

Abstract
Polymorphonuclear neutrophils (PMNs) generate reactive oxygen species (ROS) during phagocytosis and in response to soluble agonists. This functional response, termed oxidative burst, contributes to host defense, but it can also result in collateral damage of host tissues. To study this important PMN response, different methods have been developed that are based on the assessment of oxidative burst by measuring intracellular ROS production or formation of ROS in the extracellular space. Among the different methods that were developed, the following two are particularly widely used because of their convenience and accuracy. The first method depends on the reduction of cytochrome c, which can be assessed by photometry, while the second method relies on changes in the fluorescence properties of dihydrorhodamine 123, which can be assessed by flow cytometry.
Keywords: Oxidative burst, Polymorphonuclear neutrophils, fMLP, NADPH oxidase activity, Cytochrome c reduction, DHR oxidation
1. Introduction
Effective polymorphonuclear neutrophil (PMN) activation is important for a successful host defense. Oxidative burst, cell migration, and degranulation are some of the key functional responses that enable PMNs to accomplish their tasks in host defense. These functional responses can be triggered by receptors that recognize bacterial peptides, such as N-formyl-Met-Leu-Phe (fMLP) or inflammatory mediators, such as C5a and IL-8 (1–3). Many of these receptors, such as formyl peptide receptors (FPRs) are G proteincoupled receptors (GPCRs) that induce rapid downstream signaling responses leading to the assembly of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. This multiprotein oxidase complex is dormant in quiescent cells, where its components are segregated into the cytosolic and membrane compartments. In response to stimulation, these components rapidly assemble at cell membranes and the enzyme becomes activated, allowing it to catalyze NADPH-dependent reduction of O2 to form superoxide anions (O2−.) and reactive oxygen species (ROS) derived from this radical, including hydrogen peroxide (H2O2), hydroxyl radical (OH.), and hypochlorous acid (HClO) (4). This process termed oxidative burst is also referred to as respiratory burst, which plays an important role in innate immunity against invading microorganisms. There are several inherited disorders or deficiencies in oxidative burst. For example, chronic granulomatous disease (CGD) is caused by X-linked or autosomal-recessive inheritance and results in an inability of neutrophils to properly assemble the NADPH oxidase complex and mount appropriate oxidative burst (5). Although ROS production is critical for the killing and degradation of internalized bacteria and particles, it can also contribute to inflammatory damage of host tissue (6). In order to study the role of oxidative burst in host protection, it is important to develop efficient, simple, and highly reproducible techniques to quantify ROS generation by PMN. For instance, assessing ROS production is used to evaluate PMN function and to diagnose CGD (5). We have been using several different assays to evaluate the role of PMN and ROS in the immune response to severe trauma, shock, and sepsis (7). Below, we describe different methods we have optimized to measure oxidative burst of isolated PMN and PMN suspended in heparinized whole blood.
2. Materials
2.1. Human PMN Isolation (seeNote 1)
Vacutainer plasma tubes with spray-coated sodium heparin.
Normal saline (IVNS), 0.9% sodium chloride solution, sterile, injection grade.
Hank’s balanced salt solution (HBSS) with calcium and magnesium.
20% Dextran to make 5% dextran solution, dilute 1:4 with HBSS.
Percoll (GE Heathcare, New York).
Sodium chloride (NaCl), tissue culture grade heat treated to render pyrogen free: Add 2.2 g NaCl to previously heat-treated 250-ml glass bottle; cap with two layers of aluminum foil; heat bottle with salt at 450°C overnight to render pyrogen free.
Percoll (100%): Add 50 ml of Percoll to bottle containing 2.2 g heat-treated NaCl; gently swirl until salt is completely dissolved, and sonicate if necessary; add remaining 200 ml of Percoll and thoroughly mix. Cap with a sterile cap and store at 4°C until use.
