

Abstract
A previous meta-analysis of genome-wide association data by the Cohorts for Heart and Aging Research in Genomic Epidemiology and CKDGen consortia identified 16 loci associated with eGFR. To define how each of these single-nucleotide polymorphisms (SNPs) could affect renal function, we integrated GFR-associated loci with regulatory pathways, producing a molecular map of CKD. In kidney biopsy specimens from 157 European subjects representing nine different CKDs, renal transcript levels for 18 genes in proximity to the SNPs significantly correlated with GFR. These 18 genes were mapped into their biologic context by testing coregulated transcripts for enriched pathways. A network of 97 pathways linked by shared genes was constructed and characterized. Of these pathways, 56 pathways were reported previously to be associated with CKD; 41 pathways without prior association with CKD were ranked on the basis of the number of candidate genes connected to the respective pathways. All pathways aggregated into a network of two main clusters comprising inflammation- and metabolism-related pathways, with the NRF2-mediated oxidative stress response pathway serving as the hub between the two clusters. In all, 78 pathways and 95% of the connections among those pathways were verified in an independent North American biopsy cohort. Disease-specific analyses showed that most pathways are shared between sets of three diseases, with closest interconnection between lupus nephritis, IgA nephritis, and diabetic nephropathy. Taken together, the network integrates candidate genes from genome-wide association studies into their functional context, revealing interactions and defining established and novel biologic mechanisms of renal impairment in renal diseases.
Keywords: pathophysiology of renal disease and progression, transcriptional profiling, renal progression, transcription regulation, CKD, chronic kidney failure
CKD affects >13% of the Unites States population and is a major contributor to cardiovascular morbidity and mortality.1,2 Defining the pathophysiology of CKD is critical to identify predictors of the disease course and therapeutic targets. Diverse mechanisms have been linked with the development and progression of CKD using experimental models and renal tissues based expression studies.3–6
The CKDGen and CHARGE consortia were able to use genome-wide association studies (GWASs) to identify preexisting genetic risk factors for renal function decline.7,8 However, GWASs, just like renal tissue gene expression profiling studies, only capture one aspect of the underlying pathophysiology of CKD. A combined effort, linking the knowledge of genetic and transcriptomic alterations with clinical phenotype information in CKD, is now feasible and offers the opportunity for an integrated understanding of the intrarenal drivers of CKD.
This study used a sequential strategy to construct and interpret a network of CKD-associated pathways that combines distinct but complementary sources of data: GWAS candidate genes, renal biopsy-derived transcriptional profiles with matching clinical information, and literature-derived knowledge of molecular pathways.
The CKDGen GWAS identified genetic loci associated with eGFR. Intrarenal gene expression levels generated from an independent population of subjects were tested for correlation with eGFR. Genes with evidence for eGFR association from those two independent lines of evidence (genetic and transcriptomic) were evaluated for their molecular functional context in CKD using a coexpression strategy. For each gene in the intersection, mRNAs with stringent coexpression in the renal tissues of subjects with CKD were identified. The functional context of coexpressed gene sets was defined using prior biologic knowledge derived from comprehensive pathway databases, linking each gene from GWAS through gene expression and eGFR correlation to a set of molecular pathways. Connections between these pathways indicate that they share at least one transcript correlated with one or more GWAS-derived candidate genes, allowing the construction of a network of interacting pathways in CKD. For each renal disease included in the CKD dataset, the disease-specific interplay of CKD-related pathways was identified and used to define a transcript-based similarity matrix between the glomerular diseases studied.
The combination of genetic, molecular, and clinical datasets in individuals with CKD allows for defining renal diseases on the basis of their shared and specific molecular mechanism. Identification of CKD pathways and their interplay provides starting points for experimental studies to define the biologic mechanisms underlying chronic renal failure.
