portions of this stress regulation are not seen in resilient animals. Moreover, these analyses have demonstrated that resilience is associated with regulation of many genes that are not altered in susceptible individuals, suggesting that resilience is not simply the absence of stress-induced changes, but, rather, represents an active process. Interestingly, there is considerable overlap between genes regulated by antidepressant medications and those regulated in resilient animals. This raises the additional interesting notion that antidepressants work in part by inducing in sick individuals some of the same gene and chromatin changes that occur naturally in more robust, resilient individuals. This in turn suggests a novel strategy for the development of new antidepressants: in addition to searching for drugs that block the bad effects of chronic stress, it should also be possible to identify drugs that mimic the unique mechanisms of resilience. The vast network of long-lasting changes in gene and chromatin regulation being revealed by this work is defining many molecular pathways that can now be exploited in drug discovery efforts. One example is the recent findings that HDAC inhibitors, when given systemically or directly into the NAc or hippocampus, exert potent antidepressant-like effects in the social defeat and other depression models. These results are now driving efforts to synthesize HDAC inhibitors that better penetrate the brain and more selectively target subtypes of HDACs that are enriched in the NAc, hippocampus, and other brain regions of interest.
The discovery that inbred mice, raised under identical environmental conditions, exhibit dramatically different adaptations (i.e., susceptibility vs. resilience) to chronic social defeat stress raises the possibility that epigenetic mechanisms are involved. According to this notion, the inherent activity of genes is influenced by stochastic processes during development, leading to very different sensitivities of individuals to chronic stress. A major challenge of current research is to identify the specific genes whose epigenetic modifications underlie these vast behavioral differences.
Another animal model used to study depression involves categorizing young mouse or rat pups based on the quality of the maternal care they receive early in development. Dr. Michael Meaney and colleagues at McGill University has defined epigenetic changes in the brain as the basis for long-lasting behavioral differences induced in the pups by different levels of maternal care, as well as the mechanism by which certain behavioral traits are passed from one generation to the next. In rats, some mothers naturally display high levels of nurturing behaviors, such as licking, grooming, and arched-back nursing (active mothers), while others have low levels of such behaviors (passive mothers). Offspring of active mothers are less anxious, have attenuated release of corticosterone (a glucocorticoid stress hormone homologous to cortisol in humans) in response to stressful stimuli, and display increased expression of glucocorticoid receptor (GR) mRNA and protein in hippocampus when compared to pups of passive mothers. In addition, female offspring of active mothers themselves become active mothers, while female offspring of passive mothers become passive mothers, based partly on the differences in GR expression levels in hippocampus. This paradigm therefore exemplifies the trans-generational transmission of maternal behavior and stress responses.
The basis for this trans-generational transmission via maternal behavior is mediated partly through epigenetic mechanisms (Fig. 3). The promoter that drives expression of a brain specific variant of the GR is activated upon binding a transcription factor called nerve-growth-factor-inducible factor (NGFI-A). NFGI-A binds to the promoter at a specific response element, and this DNA sequence shows significantly higher rates of methylation at CpG sites in the hippocampus of passive mothers compared to active mothers. This difference in methylation emerges in the first week of life as a result of the type of maternal care a pup receives and it persists throughout adulthood. As adults, the offspring of mothers with low levels of nurturing behavior are at a molecular disadvantage–their methylated GR gene promoter prevents binding of NGFI-A, effectively disrupting the normal transcriptional regulation of the GR gene, leading to passive mothering behavior and inflicting equivalent epigenetic changes in their offspring. Although this epigenetic methylation is long-lasting, it can be reversed by infusion of an HDAC inhibitor, which increases levels of histone acetylation and thereby disrupts gene repressive complexes, decreases DNA methylation, and increasing the binding of NGFI-A at the GR gene promoter in the offspring of passive mothers, thus restoring the molecular and behavioral profiles of these animals to those seen in the offspring of active mothers. Of course, GR is just one of many putative genes involved in these phenomena. For example, Dr. Frances Champagne of Columbia University has implicated similar epigenetic changes at the gene encoding the estrogen receptor in mediating the behavioral differences between offspring of active and passive mothers, and there are likely many other genes involved as well.
Figure 3.

Epigenetic mechanisms of stress and depression. The figure illustrates a mechanism by which the style of maternal behavior experienced by a rat pup can be transmitted to subsequent generations through chromatin mechanisms..
Epigenetic regulation in other brain disorders
While the above narrative focuses on drug addiction and depression, equivalent evidence exists for a role of epigenetic mechanisms in several other neuropsychiatric conditions. Chromatin modifications at specific neural genes, specifically, altered methylation of the gene promoters in regions of prefrontal cortex, have been implicated in animal models of schizophrenia, and some of these modifications have been validated in human brain tissue examined postmortem. Maternal diet during pregnancy, both starvation and consumption of high fat foods, has been shown to exert life-long effects on the feeding habits of the offspring in both animals and humans. Recent data suggest that the epigenetic modification of specific genes in hypothalamus, which control feeding behavior, is involved. Mutations of several of the genes that control epigenetic mechanisms (i.e., the writers, erasers, and readers) have over the past decade been shown to cause mental retardation and related syndromes. Mutation of CBP, for example, causes Rubinstein-Taybi syndrome, while mutation of MeCP2 (a type of methyl domain binding protein; see Box 1) causes Rett Syndrome. Together, this work illustrates how the field of epigenetics has captured the imagination of neuroscientists in diverse fields, and supports the increasing attention given to chromatin regulation in efforts to understand the causes of neuropsychiatric disorders as well as to develop improved treatments.
