

Abstract
The balanced self-renewal and differentiation of nephron progenitors are critical for kidney development and controlled, in part, by the transcription factor Six2, which antagonizes canonical Wnt signaling-mediated differentiation. A nuclear factor, Sall1, is expressed in Six2-positive progenitors as well as differentiating nascent nephrons, and it is essential for kidney formation. However, the molecular functions and targets of Sall1, especially the functions and targets in the nephron progenitors, remain unknown. Here, we report that Sall1 deletion in Six2-positive nephron progenitors results in severe progenitor depletion and apoptosis of the differentiating nephrons in mice. Analysis of mice with an inducible Sall1 deletion revealed that Sall1 activates genes expressed in progenitors while repressing genes expressed in differentiating nephrons. Sall1 and Six2 co-occupied many progenitor-related gene loci, and Sall1 bound to Six2 biochemically. In contrast, Sall1 did not bind to the Wnt4 locus suppressed by Six2. Sall1-mediated repression was also independent of its binding to DNA. Thus, Sall1 maintains nephron progenitors and their derivatives by a unique mechanism, which partly overlaps but is distinct from that of Six2: Sall1 activates progenitor-related genes in Six2-positive nephron progenitors and represses gene expression in Six2-negative differentiating nascent nephrons.
The nephron is a basic functional unit of the kidney, which includes the glomerulus, proximal and distal renal tubules, and the loop of Henle. The mammalian kidney, the metanephros, is formed by reciprocally inductive interactions between two precursor tissues, namely the metanephric mesenchyme and the ureteric bud. The mesenchyme contains nephron progenitors that express a transcription factor, Six2. When Six2-positive cells are labeled using Six2GFPCre, a mouse strain expressing Cre recombinase fused to green fluorescent protein (GFP) under the control of the Six2 promoter, they give rise to nephron epithelia in vivo.1 Six2 opposes the canonical Wnt-mediated differentiation evoked by ureteric bud-derived Wnt9b, thereby maintaining the self-renewal of nephron progenitors.2–4 However, the progenitors gradually lose Six2 expression and start to differentiate. These differentiating cells express Wnt4, which further enhances the differentiation. Six2 binds to the Wnt4 enhancer to ensure that only a subset of progenitors differentiate at each time point. Through this balance between self-renewal and differentiation, progenitors sequentially transit to pretubular aggregates, renal vesicles, and C- and S-shaped bodies, which eventually develop into nephron epithelia.
spalt (sal) was first isolated from Drosophila as a region-specific homeotic gene, and it encodes a nuclear protein characterized by multiple double zinc finger motifs.5 Humans and mice each have four known sal-like genes (known as SALL1–4 in humans and Sall1–4 in mice). Mutations in SALL1 and SALL4 have been associated with Townes–Brocks and Okihiro syndromes, respectively, both of which are autosomal dominant diseases that involve abnormalities in various organs, including ears, limbs, heart, and kidneys.6,7 We have shown that Sall1 is expressed in the metanephric mesenchyme and that Sall1 knockout mice exhibit kidney agenesis resulting from failure of ureteric bud attraction to the mesenchyme at day 11.5 of gestation (E11.5).8 However, Sall1 should have additional roles, because it continues to be expressed in the metanephric mesenchyme after ureteric bud invasion. We previously showed the presence of nephron progenitors in Sall1-positive mesenchyme by establishing a novel colony assay.9 Because Six2 is expressed in Sall1-high mesenchymal cells, the Sall1-high and Six2-positive mesenchyme represents a nephron progenitor population in the embryonic kidney.10 However, the role of Sall1 in the progenitors remains unknown. Therefore, we generated mice lacking Sall1 in Six2-positive progenitors and their derivatives and found that Sall1 is, indeed, essential for maintenance of these populations.
Results
Sall1 Deletion Causes Depletion of Nephron Progenitors Accompanied by Reduction of Nephron Structures
To gain insights into the roles of Sall1 in nephron progenitors, we crossed the floxed allele of Sall1 with Six2GFPCre BAC transgenic mice expressing a fusion protein of GFP and Cre recombinase in the progenitor population.1 Six2GFPCre;floxed Sall1 mice were born at Mendelian frequency, but all of them died shortly after birth with abnormally small kidneys (Figure 1, A and B). The mutant kidneys contained multiple glomerular cysts, dilated renal tubules, and thin cortexes (Figure 1, C and D). Six2-positive nephron progenitors were undetectable (Figure 1, E and F), and development of the nephron components, including glomerular podocytes, proximal renal tubules, the loop of Henle, and distal renal tubules, was significantly impaired (Figure 1, G–N).
Figure 1.

Sall1 deletion depletes nephron progenitors and their derivatives. (A) Kidneys in a newborn control mouse (P0). ad, Adrenal gland; bl, bladder; kid, kidney; ov, ovary. (B) Kidney size is reduced in a newborn Six2GFPCre;Sall1flox/flox mouse (P0). te, Testis. (C and D) Hematoxylin-eosin staining of newborn kidneys. Severe dysgenesis is observed in the Six2GFPCre;Sall1flox/flox mouse kidney. Scale bar, 100 μm. (E–N) Immunostaining for Six2 (nephron progenitor), Wt1 (nephron progenitor and podocyte), LTL (proximal renal tubule), THP (the loop of Henle), and NCC (distal renal tubule). Development of the nephron components is significantly impaired in the Six2GFPCre;Sall1flox/flox mouse kidney.