Percoll-gradient components: To obtain 73% Percoll (bottom layer), add 36.5 ml Percoll (100%) to 13.5 ml IVNS. To obtain 55% Percoll (top layer), add 27.5 ml Percoll (100%) to 22.5 ml IVNS.
Beckman GPR centrifuge with GH-3.8 rotor (or similar); switch off cooling function to avoid repeated temperature fluctuations during centrifugation.
2.2. Measurement of Oxidative Burst Using Cytochrome c Assay
Cytochrome c from bovine heart (purity ≥ 95%): Dissolve cytochrome c in HBSS to obtain a final concentration of 2 mM; aliquot and store at −80°C until use.
fMLP (Sigma, purity ≥97% by HPLC): Dissolve fMLP in sterile tissue culture-grade dimethyl sulfoxide (DMSO) to a final concentration of 10 mM; aliquot and store at −80°C until use.
Use aliquots of this fMLP stock solution in DMSO to prepare 100 nM fMLP working solutions in HBSS just before use. Use polypropylene rather than polystyrene tubes to minimize loss of fMLP due to adsorption.
DMSO (purity ≥99.7%, sterile filtered).
Tissue culture plates (96-well).
Superoxide dismutase (SOD) powder; prepare solution by dissolving in HBSS at a final concentration of 5,000 U/ml, sterile filter, aliquot, and store at −80°C until use.
2.3. Measurement of Oxidative Burst Using Dihydrorhodamine 123
Dissolve dihydrorhodamine 123 (DHR) to a final concentration of 30 mM in DMSO; aliquot and store at −80°C until use.
FACS fixing solution: Flow cytometry sheath fluid (J&S Medical Association, Inc. Framingham, MA) containing 0.5% formaldehyde (prepared with 37% formaldehyde solution, w/w).
2.4. Measurement of Oxidative Burst in Human Whole Blood
BD FACS™ lysing solution, 10× concentrated: Dilute (1:10) with distilled water.
2.5. Measurement of Oxidative Burst in Mouse Whole Blood Using DHR
RBC lysis buffer.
Heparin sodium injection, USP (1,000 USP units/ml).
W-peptide (WKYMVM; Phoenix Pharmaceuticals).
3. Methods
Several methods are available to measure PMN oxidative burst (8). Some conventional tests, such as chemiluminescence (9) and the reduction of cytochrome c (10), require the isolation of PMN and therefore relatively large amounts of blood. These assays allow monitoring of the production and release of extracellular superoxide anions using photometric or luminometric equipment. More recently, flow cytometry has become widely available to many researchers and therefore assays suitable for this type of equipment have been developed to assess oxidative burst. The assays based on flow cytometry have the advantage that tens of thousands of cells can be assessed in a very short period of time using small volumes of whole blood or isolated PMN (11). Here, oxidation of specific probes, such as 2′, 7′-dichlorofluorescein diacetate (DCFH) or DHR, to fluorescent derivatives is used to detect superoxide formation in individual cells (Fig. 1).
Fig. 1.