Results
eGFR Correlation
A meta-analysis by the CKDGen and CHARGE consortia identified 16 loci associated with renal function.7 A follow-up study identified 13 additional loci for renal function and CKD in the same regions.8 Forty candidate genes were located in proximity (±60 kb) of 16 single-nucleotide polymorphisms (SNPs) and evaluated for differential regulation in CKD (Table 1). Of them, 29 transcripts were found to be expressed above background in transcriptional expression profiles of renal biopsies of 157 subjects (Figure 1, step 1). These biopsies were from subjects diagnosed as having one of nine different chronic renal diseases (FSGS, membranous GN [MGN], minimal change disease [MCD], diabetic nephropathy [DN], hypertensive nephropathy [HTN], IgA nephritis [IgAN], lupus nephritis [LN], and thin-membrane disease) or histologically unaffected parts of tumor nephrectomies. Renal biopsies from 10 living kidney donors (LDs) served as controls to test for disease-specific regulation. Disease cohort sizes ranged from 4 to 30 patients, with LN (30 patients) and IgAN (24 patients) being the largest subcohorts. The mean age at the time of biopsy was 46±17 years (mean±SD), sex ratio was 90:67 (men/women), and the average eGFR was 70±36 ml/min per 1.73 m2, covering a range from 44 to 101 ml/min per 1.73 m2 (Table 2). The validation cohort, although different in disease composition, had comparable clinical characteristics (Table 2).
Table 1.
List of 16 SNPs associated with CKD originally identified by Köttgen et al.7,8 and candidate genes (Gene Symbol and Entrez GeneID) within a 60-kb range, gene expression correlation with eGFR within the original cohort (n=157) for candidate genes correlated with eGFR (FDR<0.01), and log2 fold change of candidate genes in all patients with CKDs compared with LD controls (FDR was NS)
| SNP/Genes within ±60 kb (Gene Symbol) | Entrez GeneID | eGFR Correlation r Original Cohort (FDR<0.01) | log2 Fold Change All CKD Versus LD (FDR) |
|---|---|---|---|
| rs17319721 | |||
| SHROOM3 | 57619 | ||
| CCDC158 | 339965 | ||
| rs10109414 | |||
| STC1 | 6781 | ||
| rs12917707 | |||
| ACSM5 | 54988 | 0.44 | −0.18 (0.1) |
| GP2 | 2813 | 0.32 | −0.11 (0.11) |
| PDILT | 204474 | ||
| UMOD | 7369 | 0.37 | 0.14 (0.29) |
| rs267734 | |||
| LASS2 | 29956 | 0.39 | −0.08 (0.17) |
| ANXA9 | 8416 | 0.61 | −0.04 (0.28) |
| SETDB1 | 9869 | ||
| FAM63A | 55793 | ||
| BNIPL | 149428 | ||
| PRUNE | 58497 | 0.27 | 0.14 (0.03) |
| rs1260326 | |||
| GCKR | 2646 | ||
| FNDC4 | 64838 | ||
| IFT172 | 26160 | ||
| rs13538 | |||
| ALMS1 | 7840 | −0.38 | 0.11 (0.06) |
| NAT8 | 9027 | 0.29 | 0.11 (NS) |
| NAT8B | 51471 | 0.57 | 0.41 (0.16) |
| rs347685 | |||
| TFDP2 | 7029 | 0.45 | −0.03 (0.27) |
| rs11959928 | |||
| DAB2 | 1601 | 0.28 | 0.13 (0.34) |
| C9 | 735 | ||
| rs6420094 | |||
| SLC34A1 | 6569 | 0.55 | −0.42 (0.06) |
| F12 | 2161 | −0.43 | 0.22 (0.07) |
| GRK6 | 2870 | ||
| LMAN2 | 10960 | −0.38 | 0.18 (0.01) |
| PFN3 | 345456 | ||
| PRR7 | 80758 | ||
| RGS14 | 10636 | ||
| rs881858 | |||
| VEGFA | 7422 | 0.59 | −0.33 (0.01) |
| rs7805747 | |||
| PRKAG2 | 51422 | ||
| rs4744712 | |||
| PIP5K1B | 8395 | 0.3 | −0.14 (0.1) |
| FAM122A | 116224 | ||
| rs653178 | |||
| ATXN2 | 6311 | ||
| rs626277 | |||
| DACH1 | 1602 | ||
| rs1394125 | |||
| UBE2Q2 | 92912 | ||
| FBXO22 | 26263 | 0.27 | −0.04 (0.23) |
| rs12460876 | |||
| TDRD12 | 91646 | ||
| CCDC123 | 84902 | ||
| SLC7A9 | 11136 | 0.39 | −0.18 (0.12) |
Association between eGFR and gene expression levels of 18 candidate genes analyzed further remains significant after correction of age and sex.