Sall1 mutant kidneys were already smaller than controls at E14.5 (Figure 2, A and B). Six2 was still expressed in the Sall1 mutants, but the number of Six2-positive cells was significantly less (Figure 2, C and D). Sall1 was expressed in not only the Six2-positive nephron progenitor region but also, the differentiating nephrons located deeper inside of the kidney (Figure 2E). Sall1 expression in both populations was significantly less in Sall1 mutants, although expression in the stroma, the outermost layer of the kidney, was not affected (Figure 2F, asterisk). This finding reflects the spatially restricted activity of Cre recombinase in Six2GFPCre mice.
Figure 2.

Nephron progenitors are depleted in midgestation embryos. (A and B) Hematoxylin-eosin staining of E14.5 kidneys. Scale bar, 100 μm. (C and D) Immunostaining for Six2. There are fewer Six2-positive cells relative to controls. (E and F) Immunostaining for Sall1. There is much less expression of Sall1 in the mutant nephron components. *Sall1 in the stroma remains expressed. (G–J) Expansion of nephron progenitors in organ culture. (G and H) Kidneys at the beginning of the culture (E12.5). (I and J) Kidneys after 2 days of the culture. The GFP signal in the mutant kidney becomes weaker during the course of the culture. Time-lapse videos are shown in Supplemental Video 1 (Six2GFPCre) and Supplemental Video 2 (Six2GFPCre;Sall1flox/flox). Scale bar, 100 μm. (K and L) FACS analysis of the GFP-positive cells. Kidneys were isolated at E12.5 and cultured for 2 days. The proportion of GFP-positive progenitors is significantly less in the mutant. Average (SD) of three samples. A, area; FSC, forward scatter; PI, propidium iodide. (M and N) Cell cycle analysis of GFP-positive cells. There is no difference between the Sall1 mutant and the control. Average (SD) of three samples. (O–T) Dual immunostaining of Sall1 and Six2 in Sall1flox/flox and Six2GFPCre;Sall1flox/flox kidneys at E13.5. Sall1 expression (red) is reduced in some of the nephron progenitors (arrowheads) and differentiating nephrons (arrows). The expression of Sall1 is not affected in the stroma (*). There are fewer Six2-positive cells (green), but most of them are negative for Sall1. ub, Ureteric bud. Scale bar, 40 μm.
We next isolated kidneys at E12.5 and cultured them for 3 days in vitro. Because Six2GFPCre mice also express GFP driven by the Six2 promoter, Six2-positive nephron progenitors could be monitored by time-lapse confocal microscopy. Sall1 mutants showed a comparable GFP signal at the beginning of the culture (Figure 2, G and H). During the culture, the nephron progenitors in the Six2GFPCre kidney rapidly expanded around the branching ureteric bud tips (Figure 2I, Supplemental Video 1). However, in the Sall1 mutants, the signal became almost undetectable within 48 hours of culture (Figure 2J, Supplemental Video 2). We also confirmed the reduction of GFP-positive progenitors by FACS analysis (Figure 2, K and L). Thus, progenitor depletion occurs between E12.5 and E14.5 in the absence of Sall1. Cell cycle analysis of the GFP-positive populations did not show any significant differences (Figure 2, M and N), indicating that progenitor depletion is unlikely caused by proliferation defects.
Sall1 Deletion Impairs Self-Renewal of Nephron Progenitors and Induces Apoptosis in the Differentiating Nephrons
We then used lineage tracing to examine the fate of Sall1 mutant nephron progenitors. We crossed Six2GFPCre mice with a reporter strain, in which the CAG promoter, floxed stop sequences, and the tandem dimer Tomato coding sequence have been inserted into the Rosa26 locus.11 As reported previously,1 Six2-positive progenitors gave rise to most components of the nephron (Figure 3A). In contrast, the differentiation of nephrons was severely impaired in Sall1 mutants at birth (Figure 3B), most profoundly in the proximal tubules and the loop of Henle. At E14.5 in the control, a significant portion of progenitor descendants was still located in the Six2-positive region where they were originated (arrowheads in Figure 3, C and C′), whereas the remaining cells had joined the neural cell adhesion molecule (NCAM)-positive differentiating population (arrows in Figure 3, C and C′). In the Sall1 mutants, the number of labeled cells located in the Six2-positive region was significantly smaller (arrowheads in Figure 3, D and D′), suggesting that the self-renewal capacity of Sall1 mutant progenitors is impaired. There was a small contribution of labeled cells to the differentiating nephrons, but the morphology of these structures was different from the well organized S-shaped bodies seen in the controls (arrows in Figure 3, C′ and D′). Nevertheless, cell fates were restricted to the NCAM-positive nephron lineages, and no signs of transdifferentiation to other lineages were detected (Figure 3D). At E13.5, we detected apoptotic cells in the Sall1-deficient differentiating nephrons (arrows in Figure 3, E and F). Apoptosis in the progenitors was less prominent (arrowheads in Figure 3, E and F), and proliferation defects were also undetectable (Figure 3, G and H), which is consistent with Figure 2N. Considering the absence of apparent apoptosis, proliferation defects, or aberrant lineage conversion in Sall1-deificient nephron progenitors, depletion of these progenitors could result from the skewed balance to differentiation versus self-renewal, which is followed by apoptosis in the differentiating nephrons.
Figure 3.

Sall1 deletion impairs self-renewal of nephron progenitors and induces apoptosis in differentiating nephrons. Lineage trace analysis of the nephron progenitors. Scale bar , 100 μm. (A and B) Tandem dimer Tomato (tdTomato) is stained by the anti–red fluorescent protein antibody (blue). The development of nephrons, especially proximal tubules (*) and the loop of Henle (**), is severely impaired in the Sall1 mutant at P0. (C and D) Immunostaining at E14.5 shows the reduction of tdTomato-positive (red) nephron progenitors (arrowheads) and the NCAM/tdTomato-positive (yellow) differentiating nephrons (arrows). C′ and D′ show higher magnification. (E and F) Immunostaining for cleaved caspase 3 (brown) at E13.5. Apoptotic cells are detected in the differentiating nephrons of the Sall1 mutants. (G and H) Immunostaining for phosphohistone-H3 (PHH3 [brown]). Proliferation defects are not observed in the Sall1 mutant. ub, ureteric bud.