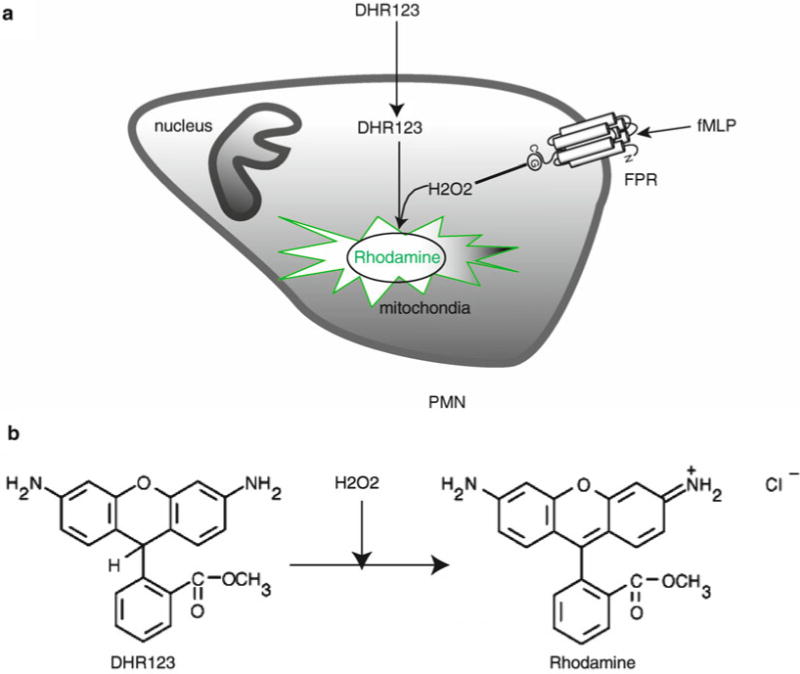
Principle of assaying oxidative burst with DHR. (a) The freely permeable, nonfluorescent DHR 123 enters cells. Upon cell stimulation, DHR 123 is oxidized by hydrogen peroxide (H2O2) formed from superoxide, resulting in the formation of fluorescent dye localized in mitochondria (11). (b) Chemical structures of DHR 123 and the product rhodamine.
Besides the methods mentioned above, some additional assays have been described to assess oxidative burst. For example, 3′, 3′-diaminobenzidine (DAB) oxidation and p-nitroblue tetrazolium (NBT) reduction are two simple methods to measure intracellular oxygen radicals or superoxide anions through precipitation reactions (8, 12). However, these methods are comparatively cumbersome and therefore they are rarely used.
Because they are most reliable in our hands, we have extensively used and optimized the DHR method described above as well as the more traditional SOD-inhibitable reduction of cytochrome c to characterize oxidative burst activity in PMN (7, 13).
3.1. Human PMN Isolation
Draw blood using heparinized vacutainer plasma tubes (seeNote 2).
Dextran sedimentation: Add 3 ml of 5% dextran per 10 ml of blood in sterile 50-ml centrifuge tube. Mix gently by inverting tubes several times. Let cells settle at room temperature for 30 min (seeNote 3).
While the sedimentation is taking place, set up the Percoll centrifugation gradients in sterile 15-ml centrifuge tubes. You need one tube for each 10-ml aliquot of blood. Place 4 ml of 73% Percoll in a 15-ml tube and carefully layer 4 ml of 55% Percoll on top using a 5-ml serological pipette.
After dextran sedimentation of the blood, harvest the supernatant that contains plasma and white cells; place in fresh 50-ml centrifuge tubes, fill with HBSS, mix gently, and centrifuge at 1,500 × g for 10 min to wash cells.
Remove supernatant and gently disrupt cell pellet (seeNote 3). Then, add 3 ml of HBSS per 10-ml blood aliquot and carefully layer 3 ml of this cell suspension onto the Percoll gradient in the 15-ml tubes; then, spin at 400 × g for 20 min at room temperature.
Remove top phase to just above the cell layer containing the PMN using a sterile, heat-treated Pasteur pipette and vacuum suction system. Use a serological pipette to remove the PMN layers and combine cell suspension in fresh 50-ml centrifuge tubes using no more than 10 ml of cell suspension in each 50-ml tube.
Fill tubes with HBSS and centrifuge at 1,500 × g for 10 min to wash cells for the first time.
Remove supernatants and gently disrupt cell pellets (seeNote 3). Fill 50-ml tubes with fresh HBSS and centrifuge at 220 × g for 10 min for a second wash.
Gently disrupt pellet, add HBSS, resuspend and count cells, and then adjust cell concentration to 1 × 107/ml using fresh HBSS. Store cells at room temperature until use. Use cells as soon as possible.
3.2. Measurement of Oxidative Burst in Human PMN Using Cytochrome c in Tissue Culture Plates
Prepare two wells for each sample: one well for sample without and one well for sample with SOD (seeNote 4).
PMNs (15 μl of 1.33 × 107/ml cell suspension) are added in each well of 96-well tissue culture plates prewarmed for 1 h at 37°C in a water bath.