Figure 1.
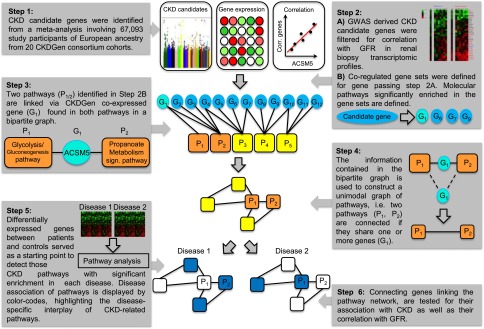
Integration of GFR-associated loci with regulatory pathways allowed for the generation of a molecular map of CKD. Schematic illustration of the strategy to identify CKD-associated pathways and their connections to molecular mechanisms of renal function.
Table 2.
Clinical characteristics of subjects per disease group analyzed by oligonucleotide array-based gene expression profiling and correlation analysis with clinical parameters
| Condition | N | Sex (men/women) | Age (yr) | GFR (ml/min per 1.73 m2) | Proteinuria (g/d) |
|---|---|---|---|---|---|
| Original cohort | |||||
| LD | 10 | 9:1 | 46±13 | 81±16 | <0.2 |
| DN | 17 | 12:5 | 58±11 | 44±25 | 3.1±2.7 |
| FSGS | 16 | 7:9 | 46±18 | 73±38 | 4.4±2.7 |
| HTN | 20 | 15:5 | 57±12 | 44±25 | 1.4±1.5 |
| IgAN | 24 | 18:6 | 36±15 | 76±38 | 2.4±2.4 |
| MCD | 12 | 8:4 | 36±17 | 101±34 | 6.7±5.8 |
| MGN | 18 | 10:8 | 53±19 | 89±41 | 4.6±3.2 |
| LN | 30 | 7:23 | 35±13 | 64±29 | 3.1±3.3 |
| Thin-membrane disease | 6 | 4:2 | 46±14 | 93±29 | 0.5±0.6 |
| Tumor nephrectomy | 4 | 0:4 | 65±7 | 60±10 | <0.2 |
| Total | 157 | 90:67 | 46±17 | 70±36 | 3.1±3.3 |
| Validation cohort | |||||
| FSGS | 4 | 4:0 | 57±13 | 51±32 | 3.3±3.4 |
| IgAN | 6 | 3:3 | 50±14 | 60±35 | 2.2±2.0 |
| Immuntactoid GN | 1 | 1:0 | 71 | 40 | 0.5 |
| LN | 7 | 1:6 | 34±11 | 76±30 | 4.6±2.4 |
| MCD | 1 | 1:0 | 27 | 152 | 0 |
| MGN | 14 | 6:8 | 46±14 | 92±37 | 5.0±4.0 |
| HTN | 2 | 1:1 | 55±14 | 47±47 | 0.5±0.6 |
| HTN/DN | 1 | 1:0 | 71 | 70 | 15 |
| Vasculitis | 1 | 1:0 | 66 | 22 | 2.1 |
| Amyloidosis | 4 | 2:2 | 53±13 | 49±19 | 2.2±2.0 |
| DN | 2 | 1:1 | 50±14 | 39±13 | 6.0±1.7 |
| Total | 43 | 22:21 | 48±15 | 71±38 | 3.9±3.6 |
GFR and proteinuria are displayed as average values±SD.