Inducible Sall1 Deletion Phenocopies the Conditional Sall1 Mutant
Cre activity in the Six2GFPCre mice is mosaic, and it takes several days to completely delete Sall1 in the entire progenitor-derived populations.1 Dual immunostaining for Sall1 and Six2 at E13.5 showed that Six2 was retained in the Sall1-null cells (Figure 2, O–T), suggesting that Sall1 reduction does not lead to an immediate loss of Six2. We intermittently observed residual nephron formation in mutant mice, but these structures were always positive for Sall1 (Supplemental Figure 1A), indicating that only escapers from Six2GFPCre-mediated deletion can form nephrons. To identify the direct molecular events downstream of Sall1, we analyzed inducible Sall1 mutants (Sall1CreER/flox). On tamoxifen treatment at E12.5, this mutant strain had smaller kidneys at birth, which we reported previously.12 We found that Six2-positive nephron progenitors were lost in the newborn mutant (Figure 4, A and B), and the development of most nephron components was significantly impaired (Figure 4, C–H). The differentiating nephrons exhibited apoptosis at E14.5 (Figure 4, I–K). Thus, the inducible Sall1 mutant strain phenocopied the Six2GFPCre-dependent Sall1 deletion. Sall1 expression was already reduced at E13.5, 1 day after tamoxifen treatment, whereas Six2 was retained in the Sall1-null cells (Figure 4, L–Q), which is consistent with the results shown in Figure 2, O–T. At E14.5, Six2 expression still remained (Figure 4, R–U). Although FGF9 and FGF20 maintain nephron progenitors,13 Etv4, with expression in the mesenchyme that correlates with FGF activity,14,15 showed normal expression (Supplemental Figure 1, B and C). Furthermore, addition of FGF2 to the Six2GFPCre;Sall1flox/flox kidney in organ culture did not rescue the phenotypes (Supplemental Figure 1, D and E). Lef1 is an indicator of Wnt activity, and it is normally expressed in differentiating nephrons but not progenitors.15 There was no upregulation of Lef1 in the progenitor regions 2 days after tamoxifen treatment (Supplemental Figure 1, F and G). In addition, expression patterns of Wnt9b (in the ureteric bud stalk) and Wnt11 (in the ureteric bud tip) were not significantly altered (Supplemental Figure 1, H–K). In contrast, Six2-positive progenitors were significantly reduced at E15.5, and many nascent nephrons were formed simultaneously (Figure 4, V, V′, W, and W′). These renal vesicles were aberrantly large considering the reduced progenitor population. Therefore, Sall1 deletion is likely to accelerate premature differentiation that leads to the progenitor depletion. Concomitant apoptosis in the differentiating nephrons could cause the nephron loss at birth. However, ectopic Lef1 expression was not observed in the progenitors (arrowheads in Figure 4, X and Y), suggesting that the premature differentiation is not caused by the Six2/Wnt-mediated mechanism.
Figure 4.

Inducible Sall1 deletion phenocopies the conditional Sall1 mutant. Tamoxifen was injected at E12.5 and analyzed later. (A–H) Immunostaining for Six2 (nephron progenitor), WT1 (nephron progenitor and podocyte), LTL (proximal renal tubule), and THP (the loop of Henle) at P0. Development of the nephron components is significantly impaired in the Sall1CreER/flox mouse kidney. Scale bar, 100 μm. (I–K) Terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (red) and NCAM (green) staining at E14.5. (J and K) Apoptotic cells are detected in differentiating nephrons in the mutant. (K) Apoptosis is more prominent in the differentiating nephrons located deeper inside of the kidney. Arrows, differentiating nephrons; arrowheads, nephron progenitors. Scale bar, 40 μm. (L–Q) Dual immunostaining for Sall1 and Six2 at E13.5. Sall1 expression (red) is reduced in some of the nephron progenitors (arrowheads) and differentiating nephrons (arrows). Most of the Six2-positive cells (green) are negative for Sall1 in the mutant. *Stroma. Scale bar, 40 μm. (R–U) Immunostaining for Sall1 and Six2 at E14.5. Scale bar, 100 μm. (V and W) Immunostaining for Six2 at E15.5. Note that the differentiating nephrons (renal vesicles; arrows) were aberrantly large considering the reduced progenitors (arrowheads) in the Sall1 mutant. Six2 was slightly overstained, and therefore, the nascent nephrons expressing Six2 weakly were detectable. Scale bar, 40 μm. V′ and W′ show higher magnification. (X and Y) Immunostaining for Lef1 (Wnt activity) at E15.5. Lef1 is expressed in the differentiating nephrons (arrows) and excluded from the progenitors (arrowheads; dotted regions) in both Sall1+/flox and Sall1CreER/flox kidneys. Scale bar, 40 μm. ub, ureteric bud.
It is noteworthy that Sall1 expression in the inducible strain was reduced in not only nephron progenitor-derived lineages but also, the cortical stroma (compare Figure 4Q with Figure 2T). However, the phenotype similarities of the two mutant strains indicate that Sall1 expressed in the nephron lineage has a major role in kidney development, at least until birth. The large renal vesicles in the inducible strain are likely to result from the simultaneous Sall1 deletion, which is in contrast to the gradual Six2GFPCre-mediated deletion, although it is formally possible that Sall1 in the stroma could also play a role.