Future directions: Is heritable epigenetic regulation involved in psychiatry?
Work to date has focused on the role of epigenetic changes that occur within neurons of discrete brain regions in mediating the ability of environmental perturbations to produce long-lasting changes in behavior, in other words, to mediate nature-nurture interactions. In all of these situations, such epigenetic modifications in brain, by altering an individual’s behavior in a life-long manner, can underlie transmission of behavioral traits from one generation to another, as exemplified by the maternal care experiments described above.
A related question is whether environmental perturbations cause epigenetic modifications not only in brain, but in germ (sperm and egg) cells too, which might then be transmitted directly to subsequent generations. According to this scheme, environment-induced epigenetic changes in germ cells would persist into the next generation and help prescribe modifications to a gene’s relative activity or repression within a particular brain region or other organ that can influence behavior (e.g., an endocrine gland). It is not far fetched to think of a drug of abuse or stress altering genes in sperm or egg cells—drugs and stress hormones flood the entire body including gonads. Moreover, the genes affected in germ cells need not be those that are altered in a particular region of brain. It is, however, much more far fetched to think of such a change in germ cells persisting into adulthood and influencing expression of functionally relevant genes within brain or endocrine organs.
Nevertheless, there are early indications from several fields that trans-generational epigenetic transmission might occur. Diethylstilbestrol, a non-steroidal estrogen receptor agonist, was used decades ago to prevent miscarriages, but later withdrawn due to evidence that it promotes ovarian cancer and miscarriages not only in the daughters of women given the drug, but in granddaughters too. Similar data have been reported in mice. Starvation in animals, prior to gestation, has been shown to result in offspring with lower body and brain weights, which is seen in the next generation as well. This coincides with reports of metabolic derangements in grandchildren of Holocaust victims. There are similar reports, in animals and humans, of deleterious effects of high fat diet-induced obesity persisting through at least two generations. Likewise, several groups have presented preliminary evidence that rodents that are chronically stressed subsequently yield offspring that exhibit greater vulnerability to stress. For example, Dr. Isabelle Mansuy of the University of Zurich subjected mouse pups to maternal separation during their first two weeks of life. When the male mice grew to maturity, they exhibited several depression-like behaviors, very similar to those observe in the maternal care experiments outlined above. The males were then bred with normal female mice. The resulting offspring showed some depression-like behaviors when they were adults even though they were not subjected to stress during their upbringing. This transmission of stress vulnerability was correlated with altered levels of DNA methylation of several specific genes in both sperm and brain.
However, at this point in time, we must emphasize the limitations of all of these data. It has not yet been possible to demonstrate definitively that any of the examples of trans-generational transmission cited above are mediated via epigenetic modifications in germ cells as opposed to better established modes of transmission (e.g., behavioral). For example, finding altered levels of gene methylation in sperm is correlative: it does not provide direct causal evidence that altered methylation of a particular gene or set of genes is responsible for the trans-generational transmission observed. The field must now develop the experimental tools that will make it possible to, not only identify epigenetic modifications of genes in germ cells, but to establish that such modifications are both necessary and sufficient to induce the transmission of traits observed. It will also be important to explore for how many generations does such transmission, if it indeed occurs, persists. Nevertheless, while far from bona fide, these early studies are very provocative indeed.
In the late 1700s, Jean-Baptiste Lamarck of the French Academy of Sciences was known for his theory of inheritance of acquired characters. According to this idea, an adult’s traits (for example, an exercised muscle) can be passed on to his or her offspring. More than two centuries of research has discredited the Lamarckian perspective. We now know definitively that an individual’s genes play a dominant role in determining all aspects of his or her functioning. Likewise, environmental exposures throughout development and adulthood are essential in modifying the manifestation of most of those genetic traits. As discussed in this review, epigenetic modification of genes plays a key role in mediating this nature vs. nurture interplay, that is, the effects of the environment on shaping the output of an individual’s genetic potential. We also now know that random epigenetic modifications that occur during development further modify that genetic potential and environmental influence. The very recent work discussed in the previous paragraph suggests that perhaps true epigenetic inheritance may also contribute, if only to a small extent under most circumstances, to the heritability of behavioral traits and susceptible to mental illness. Future research is needed to define the magnitude of such contributions and the specific circumstances when it is most important.
Suggested readings
- Cameron NM, Shahrokh D, Del Corpo A, Dhir SK, Szyf M, Champagne FA, Meaney MJ. Epigenetic programming of phenotypic variations in reproductive strategies in the rat through maternal care. J Neuroendocrinol. 2008;20:795–801. doi: 10.1111/j.1365-2826.2008.01725.x. [DOI] [PubMed] [Google Scholar]
- Franklin TB, Russig H, Weiss IC, Gräff J, Linder N, Michalon A, Vizi S, Mansuy IM. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68:408–415. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- Maze I, Covington HE, III, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren YH, Sampath SC, Hurd YL, Greengard P, Tarakovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2009;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nature Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]