

Protein–protein interactions are essential for transmitting signals from extracellular space to cell nuclei and also for cell–cell communication. Small molecules that target protein–protein interactions, therefore, are great research tools for dissecting the biological functions of a given protein–protein interaction and potential therapeutics for many different human diseases.[1] However, targeting protein–protein interactions by small molecules remains a significant challenge.[1, 2]
Cyclic-AMP response element (CRE) binding protein (CREB) belongs to a large family of basic leucine zipper (bZIP)-containing transcription factors.[3, 4] It is phosphorylated at Ser133 by mitogen- and stress-activated protein kinases.[4] The phosphorylated CREB (p-CREB) then binds the mammalian transcription coactivator, CREB-binding protein (CBP) via the kinase-inducible domain (KID) in CREB and the KID-interacting (KIX) domain in CBP.[5] This binding event will further recruit other transcriptional machinery to the gene promoter to activate CREB-dependent gene transcription.[4] Recent studies have revealed that CREB is overexpressed in many different types of cancers including prostate cancer,[6] breast cancer,[7] non-small-cell lung cancer,[8] and acute myeloid leukemia.[9] Therefore, small-molecule inhibitors of CREB–CBP are potential anticancer agents. Herein we describe our discovery of naphthol AS-E (1) as a cell-permeable small-molecule inhibitor of the KIX–KID interaction by using a novel Renilla luciferase complementation assay.
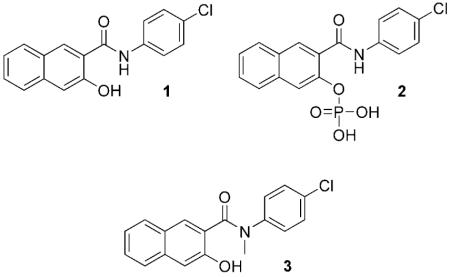
Naphthol AS-E phosphate (2) was identified by a medium-throughput NMR screening as a candidate compound to inhibit the KIX–KID interaction with an IC50 of ~90 μm.[10] Yet this same compound displayed a much higher potency in a cell-based assay to inhibit CREB-mediated gene transcription with an IC50 in the low-mm range. The reason for the huge potency difference remains to be resolved. We hypothesized that the cellular activity seen with compound 2 might come from compound 1 being generated in situ by dephosphorylation. Consistent with this hypothesis, compound 2 was found to be dephosphorylated in regular tissue culture media and not cell-permeable (Figure S1 in the Supporting Information). However, it is unclear whether compound 1 has any biological activity in vitro or in cells.
In order to investigate whether compound 1 inhibits the CREB-CBP interaction, a Renilla luciferase complementation assay[11-15] was designed to monitor the KIX–KID interaction (Figure 1). In this assay, KID and KIX were fused to the N- and C-terminal fragments of Renilla luciferase, respectively. On their own, the individual fragments do not present luciferase activity ; however, once KIX binds phosphorylated KID, the Renilla luciferase fragments are brought together to reconstitute active Renilla luciferase. Therefore, measurement of Renilla luciferase activity will register the binding interaction between KIX and KID.
Figure 1.
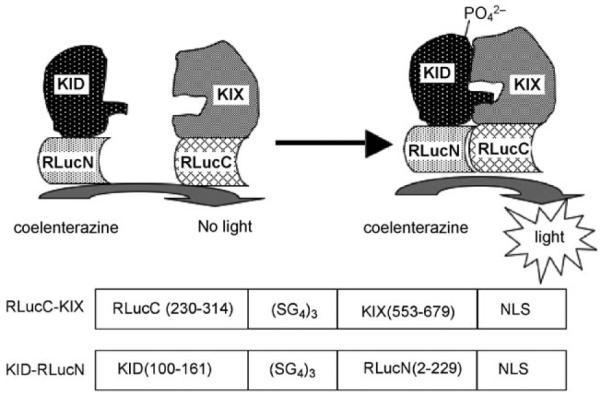
Design principle of and fusion constructs for the KIX–KID Renilla luciferase complementation assay. NLS : nuclear localization sequence.