Sall1 Is an Activator in Nephron Progenitors and a Repressor in Differentiating Nephrons
To identify downstream targets of Sall1, we performed microarray analysis using the kidneys of inducible Sall1 mutant mice, along with controls, harvested 24 and 48 hours after tamoxifen treatment at E12.5. We also performed microarray analysis using kidneys from Six2GFPCre;Sall1flox/flox mice and controls at E14.5. We then compared gene expression among the four genotypes and picked up genes that overlapped in all these comparisons; 31 genes (41 probes) were downregulated immediately on Sall1 deletion (Figure 5A, Supplemental Table 1). We further performed microarray analysis using sorted Six2GFP-positive progenitors from embryonic kidneys and found that 25 of 31 genes (31 of 41 probes) were more abundantly expressed in the Six2-positive progenitors, indicating that Sall1 positively regulates these progenitor-related genes. This list includes Cited1, Osr1, and Robo2, which are expressed in the Six2-positive domains and have important roles in kidney development,16–19 and also, heretofore unappreciated genes, such as Megf9. Quantitative RT-PCR and histologic analysis confirmed marked reduction of Cited1 and a slight decrease of Osr1 (Figure 5, B–E, Supplemental Figure 1L).
Figure 5.
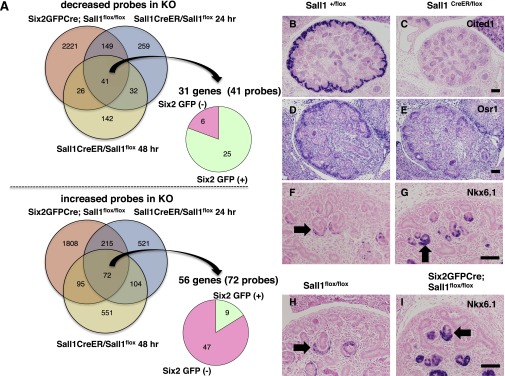
Sall1 is an activator in nephron progenitors and a repressor in differentiating nephrons. (A) Venn diagrams of the left show the overlap of decreased (upper panels) or increased (lower panels) probes in Six2GFPCre;Sall1flox/flox kidneys and Sall1CreER/flox kidneys 24 and 48 hours after tamoxifen treatment. The circle graphs on the right show the distributions of the decreased or increased genes in Six2-GFP–positive or -negative cells. Gene numbers are smaller than probe numbers because of the overlaps of the probes. KO, knockout. (B and C) Immunostaining for Cited1. Tamoxifen was injected at E12.5 and analyzed at E14.5. The expression of Cited1 is significantly decreased in the Sall1CreER/flox kidney. Scale bar, 100 μm. (D and E) In situ hybridization of Osr1. The expression of Osr1 is mildly reduced in the Sall1CreER/flox kidney at E14.5. (F–I) Immunostaining for Nkx6.1. The expression of Nkx6.1 in the differentiating nephrons (arrows) is significantly increased in both the Sall1CreER/flox kidney treated with tamoxifen and the Six2Cre;Sall1flox/flox kidney at E14.5.
The microarray analysis also identified 56 genes (72 probes) that were upregulated immediately on Sall1 deletion (Figure 5A, Supplemental Table 2). Interestingly, 47 of 56 genes (56 of 72 probes) were more abundantly expressed in the Six2-negative population. Because Sall1 is expressed in not only Six2-positive progenitors but also, the Six2-negative differentiating nephrons, Sall1 may negatively regulate a subset of genes in the latter population. This gene list included Nkx6.1, with function in the kidney that has not been identified. The significant increase of this gene in the Sall1 mutants was confirmed by quantitative RT-PCR (Supplemental Figure 1L). Immunostaining showed that Nkx6.1 was, indeed, increased in the differentiating nephrons of the inducible Sall1 mutants, whereas it was expressed only weakly in the control (Figure 5, F and G). This increase was also confirmed in the Six2GFPCre-dependent Sall1 deletion (Figure 5, H and I); therefore, Sall1 could function as a negative regulator in the differentiating nephrons, while functioning as a positive regulator in the progenitors.
Progenitor-Related Loci but Not Differentiation-Related Loci Are Co-Occupied by Sall1 and Six2
We next performed chromatin immunoprecipitation (ChIP) followed by sequencing (ChIP-Seq) analysis using embryonic kidneys. We found that Sall1 and Six2 co-occupy the loci encoding the genes that are downregulated in the absence of Sall1, such as Osr1, Robo2, and Megf9. This result indicates that these genes are the direct Sall1 targets (Figure 6A, Supplemental Table 1), although the Sall1 peaks in the Cited1 locus were equivocal. In addition, many loci containing genes essential for kidney development, such as Eya1, Pax2, Wt1, and Gdnf, as well as Sall1 and Six2 themselves are co-occupied by both Sall1 and Six2 (Figure 6A, Supplemental Figure 2A). One of the major exceptions is the Hox cluster, which is occupied only by Sall1 (Supplemental Figure 2B). To rule out nonspecific binding of the Sall1 antibody, we generated another mouse strain that contains Flag-tagged Sall1 (Sall1Flag) by homologous recombination (Supplemental Figure 3A). The expression pattern of Sall1Flag was identical to that of endogenous Sall1 proteins (Supplemental Figure 3B), and we confirmed the Sall1 binding by ChIP-quantitative PCR (Supplemental Figure 3C).
Figure 6.