Meanwhile, prepare 100 μM cytochrome c in HBSS with or without 100 nM fMLP, and prewarm to 37°C.
Add 5 μl SOD solution in SOD reference wells and 5 μl HBSS in sample wells. Add 80 μl prewarmed cytochrome c solution to each well and incubate at 37°C in a water bath.
After 10 min at 37°C, optical density changes are measured with a plate reader at a wavelength of 550 nm.
Calculate the relative amount of O2− generated using positive and negative controls.
3.3. Measurement of Oxidative Burst in Human PMN Using Cytochrome c in Cuvettes
Prepare two tubes (e.g., sterile 1.5-ml Eppendorf centrifuge tubes) for each sample: one for each sample and another one for each SOD reference (seeNote 4).
PMNs (100 μl of 1 × 107/ml cell suspension) are added to each tube and HBSS is added to result in a final total volume of 1 ml per tube. The tubes are prewarmed at 37°C in a water bath for 30 min to 1 h.
Meanwhile, prepare 100 μM cytochrome c solution in HBSS with or without 100 nM fMLP.
Add 50 μl SOD solution in SOD reference tubes; add 100 μl cytochrome c solution to each tube.
After 10 min, stop reactions by placing tubes into an ice bath for at least 10 min. Centrifuge samples at 665 × g for 5 min at 4°C in an Eppendorf centrifuge and transfer supernatants to fresh Eppendorf tubes kept in an ice bath. Transfer supernatants into a cuvette with a 1-cm path length and measure optical density differences using a spectrophotometer at a wavelength of 550 nm.
- Calculate the molar amount of O2− generated using the following formula (seeNote 5):
3.4. Measurement of Oxidative Burst Using Dihydrorhodamine 123 and Flow Cytometry
Mix isolated PMN (100 μl of 1 × 107/ml cell suspension), stimuli, any other agents of interest, and HBSS in sterile 1.5-ml Eppendorf centrifuge tubes to achieve a final volume of 500 μl per tube. Prewarm tubes in water bath at 37°C for 1 h (seeNote 6).
Open lids of tubes and add 2.8 μl DHR or HBSS as control, 25 μl fMLP or HBSS as control, onto the lid of each tube. Gently close the lids without disturbing the drops attached to the inside of the lids.
Using appropriate racks, convert all tubes at the same time to allow mixing of cell suspensions with DHR and stimuli added to the lids. Gently flick rack to ascertain that cell suspensions return to the bottom of each centrifuge tube, and then place tubes back into the water bath and incubate at 37°C for 20 min.
Place racks with tubes into an ice bath and incubate for 10 min to stop all reactions (seeNote 7). Centrifuge tubes at 425 × g for 5 min at 4°C, remove supernatants, wash one more time with HBSS, resuspend cells in 300 μl FACS fixing solution, and keep on ice until analysis.
Measure samples in flow cytometer (FL1 channel with 488-nm laser) as soon as possible.
3.5. Measurement of Oxidative Burst in Human Whole Blood Using Flow Cytometry
Mix heparinized whole blood (100 μl), stimuli or other agents of interest, and HBSS in sterile 1.5-ml Eppendorf centrifuge tubes resulting in a final volume of 200 μl. Prewarm tubes in water bath at 37°C for 5 min (seeNote 6).
Add 2.8 μl DHR or HBSS, 25 μl fMLP or HBSS, onto lids of tubes, mix contents as described above, and incubate at 37°C for 20 min.
Place tubes on ice for 10 min to stop reactions (seeNote 7). Add 1 ml FACS lysing solution per tube and keep tubes on ice for 20 min to lyse completely erythrocytes.
Centrifuge tubes at 425 × g for 5 min at 4°C. Remove supernatants and wash cells twice using 1 ml HBSS. Resuspend cell pellets in 300 μl FACS fixing solution and keep on ice until analysis.