The constructs RLucC–KIX and KID–RLucN for the Renilla luciferase complementation assay were created by fusing the C-terminal Renilla luciferase fragment (230–314) to KIX (553–679) and KID (100–161) to N-terminal fragment of Renilla luciferase (2–229) via a flexible linker (Figure 1, bottom). The splitting point of Renilla luciferase was based on previous studies.[11] The relative orientation of the different fragments was derived after careful inspection of the NMR structure of the KIX–KID complex.[5] To test whether this assay was working as expected, HEK293T cells were transfected with these two constructs, and increasing concentrations of forskolin were then added to the cells to stimulate phosphorylation of KID. As presented in Figure 2A, forskolin dose-dependently stimulated Renilla luciferase activity ; this suggests that the KIX–KID interaction was induced inside the cells upon forskolin stimulation.
Figure 2.

Compound 1 inhibits KIX–KID interaction in living cells. A) Forskolin dose-dependently stimulates KIX–KID interaction in cells. HEK293T cells were transfected with RLucC–KIX and KID–RLucN. The transfected cells were then treated with increasing concentrations of forskolin for 2 h. The Renilla luciferase activity was then measured by adding coelenterazine to the cells. B) Compound 1 inhibits KIX–KID interaction in cells. Same as in (A), except that the transfected cells were pretreated with increasing concentrations of 1 (▲) or 3 (●) before the addition of forskolin (2.5 μm).
Having established the Renilla luciferase complementation assay, we then asked whether compound 1 could inhibit the KIX–KID interaction in living cells. Therefore, RLucC–KIX- and KID–RLucN-transfected HEK293T cells were treated with compound 1 before stimulation with forskolin (2.5 μm). The data shown in Figure 2B demonstrate that compound 1 dose-dependently inhibited Renilla luciferase activity with an IC50 of 2.26 ± 0.65 μm. Compound 3, an N-methylated analogue of 1, showed significantly reduced inhibitory activity (IC50 = 24.54 ± 5.46 μm) and therefore serves as a negative control. In a similar Renilla luciferase complementation assay using Id–MyoD (inhibitor of DNA binding–myogenic differentiation) as a protein interaction pair,[11] no inhibition was observed in the presence of compound 1 (Figure S2).
To rule out the possibility that the observed decrease in Renilla luciferase activity is due to decreased phosphorylation of KID, HEK293T cells were treated with compound 1 followed by forskolin stimulation. The phosphorylation status of CREB was evaluated by Western blot in the crude cell lysates. No change in the level of phosphorylated CREB was observed (Figure S3), thus indicating that the decreased luciferase activity was indeed due to inhibition of the KIX–KID interaction in living cells. The reconstituted Renilla luciferase could not be completely inhibited by compound 1 (Figure 2B). Incomplete inhibition of Renilla luciferase activity was also observed with HSP90 (heat shock protein 90) inhibitors in inhibiting HSP90 and p23 interaction in a similar assay format.[16] Two possibilities, not mutually exclusive, could cause the residual activity. First, the residual activity might reflect a basal level of KIX–KID interaction that is not inhibited by compound 1. Second, the residual activity might result from a fraction of complexes whose luciferase fragments still associate with each other even though KIX–KID is separated by compound 1.