Tubulointerstitial and glomerular gene expression profiles were used to compute the correlation of log eGFR with the log-transformed steady-state expression levels of 29 candidates within each subject (Figure 1, step 2A). In the tubulointerstitial compartment, 18 of 29 candidate genes were found to significantly correlate with renal function: VEGFA, ANXA9, NAT8B, SLC34A1, TFDP2, ACSM5, SLC7A9, LASS2, FBXO22, UMOD, PIP5K1B, NAT8, GP2, DAB2, ALMS1, LMAN2, PRUNE, and F12 (false discovery rate [FDR] ≤0.01, |r|≥0.25) (Figure 2A). Tubulointerstitial gene expression profiles for these 18 transcripts across all subjects with CKDs versus LD controls are shown in Table 1. Four of these transcripts also passed the significance filter in the glomerular compartment (VEGFA, ANXA9, NAT8B, and SLC34A1), with only DACH1 specific to the glomerular compartment. Therefore, CKDGen candidate genes were enriched for eGFR-correlated genes in the tubulointerstitial compartment compared with a random gene set (z-score for enrichment compared with random datasets: 3.86), and additional analysis focused on the tubulointerstitial gene expression datasets. The directionality of correlation of transcript levels with eGFR was conserved across all diseases.
Figure 2.
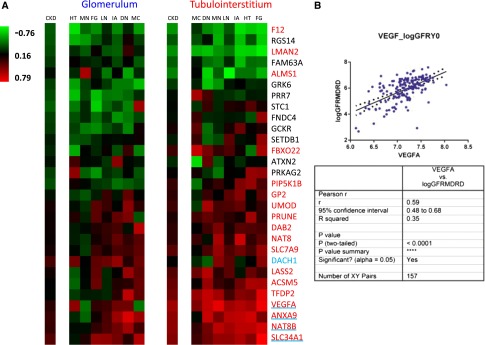
Tubulointerstitial gene expression profiles for 18 CKD candidate genes correlate with log eGFR across all subjects with CKD. (A) Heat map of the correlation of logarithmic eGFR with mRNA expression for 29 CKDGen candidate genes (r values ranging from −0.76 to 0.79). Includes 157 subjects from seven individual CKDs. The CKD column displays the aggregate correlation across all diseases. Glomerulum and tubulointerstitium of renal biopsies were analyzed separately. Gene names in red indicate significance in the tubulointerstitial expression dataset (FDR≤0.01, |r|≥0.25). Blue underlining or blue font indicates significance in the glomerular expression dataset. Red gene names with blue underlining indicate significant coregulation in both renal compartments. FG, FSGS; HT, hypertensive nephropathy; IA, IgA nephritis; LN, lupus nephritis; MC, minimal change disease; MN, membranous GN. (B) Exemplary graph showing the correlation of VEGFA expression with corresponding eGFR data across 157 subjects and statistical assessment.
Gene Coexpression Pathways
The 18 CKDGen-associated and eGFR-correlated candidate genes were evaluated for their functional context using a coexpression strategy (Figure 1, step 2B). For 14 of 18 candidate genes, the following number of transcripts correlated with the expression of the candidate gene (FDR≤0.01, |r|>0.5), providing a basis for the detection of enriched pathways among coexpressed genes: SLC7A9 (1078), VEGFA (988), ACSM5 (925), SLC34A1 (839), NAT8B (811), ANXA9 (690), LASS2 (519), DAB2 (365), NAT8 (283), GP2 (174), TFDP2 (118), UMOD (83), LMAN2 (57), and F12 (22). The resulting 14 coexpressed gene sets show significant enrichment of 147 unique canonical pathways. Of these pathways, 97 pathways were identified in at least two candidate-correlated gene sets.