Progenitor-related loci but not differentiation-related loci are co-occupied by Sall1 and Six2. (A) ChIP-Seq analysis of Sall1 and Six2 within the progenitor-related loci (mm9 coordinates). Asterisks show peaks co-occupied by Sall1 and Six2, and diamonds show peaks reported in a study by Park et al.3 Red asterisks correspond to the regions used for the EMSA in Figure 7. The co-occupied peak in the Osr1 locus was not detected in the stringent peak calls, but Sall1 binding was verified by an EMSA. (B) Venn diagram showing the number of peaks bound by Sall1 or Six2 across the whole genome. Sall1 binding peaks within 500 bp from Six2 binding peaks are classified as co-occupied. (C) The enriched de novo Sall1 binding motif recovered from ChIP-Seq peak regions. (D and E) No Sall1 binding peaks in the Wnt4 or Nkx6.1 loci. Diamond shows the peak reported in a study by Park et al.3
Co-occupancy of Sall1 and Six2 was mainly limited to progenitor-related genes and not detectable at the gene loci related to differentiation, extracellular matrix, or ureteric bud attraction (Supplemental Table 3). Sall1 binding sites significantly overlapped within 500 bp from the Six2 binding sites (Figure 6B). The Six2-bound regions were similar to those regions reported by Park et al.,3 including Six2 and Wnt4 enhancers (Figure 6, Supplemental Tables 1 and 3). Extracted Six2 binding consensus sequences were also identical (GNAACNNNANNC). In contrast, Sall1 binding consensus sequences were enriched with A and T (Figure 6C), which is consistent with previous reports, including our own work.20,21 Furthermore, we confirmed the binding of Sall1 to the loci mentioned above by an electrophoretic mobility shift assay (EMSA) (Figure 7A). In addition, the immunoprecipitation assay using Sall1Flag embryonic kidneys as well as overexpression analysis showed that Sall1 bound to Six2 (Figure 7, B and C). This interaction was still observed in the presence of deoxyribonuclease and ribonuclease and confirmed by the recombinant proteins generated in vitro (Figure 7, C and D). Thus, these two proteins bind to each other directly.
Figure 7.

Sall1 binds to the progenitor-related loci and biochemically associates with Six2 and Mi2/NuRD. (A) EMSA of Sall1 showing Sall1 binding to the progenitor-related loci. Competitor MT, mutated competitor oligonucleotides; competitor WT, wild-type competitor oligonucleotides; IgG, negative control for the supershift (*) by the anti-Sall1 antibody (Sall1 ab); Sall1, in vitro-translated Sall1 proteins; vector, in vitro-translated lysates from the empty vector. (B) Binding of Sall1 with Six2 (upper panel) or the Mi2/NuRD complex (lower panel) in the kidney. Control and Sall1Flag E15.5 kidneys were immunoprecipitated using the anti-Flag antibody and then blotted with the indicated antibodies. IP, immunoprecipitation. (C) Binding of Sall1 with Six2 independent of DNase and RNase treatment. Flag-Sall1 and myc-Six2 were overexpressed in human embryonic kidney 293 cells followed by IP using the anti-Flag antibody. Endogenous HDAC2 also bound to overexpressed Sall1. (D) Direct binding of the recombinant Sall1 and Six2 proteins generated in vitro. Recombinant proteins prepared by the rabbit reticulocyte lysate system were mixed, immunoprecipitated, and blotted with the indicated antibodies. DNase, deoxyribonuclease; RNase, ribonuclease; Six2, in vitro-translated Six2 proteins.
It is reported that Six2 binds to differentiation-related gene loci, such as Wnt4, Fgf8, and Bmp7, and suppresses these genes in the nephron progenitors.3 However, we did not detect any Sall1 binding throughout a few hundred kilobases of these loci (Figure 6D, Supplemental Figure 2C), indicating that Sall1 functions independently of the inhibitory roles of Six2. Thus, the cooperation between Sall1 and Six2 could be limited to the progenitors.
Sall1 binding was also undetectable in most of the derepressed loci, including the Nkx6.1 locus (Figure 6E, Supplemental Table 2), suggesting that Sall1-mediated repression is independent of direct binding to DNA. We detected binding of Sall1 with endogenous HDAC2 and Mi2β, which are components of the histone deacetylase (HDAC)-containing Mi2/nucleosome remodeling deacetylase (NuRD) complex, in Sall1Flag embryonic kidneys and the overexpression analysis (Figure 7, B and C). These observations are consistent with the hypothesis that Sall1 could function as a repressor in the differentiating nephrons where Six2 expression has disappeared. There could exist another DNA binding molecule that bridges the Sall1 and Mi2/NuRD complex with the Nkx6.1 locus.
Discussion
Sall1 is expressed in Six2-positive progenitors as well as differentiating nascent nephrons, and Sall1 deletion results in depletion of both populations. Sall1 maintains nephron progenitors and their derivatives by a mechanism that partly overlaps but is distinct from that of Six2. Sall1 activates progenitor-related genes in Six2-positive nephron progenitors but is not involved in Six2-mediated suppression of the Wnt4/Fgf8 pathway.
The expression changes of the Sall1 target genes were unexpectedly mild, considering the severe phenotypes. We previously showed that Sall4 is essential for the maintenance of embryonic stem (ES) cells.22 Sall4 forms a network with other nuclear factors by both protein–protein interaction and mutual transcriptional activation, thereby maintaining pluripotency.21,23 In this type of network, deletion of Sall4 alone leads to mild changes in a subset of the components but still results in stem cell depletion. Likewise, Sall1 binds to many progenitor-related gene loci, but in terms of transcription, a subset of them is affected moderately. The additive effects of these changes could still impair the self-renewal of nephron progenitors. This network view of stem cells may explain why Sall1 deletion does not cause an immediate loss of Six2, despite Sall1 binding to the Six2 enhancer.