To further demonstrate that compound 1 inhibits the KIX–KID interaction, a biochemical Renilla luciferase complementation assay was designed. A His6-tagged RLucC–KIX protein was expressed and purified from E. coli (Figure S4); meanwhile, phosphorylated KID–RLucN was expressed from HEK293T cells, and crude cell lysates were used directly for complementation. As presented in Figure 3A, Renilla luciferase activity was specifically reconstituted when RLucC–KIX was mixed with KID–RLucN. No significant increase in luciferase activity was observed when RLucC–KIX was combined with Id–RLucN, which contains the same N-terminal Renilla luciferase fragment fused to a non-KIX-interacting protein partner Id. With both the interacting partners in hand, an in vitro biochemical assay was performed to evaluate the ability of different compounds to inhibit the KIX–KID interaction. As presented in Figure 3B, compound 1 dose-dependently inhibited KIX–KID interaction with an IC50 of 2.90 ± 0.81 μm. On the other hand, no significant inhibition of the KIX–KID interaction was observed with compound 2 or 3 at concentrations up to 50 μm; this suggests that they are rather weak inhibitors of the KIX–KID interaction.
Figure 3.

Compound 1 inhibits the KIX–KID interaction in vitro. A) Renilla luciferase activity was specifically reconstituted upon mixing RLucC–KIX and KID–RLucN. B) Compound 1 inhibits the KIX–KID interaction in vitro. RLucC–KIX and KID–RLucN were combined in the presence of varying concentrations of 1 (●), 2 (■) or 3 (▲). The mixture was then incubated on ice for 24 h, and the remaining luciferase activity was measured.
To investigate whether compound 1 could directly bind KIX to inhibit the KIX–KID interaction, it was incubated with various concentrations of purified His6KIX immobilized on Ni–Sepharose beads. The remaining unbound compound 1 was then analyzed by HPLC (Figure 4A). The concentration of unbound 1 decreased with increasing concentration of His6KIX. The apparent Kd of binding compound 1 to KIX was ~8.6 μm. On the other hand, negligible binding of compound 2 to His6KIX was observed under the same experimental conditions. These data are consistent with the in vitro KIX–KID interaction inhibition results (Figure 3B).
Figure 4.

Compound 1 binds KIX and inhibits CREB-mediated gene transcription. A) Compound 1 binds His6KIX. Different concentrations of His6KIX were immobilized on Ni–Sepharose beads and incubated with either 1 (□) or 2 (■; 50 μm). The fraction of unbound compounds was analyzed by HPLC. B) Compound 1 inhibits CREB-mediated gene transcription in HEK293T cells. CRE-RLuc-transfected HEK293T cells were treated with increasing concentrations of 1 (□) or 2 (■). The Renilla luciferase activity was measured 4.5 h after forskolin stimulation.
The above studies clearly demonstrated that compound 1 inhibits the KIX–KID interaction both in cells and in vitro. To investigate the functional consequences of inhibiting the KIX–KID interaction in cells, a Renilla luciferase (RLuc) reporter assay was employed to look at the ability of compound 1 to inhibit CREB-mediated gene transcription in living HEK293T cells. HEK293T cells were transfected with CRE-RLuc, which expresses RLuc under the control of three iterative copies of CRE. RLuc was used instead of firefly luciferase (FLuc) because we found that compound 1 inhibits FLuc’s enzymatic activity (Figure S5). The transfected cells were then treated with compound 1 followed by forskolin (10 μm) stimulation. As presented in Figure 4B, compound 1 dose-dependently inhibited CREB-mediated gene transcription with an IC50 in the low-μm range, which is comparable to its in vitro and cellular KIX–KID inhibitory potency (Figures 2B and 3B). Compound 2 inhibited CREB reporter transcription with reduced potency. This activity was a result of compound 1 being generated in situ by partial dephosphorylation of 2 during the experiment (5 h, Figure S1) because compound 2 is not cell permeable. Control compound 3 only weakly inhibited CREB-mediated gene transcription (IC50 > 50 μm; Figure S6). To assess the role of the KIX–KID interaction in mediating the inhibition of CREB reporter transcription by compound 1, a constitutively active CREB mutant, CREB-VP16 whose transcription activity is independent of KID,[17] was cotransfected with CRE-RLuc into HEK293T cells. No significant inhibition of CREB-VP16-mediated gene transcription was observed with compound 1 (Figure S7); this suggests that KIX–KID interaction inhibition is required to inhibit CREB-mediated gene transcription by 1.