Measure samples using flow cytometer (FL1 channel with 488-nm laser) as soon as possible. Adjust the fluorescence gain properly using unstimulated control samples (shaded curve, Fig. 2a) and positive fMLP (100 nM) control samples (open curve, Fig. 2a). Figure 2b shows a sample scatter plot after proper adjustment of forward and side scatter detectors, allowing clear identification of the PMN population.
Fig. 2.
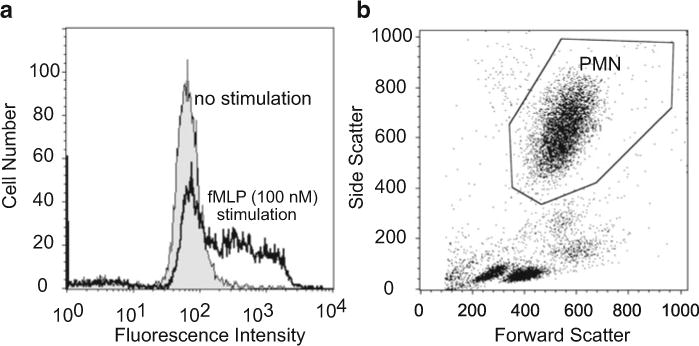
Oxidative burst measurement with flow cytometry using DHR. (a) Fluorescence histogram profiles of the oxidative metabolic response of PMN stimulated with 100 nM fMLP (open curve) or of unstimulated PMN (shaded curve). An example of the histograms 10 min after stimulation is shown. (b) Gating of PMN using forward and side scatter plot of lysed human whole blood.
3.6. Measurement of PMN Oxidative Burst in Mouse Whole Blood
Draw mouse blood by cardiopuncture into 1-ml syringe previously rinsed with heparin.
Mix heparinized whole blood (50 μl), drugs of interests, and HBSS in sterile 1.5-ml Eppendorf centrifuge tubes to achieve a final volume as 600 μl. Prewarm tubes in water bath at 37°C for 5 min (seeNote 6).
Add 3.5 μl DHR or HBSS, 6 μl w-peptide or HBSS, into tubes and mix with blood as described above. Incubate tubes in water bath for 20 min at 37°C (seeNote 8).
Place tubes in ice bath for 10 min to stop all reactions (seeNote 7). Centrifuge at 425 × g for 5 min at 4°C and remove supernatants. Then, add 1 ml RBC lysis buffer in each tube and keep tubes on ice for 4 min to lyse erythrocytes.
Centrifuge tubes at 425 × g for 5 min at 4°C, remove supernatants, and wash cell with 1 ml HBSS twice. Resuspend cell pellets in 300 μl FACS fixing solution and keep on ice until analysis.
Measure samples with flow cytometer (FL1 channel with 488-nm laser) as soon as possible. Adjust the forward and side scatter detectors in order to clearly identify PMN population and then adjust fluorescence gain using unstimulated and stimulated control samples.
Acknowledgments
This work was supported by NIH grants GM-51477, GM-60475, AI-072287, and AI-080582 and Congressionally Directed Medical Research Programs grant PR043034 (W.G.J.).
Footnotes
Pyrogen and similar contaminants in materials that come in contact with blood or PMN result in cell aggregation during isolation, high baseline activation levels, and premature cell death. Key steps to minimize such problems are to avoid pyrogen contamination by using only tissue culture-grade reagents, pyrogen-free and sterile plasticware and other consumables, and infusion-grade solutions and by removing endotoxin and pyrogen by baking glassware, salts, and other equipment when possible.
Repeated heating and cooling of cell suspensions must be avoided during cell isolation because this causes unintended cell activation. To avoid temperature fluctuations, perform all isolation steps at room temperature unless otherwise mentioned; and remember to disable cooling systems of centrifuges if necessary.
Mechanical stress activates cells and results in high baseline activation levels and cell death. To avoid such problems, always handle cells gently; after centrifugations, disrupt cell pellets before adding fluids to resuspend the cells. Whenever possible, avoid polystyrene tubes; the use of polypropylene plasticware is preferable to avoid cell activation and loss due to excessive cell adhesion.