In differentiating nephrons where Six2 expression has disappeared, Sall1 is likely to function as a repressor. It is proposed that Sall family members function as transcriptional repressors by interacting with the Mi2/NuRD complex.24–26 However, genes endogenously repressed by Sall1 in the kidney remain elusive. We show that Sall1 suppresses multiple genes, including Nkx6.1. Nkx6.1 is essential for neuron and pancreas development,27,28 and Nkx6.1 misexpression in uncommitted pancreas progenitors using the Nkx6.1OE mouse specifies an endocrine fate.29 We overexpressed Nkx6.1 in vivo in nephron progenitor-derived lineages by crossing the Six2GFPCre with the Nkx6.1OE mouse (Supplemental Figure 4). Although the expression was mosaic, two of five Six2GFPCre;Nkx6.1OE mice showed hypoplastic kidneys with scattered Six2-positive progenitors. Therefore, the increase of Nkx6.1 could affect kidney development. In ES cells, Sall4 binds to the Mi2/NuRD complex and represses aberrant expression of Cdx2, a critical transcription factor that stimulates differentiation to trophectoderm.25 Therefore, dual functions of Sall family proteins in stem/progenitor cells are well conserved. To address the role of Sall1 in differentiating nephrons more precisely, we performed Wnt4Cre-mediated Sall1 deletion. However, the phenotype was again lost in progenitors, although it was not as complete as that in Six2GFPCre-mediated deletion (Supplemental Figure 5A). This observation was caused by leaky excision in nephron progenitors (Supplemental Figure 5B). Thus, the relative importance of Sall1 as an activator versus repressor remains to be clarified.
We propose that Sall1 positively regulates targets in nephron progenitors and suppresses aberrant gene expression in the differentiating nephrons, thereby maintaining these populations. Regulation of different targets as a positive and negative regulator could depend on the interacting proteins that are available in progenitors (Six2) or differentiating nephrons (Mi2/NuRD), although identification of molecules recruiting Sall1 to the repressed loci will be necessary.
Concise Methods
Generation of the Mutant Mice
Sall1flox, Sall1CreER, and Nkx6.1OE mice were described previously.12,25,29 Six2GFPCre BAC transgenic and Wnt4GFPCre mice was provided by Andrew McMahon.1,3 The R26RtandemdimerTomato mouse was obtained from The Jackson Laboratory.11 Tamoxifen treatment was described previously.12 To generate the Sall1Flag mice, the 5′ EcoRI–HindIII Sall1 genomic fragment containing exon 3 of Sall1 fused with a Flag tag (5.5 kb) as well as the 3′ HindIII–ClaI fragment (2.8 kb) were incorporated into a vector containing Neo flanked by loxP sites and diphtheria toxin A-subunit in tandem. The targeting vector was electroporated into E14.1 ES cells, and 8 of 280 G418-resistant clones were correctly targeted as determined by Southern blotting analyses using 5′ or 3′ probes after EcoRV or HindIII digestion, respectively. The three correctly targeted ES clones were used to generate germ-line chimeras that were bred with C57BL/6J female mice at the Center for Animal Resources and Development at Kumamoto University. The Neo cassette was deleted by crossing the mutant mice with mice expressing Cre ubiquitously.30 Genotyping of the offspring was performed by PCR using a forward primer, 5′-CTGGGAACGTGGAAAAACTG-3′, and two reverse primers, 5′-CACTCTGGCAGCTTTAGCTTG-3′ and 5′-GTCATCGTCCTTGTAGTC-3′, producing products of 153 bp for the control allele and 178 bp for the mutant allele. Homozygous mice showed no apparent abnormalities. All animal experiments were performed in accordance with institutional guidelines and ethical review committees.
In Situ Hybridization and Immunostaining
Histologic examinations were performed as described previously.31 Mice were fixed in 10% formalin, embedded in paraffin, and cut into 6-μm sections. In situ hybridization was performed using an automated Discovery System (Roche) according to the manufacturer’s protocols. Templates for the probes were generated by RT-PCR and sequenced. Immunostaining was carried out automatically using a BlueMap or DABmap Kit and the automated Discovery System or manually for immunofluorescence staining. The following primary antibodies were used: anti-Six2 (Proteintech); anti-Sall122,32 (Perseus Proteomics); anti-Sall1 (AB31526; Abcam, Inc.), anti-Cited1 (Thermo Fisher Scientific); anti-Wt1 (Santa Cruz Biotechnology); LTL (Vector Laboratories); anti-THP (Santa Cruz Biotechnology); anti-NCC (EMD Millipore); anticytokeratin (Sigma-Aldrich); anti-NCAM (Developmental Studies Hybridoma Bank; EMD Millipore); anti-Nkx6.1 (R&D Systems); anticleaved caspase 3 (Cell Signaling Technology); anti–phosphohistone-H3 (EMD Millipore); anti-red fluorescent protein (Rockland); anti-Lef1 (Cell Signaling Technology); and anti-DDDDK tag (Abcam, Inc.). The monoclonal (Perseus Proteomics) and polyclonal (Abcam, Inc.) anti-Sall1 antibodies gave no background signals in Sall1 mutant kidneys. Terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling assays were performed using an ApopTag Plus fluorescein in situ apoptosis detection kit (EMD Millipore), and the signal was enhanced by Alexa 594-conjugated streptavidin (Invitrogen).