In conclusion, we have developed a novel Renilla luciferase complementation assay and discovered naphthol AS-E (1) to be a cell-permeable inhibitor of the KIX–KID interaction by binding to KIX directly. In addition, naphthol AS-E inhibits CREB-mediated gene transcription in living cells. These results could explain the observed huge potency difference (>40-fold) between in vitro and cellular assays of compound 2.[10] The structure of compound 1 represents the first KIX-binding small molecule to inhibit CREB-dependent gene transcription in living cells and a novel template for inhibiting protein–protein interactions. Recent studies have demonstrated that CREB plays a critical role in tumor progression and maintenance.[18] Therefore, the discovery of compound 1 as an inhibitor of the KIX–KID interaction might present an opportunity to develop novel transcription-based cancer therapeutics, an area that is largely underexplored.[19, 20] These studies are underway and will be reported in due course.
Supplementary Material
Acknowledgements
We thank Drs. David Dawson, Thomas Scanlan, and Jeffrey Karpen for helpful comments regarding the manuscript. This work was financially supported by Oregon Health and Science University.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.200900552.
References
- [1].Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- [2].Boger DL, Desharnais J, Capps K. Angew. Chem. 2003;115:4270–4309. doi: 10.1002/anie.200300574. Angew. Chem. Int. Ed. 2003, 42, 4138 - 4176. [DOI] [PubMed] [Google Scholar]
- [3].Vinson CR, Sigler PB, McKnight SL. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- [4].Shaywitz AJ, Greenberg ME. Annu. Rev. Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- [5].Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- [6].Wu D, Zhau HE, Huang W-C, Iqbal S, Habib FK, Sartor O, Cvitanovic L, Marshall FF, Xu Z, Chung LWK. Oncogene. 2007;26:5070–5077. doi: 10.1038/sj.onc.1210316. [DOI] [PubMed] [Google Scholar]
- [7].Chhabra A, Fernando H, Watkins G, Mansel RE, Jiang WG. Oncol. Rep. 2007;18:953–958. [PubMed] [Google Scholar]
- [8].Seo H-S, Liu DD, Bekele BN, Kim M-K, Pisters K, Lippman SM, Wistuba II, Koo JS. Cancer Res. 2008;68:6065–6073. doi: 10.1158/0008-5472.CAN-07-5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, Rao NP, Landaw EM, Sakamoto KM. Cancer Cell. 2005;7:351–362. doi: 10.1016/j.ccr.2005.02.018. [DOI] [PubMed] [Google Scholar]
- [10].Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Proc. Natl. Acad. Sci. USA. 2004;101:17622–17627. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Paulmurugan R, Gambhir SS. Anal. Chem. 2003;75:1584–1589. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Nat. Rev. Drug Discovery. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- [13].Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV. ACS Chem. Biol. 2008;3:346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- [14].Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, Michnick SW. Proc. Natl. Acad. Sci. USA. 2007;104:16916–16921. doi: 10.1073/pnas.0704257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Binkowski B, Fan F, Wood K. Curr. Opin. Biotechnol. 2009;20:14–18. doi: 10.1016/j.copbio.2009.02.013. [DOI] [PubMed] [Google Scholar]
- [16].Chan CT, Paulmurugan R, Gheysens OS, Kim J, Chiosis G, Gambhir SS. Cancer Res. 2008;68:216–226. doi: 10.1158/0008-5472.CAN-07-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- [18].Sakamoto KM, Frank DA. Clin. Cancer Res. 2009;15:2583–2587. doi: 10.1158/1078-0432.CCR-08-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Arndt H-D. Angew. Chem. 2006;118:4664–4673. Angew. Chem. Int. Ed. 2006, 45, 4552 - 4560. [Google Scholar]
- [20].Berg T. Curr. Opin. Chem. Biol. 2008;12:464–471. doi: 10.1016/j.cbpa.2008.07.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.