Association of Pathway Network with CKD
Multiple lines of evidence implicate the pathways constructed from CKDGen candidates in CKD.
Fifty-six of ninety-seven pathways are known to be associated with CKD. Supplemental Table 1 shows these pathways, an exemplar reference, and a brief summary of the proposed mechanism of CKD involvement of that pathway or group of pathways. The 41 pathways not previously associated with CKD are shown in Supplemental Table 2 ranked on the basis of the number of candidate genes connected to the respective pathways.
Because groups of genes can contribute to several disease-relevant pathways, we created a network of all 97 pathways to investigate the inter-relationships among pathways in the context of CKD. In this network, each pathway is represented by a single node, and genes shared between pathways form the edges (connections between nodes). The pathways cluster in a bowtie structure connecting two subclusters with CKD relevance: inflammation-related pathways (e.g., NRF2-mediated oxidative stress response, Cdc42 signaling, and NF-κB signaling) and metabolic pathways (e.g., fatty acid metabolism, glycolysis/gluconeogenesis, and tyrosine metabolism) (Figure 3). Transcripts from these subclusters show differential expression consistent with independent studies where the majority of genes enriched in inflammatory pathways have increased expression in CKD,9 whereas metabolism pathway genes are mainly repressed.10–12 The two subclusters are interconnected by pathways with multiple connecting genes. The pathways with the largest number of neighbors are xenobiotic signaling (53 connections), arylhydrocarbon-receptor signaling (45 connections), and NRF2-mediated oxidative stress response (44 connections) pathways.
The majority of genes shared among pairs of pathways (281 of 309 genes forming edges in the network) displays an expression pattern that correlates with log eGFR (0.26≤|r|≤0.63, FDR<0.01). Protein products of the connecting genes also form a protein–protein interaction network with a higher connectivity than randomized versions of the same network (clustering coefficient=0.27 versus 0.09, P=0.01), which suggests a strong functional interconnection of the encoded proteins (which has previously been shown for Saccharomyces cerevisiae)13 (Supplemental Figure 1).
The pathway network relationships were tested in an independent biopsy gene expression dataset obtained from the Clinical Phenotyping Resource and Biobank Core (C-PROBE) cohort, a North American CKD cohort with different environmental exposures and ethnic backgrounds but similar range of renal impairment (Table 2). Using the same strategy, 78 of 97 pathways (80%) from the original cohort were identified in the independent dataset. The validation study found 127 pathways connected by 341 genes shared among pathways compared with 313 genes shared among pathways in the original network. Among the replicated pathways, 95% of the genes connecting sets of pathways were retained in the independent cohort. The 14 candidate genes that went into additional analysis in the original cohort preserved their directionality of eGFR correlation in the independent validation cohort. Pearson correlation for genes ALMS1, ANXA9, LASS2, NAT8B, and VEGFA achieved statistical significance in this cohort.
Figure 3.
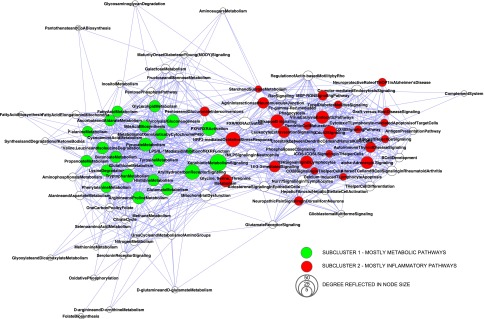
Integration of GFR-associated loci with transcriptional data allowed for the generation of a comprehensive pathway map of CKD. Network graph of 97 CKD-associated pathway nodes connected by transcripts coregulated with CKD candidate genes. Nodes display significantly (P<0.05) enriched pathways derived from two or more lists of coregulated candidate genes, and node size reflects the number of connections to other pathways (degree). A spring-embedded layout is applied, pulling together pathway nodes according to the number of shared genes among pathways. Unbiased cluster analysis of the CKD pathway network separates a subcluster of mainly inflammation-related pathways (red nodes; i.e., NRF2-mediated oxidative stress response, Cdc42 signaling, and NF-κB signaling) and a subcluster of several metabolic pathways (green nodes; i.e., fatty acid metabolism, glycolysis/gluconeogenesis, and tyrosine metabolism). Subclusters were identified by AllegroMCODE, a Cytoscape plug-in, on the original CKD pathway network.