Organ Culture of the Embryonic Kidney
E12.5 kidneys were cultured on Millicell Cell Culture Inserts (EMD Millipore) placed in glass-bottomed dishes containing DMEM media with 10% serum. Time-lapse confocal images were taken using a CellVoyager CV1000 (Yokogawa) and processed using Imaris (Bitplane). The APC BrdU flow kit (BD Pharmingen) was used for cell cycle analysis as described.25
Tamoxifen Treatment
Tamoxifen (70 mg/kg body wt) was administered intraperitoneally into pregnant female mice as described.12 Because tamoxifen treatment hindered the ability of mice to give birth, we euthanized the pregnant mother at the expected birth date.
Microarray and Quantitative RT-PCR
Two sets of Six2GFPCre;Sall1flox/flox versus Sall1flox/flox at E14.5 and three pairs of Sall1CreER/flox versus Sall1+/flox (two pairs for 24 hours and one pair for 48 hours after tamoxifen treatment at E12.5) were analyzed. Mircoarray analysis was performed by using Agilent whole-mouse genome (4×44,000; v2) or SurePrint G3 mouse gene expression (8×60,000). The data were normalized by GeneSpring GX software (Agilent Technologies). Microarray platforms were combined using Entrez Gene ID, and differentially expressed genes (>1.5-fold) were extracted. The array data have been deposited with the National Center for Biotechnology Information Gene Expression Omnibus (accession no. GSE45845). RNA was isolated from dissected kidneys using an RNeasy Plus Micro Kit (Qiagen) and then reverse-transcribed with random primers using the Superscript VILO cDNA Synthesis Kit (Invitrogen). Quantitative PCR was carried out using the Dice Real Time System Thermal Cycler (Takara Bio) and Thunderbird SYBR qPCR Mix (Toyobo). All the samples were normalized by the β-actin expression.
ChIP-Seq and Immunoprecipitation
ChIP analysis was performed as described.3,33 Briefly, 12 kidneys at E16.5 per sample were fixed for 30 minutes, sonicated, and mixed with anti-Six2 antibody (Proteintech), anti-Sall1 antibody (Abcam, Inc.), anti-Flag M2 antibody (Sigma-Aldrich), or rabbit or mouse IgG (Santa Cruz Biotechnology) at 4°C overnight followed by precipitation using Dynabeads M-280 conjugated with anti-rabbit or -mouse IgG (Invitrogen). Templates for ChIP-Seq analysis were prepared using the ChIP-Seq sample prep kit (Illumina) following the manufacturer’s instructions. Sequencing was carried out on Illumina HiSeq2000 platform, and at least 20 million 36-nucleotide single end sequences were generated for each sample. The sequence data were submitted to Genbank/DNA Data Bank of Japan (accession no. DRA000957). Sequences were mapped to the mouse genome (mm9) allowing two base mismatches. Peak call was carried out using MACS (http://liulab.dfci.harvard.edu/MACS/). Six2 peaks giving P<0.001 were filtered with the following parameters: −10×LOG10 (P value) ≥150, tags≥39, fold≥8, and false discovery rate≤1.4. Sall1 peaks giving P<0.001 were filtered with the following parameters: −10×LOG10 (P value) ≥30, tags≥35, fold≥7, and false discovery rate≤2.5. Multiple EM for Motif Elicitation-ChIP was used to identify the Sall1 and Six2 binding motif. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen). Human embryonic kidney 293T cells or kidneys at E15.5 were lysed with lysis buffer (Cell Signaling Technology) and sonicated on ice. Lysates were clarified by centrifugation and incubated with beads conjugated with the anti-Flag M2 antibody for 1 hour at 4°C. Beads were washed three times with Tris-buffered saline-1% NP40, and bound proteins were eluted with 100 mM glycine-HCl (pH 2.6) and analyzed by Western blotting using the anti-Mi2β (Santa Cruz Biotechnology) or anti-HDAC2 (Santa Cruz Biotechnology) antibodies.
EMSA
Recombinant mouse Sall1 protein was prepared by the rabbit reticulocyte lysate system (Promega) according to the manufacturer’s protocol. EMSAs were performed as described elsewhere.34 In vitro-translated Sall1 proteins (10 ng in 3 μl) were incubated at 4°C for 30 minutes with a [γ-32P]ATP-labeled double-stranded oligonucleotide in the presence or absence of unlabeled double-stranded competitors (Supplemental Table 4). Supershift assays were performed by additional incubation with an anti-Sall1 monoclonal antibody or mouse IgG for 20 minutes before electrophoresis. The bound products were resolved in a 4% nondenaturing polyacrylamide gel in 0.5×Tris-Borate/EDTA buffer, and then, they were exposed to a radioactive imaging plate and detected by an FLA-3000 laser scanner (Fuji Photo Film).
Disclosures
None.
Supplementary Material
Acknowledgments
We thank R. Matoba for microarray analysis, T. Horiuchi, K. Imamura, and M. Tosaka for chromatin immunoprecipitation and sequencing, M. Aoki for FACS analysis, and Y. Kaku, S. Inoue, and S. Fujimura for histological analysis. We also thank K. Shimamura, T. Miyata, Y. Xi, J. Kreidberg, L. O’Brien, and A.P. McMahon for helpful advice.
This study was supported by KAKENHI Grants 23390228, 25111725, and 221S0002 from Ministry of Education, Culture, Sports, Science and Technology, Japan. Research in the laboratory of M.S. was supported by National Institutes of Health Grant R01-DK68471.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013080896/-/DCSupplemental.