SOD serves as a reference to exclude any signals not due to O2−.
Use the Beer–Lambert law with an extinction coefficient (ε) of 21.1 mM−1 cm−1 for reduced cytochrome c at a wave length of 550 nm to calculate molar amounts of O2− generated by PMN (8).
Controls needed in this experiment include cells only and cells with DHR but without stimulation.
From this step on, always keep samples in an ice water bath.
W-peptide is a chemotactic peptide that activates murine FPR (14).
References
- 1.Rabiet MJ, Huet E, Boulay F. The N-formyl peptide receptors and the anaphylatoxin C5a receptors: an overview. Biochimie. 2007;89:1089–1106. doi: 10.1016/j.biochi.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 3.Capra V. Molecular and functional aspects of human cysteinyl leukotriene receptors. Pharmacol Res. 2004;50:1–11. doi: 10.1016/j.phrs.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal. 2006;8:1533–1548. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- 5.Hager M, Cowland JB, Borregaard N. Neutrophil granules in health and disease. J Intern Med. 2010;268:25–34. doi: 10.1111/j.1365-2796.2010.02237.x. [DOI] [PubMed] [Google Scholar]
- 6.Nussler AK, Wittel UA, Nussler NC, et al. Leukocytes, the Janus cells in inflammatory disease. Langenbecks Arch Surg. 1999;384:222–232. doi: 10.1007/s004230050196. [DOI] [PubMed] [Google Scholar]
- 7.Naoyuki H, Chen Y, Rusu C, et al. Whole-blood assay to measure oxidative burst and degranulation of neutrophils for monitoring trauma patients. European Journal of Trauma. 2005;31:379–388. [Google Scholar]
- 8.Dahlgren C, Karlsson A, Bylund J. Measurement of respiratory burst products generated by professional phagocytes. Methods Mol Biol. 2007;412:349–363. doi: 10.1007/978-1-59745-467-4_23. [DOI] [PubMed] [Google Scholar]
- 9.Lundqvist H, Dahlgren C. Isoluminol-enhanced chemiluminescence: a sensitive method to study the release of superoxide anion from human neutrophils. Free Radic Biol Med. 1996;20:785–792. doi: 10.1016/0891-5849(95)02189-2. [DOI] [PubMed] [Google Scholar]
- 10.Cohen HJ, Chovaniec ME. Superoxide production by digitonin-stimulated guinea pig granulocytes. The effects of N-ethyl maleimide, divalent cations; and glycolytic and mitochondrial inhibitors on the activation of the superoxide generating system. J Clin Invest. 1978;61:1088–1096. doi: 10.1172/JCI109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elbim C, Lizard G. Flow cytometric investigation of neutrophil oxidative burst and apoptosis in physiological and pathological situations. Cytometry A. 2009;5:475–481. doi: 10.1002/cyto.a.20726. [DOI] [PubMed] [Google Scholar]
- 12.Schopf RE, Mattar J, Meyenburg W, et al. Measurement of the respiratory burst in human monocytes and polymorphonuclear leukocytes by nitro blue tetrazolium reduction and chemiluminescence. J Immunol Methods. 1984;67:109–117. doi: 10.1016/0022-1759(84)90090-5. [DOI] [PubMed] [Google Scholar]
- 13.Junger WG, Hoyt DB, Davis RE, et al. Hypertonicity regulates the function of human neutrophils by modulating chemoattractant receptor signaling and activating mitogen-activated protein kinase p38. J Clin Invest. 1998;101:2768–2779. doi: 10.1172/JCI1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo JK, Choi SY, Kim Y, et al. A peptide with unique receptor specificity: stimulation of phosphoinositide hydrolysis and induction of superoxide generation in human neutrophils. J Immunol. 1997;158:1895–1901. [PubMed] [Google Scholar]