References
- 1.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP: Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G: Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 25: 5214–5228, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J-S, Ma W, O’Brien LL, Chung E, Guo J-J, Cheng J-G, Valerius MT, McMahon JA, Wong WH, McMahon AP: Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell 23: 637–651, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, Carroll TJ: Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 138: 1247–1257, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kühnlein RP, Frommer G, Friedrich M, Gonzalez-Gaitan M, Weber A, Wagner-Bernholz JF, Gehring WJ, Jäckle H, Schuh R: spalt encodes an evolutionarily conserved zinc finger protein of novel structure which provides homeotic gene function in the head and tail region of the Drosophila embryo. EMBO J 13: 168–179, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W: Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet 18: 81–83, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Kohlhase J, Heinrich M, Schubert L, Liebers M, Kispert A, Laccone F, Turnpenny P, Winter RM, Reardon W: Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet 11: 2979–2987, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Nishinakamura R, Matsumoto Y, Nakao K, Nakamura K, Sato A, Copeland NG, Gilbert DJ, Jenkins NA, Scully S, Lacey DL, Katsuki M, Asashima M, Yokota T: Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development 128: 3105–3115, 2001 [DOI] [PubMed] [Google Scholar]
- 9.Osafune K, Takasato M, Kispert A, Asashima M, Nishinakamura R: Identification of multipotent progenitors in the embryonic mouse kidney by a novel colony-forming assay. Development 133: 151–161, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Nishinakamura R: Stem cells in the embryonic kidney. Kidney Int 73: 913–917, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H: A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue S, Inoue M, Fujimura S, Nishinakamura R: A mouse line expressing Sall1-driven inducible Cre recombinase in the kidney mesenchyme. Genesis 48: 207–212, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Barak H, Huh SH, Chen S, Jeanpierre C, Martinovic J, Parisot M, Bole-Feysot C, Nitschké P, Salomon R, Antignac C, Ornitz DM, Kopan R: FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev Cell 22: 1191–1207, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AC, Adams D, de Caestecker M, Yang X, Friesel R, Oxburgh L: FGF/EGF signaling regulates the renewal of early nephron progenitors during embryonic development. Development 138: 5099–5112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mugford JW, Yu J, Kobayashi A, McMahon AP: High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev Biol 333: 312–323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard-Smith EM, Mortlock DP, Baldwin HS, de Caestecker M: Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev Biol 313: 234–245, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.James RG, Kamei CN, Wang Q, Jiang R, Schultheiss TM: Odd-skipped related 1 is required for development of the metanephric kidney and regulates formation and differentiation of kidney precursor cells. Development 133: 2995–3004, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Mugford JW, Sipilä P, McMahon JA, McMahon AP: Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev Biol 324: 88–98, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grieshammer U, Le Ma, Plump AS, Wang F, Tessier-Lavigne M, Martin GR: SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell 6: 709–717, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Yamashita K, Sato A, Asashima M, Wang P-C, Nishinakamura R: Mouse homolog of SALL1, a causative gene for Townes-Brocks syndrome, binds to A/T-rich sequences in pericentric heterochromatin via its C-terminal zinc finger domains. Genes Cells 12: 171–182, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Lim CY, Tam W-L, Zhang J, Ang HS, Jia H, Lipovich L, Ng H-H, Wei C-L, Sung WK, Robson P, Yang H, Lim B: Sall4 regulates distinct transcription circuitries in different blastocyst-derived stem cell lineages. Cell Stem Cell 3: 543–554, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Sakaki-Yumoto M, Kobayashi C, Sato A, Fujimura S, Matsumoto Y, Takasato M, Kodama T, Aburatani H, Asashima M, Yoshida N, Nishinakamura R: The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development 133: 3005–3013, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Chu J, Shen X, Wang J, Orkin SH: An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132: 1049–1061, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauberth SM, Rauchman M: A conserved 12-amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J Biol Chem 281: 23922–23931, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Yuri S, Fujimura S, Nimura K, Takeda N, Toyooka Y, Fujimura YI, Aburatani H, Ura K, Koseki H, Niwa H, Nishinakamura R: Sall4 is essential for stabilization, but not for pluripotency, of embryonic stem cells by repressing aberrant trophectoderm gene expression. Stem Cells 27: 796–805, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Denner DR, Rauchman M: Mi-2/NuRD is required in renal progenitor cells during embryonic kidney development. Dev Biol 375: 105–116, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sander M, Paydar S, Ericson J, Briscoe J, Berber E, German M, Jessell TM, Rubenstein JL: Ventral neural patterning by Nkx homeobox genes: Nkx6.1 controls somatic motor neuron and ventral interneuron fates. Genes Dev 14: 2134–2139, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M: Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 127: 5533–5540, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Schaffer AE, Freude KK, Nelson SB, Sander M: Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell 18: 1022–1029, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakai K, Miyazaki J: A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun 237: 318–324, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Fujimura S, Jiang Q, Kobayashi C, Nishinakamura R: Notch2 activation in the embryonic kidney depletes nephron progenitors. J Am Soc Nephrol 21: 803–810, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato A, Kishida S, Tanaka T, Kikuchi A, Kodama T, Asashima M, Nishinakamura R: Sall1, a causative gene for Townes-Brocks syndrome, enhances the canonical Wnt signaling by localizing to heterochromatin. Biochem Biophys Res Commun 319: 103–113, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, Suzuki Y, Sugano S, Nakaseko C, Yokote K, Koseki H, Iwama A: Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 120: 1107–1117, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Tanigawa S, Lee CH, Lin CS, Ku CC, Hasegawa H, Qin S, Kawahara A, Korenori Y, Miyamori K, Noguchi M, Lee LH, Lin YC, Steve Lin CL, Nakamura Y, Jin C, Yamaguchi N, Eckner R, Hou DX, Yokoyama KK: Jun dimerization protein 2 is a critical component of the Nrf2/MafK complex regulating the response to ROS homeostasis. Cell Death Dis 4: e921, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.