CKD Pathway Networks: Shared and Disease-Specific Features
The pathway network shows that different renal diseases share eGFR-associated pathways. By analyzing differentially regulated genes between the subjects with CKD and tissue from the healthy kidney donors, enriched pathways were detected for each disease. Additional subtle relationships among different glomerular diseases leading to CKD can be identified. The disease–pathway bipartite network (Figure 4) and one-mode projection disease network (Figure 5) compress the information shown in Figure 3 to reveal disease–pathway relationships. Using this approach, the following elements of the disease–pathway network can be displayed. Although none of the pathways are shared among all seven CKDs, the cytotoxic T lymphocyte-mediated apoptosis pathway is shared among FSGS, DN, MGN, IgAN, and LN (Supplemental Figure 2, A–G), and the NRF2-mediated oxidative stress response pathway is shared among DN, HTN, MCD, and MGN. Three diseases show unique pathways (DN: caveolar-mediated endocytosis signaling, crosstalk between dendritic cells and natural killer cells, leukocyte extravasation signaling, mitochondrial dysfunction, phospholipase C signaling, and α-adrenergic signaling; MCD: glioblastoma multiforme signaling; MGN: xenobiotic metabolism signaling), whereas 12 pathways are shared among various sets of three diseases. The most pathways from the CKD pathway network are enriched among differentially regulated genes in DN (31) and LN (22), whereas the fewest pathways were found for FSGS (5) and HTN (1). The strongest relationship among diseases as defined by Jaccard coefficient (defined as the ratio of the number of pathways shared by a set of diseases to the number of pathways associated with all diseases in the set) involves LN with both IgAN and DN (Table 3). These three diseases also share the most pathways of any set of three diseases.
Figure 4.
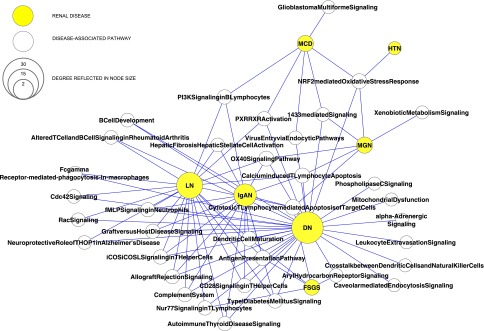
CKD associated pathways are shared between renal diseases. Bipartite network of the relationship between diseases and pathways, where a yellow node represents a disease, an open node represents a pathway, and an edge represents pathway association with a disease. Differentially regulated genes in disease versus control are mapped into pathways. For pathways, the node size reflects the number of diseases showing enrichment for these pathways genes, whereas the node size for diseases reflects the number of pathway members regulated in disease versus control.
Figure 5.

Disease-specific analyses shows a close interconnection between lupus nephritis, IgA nephritis, and diabetic nephropathy. One-mode projection of the relationships between diseases and their associated pathways, where nodes represent the individual diseases and edges represent the number of pathways shared between them. Node size reflects the number of pathways shared with other diseases.
Table 3.
Relationships among diseases as defined by the Jaccard coefficient (defined as ratio of the number of shared pathways for a set of diseases to the number of all pathways for these diseases)
| Disease Count | Diseases | Disease and Disease Group-Specific Pathways | Total Shared Pathways | Jaccard Coefficient |
|---|---|---|---|---|
| 1 | DN | Caveolar-mediated endocytosis signaling, crosstalk between dendritic cells and natural killer cells, leukocyte extravasation signaling, mitochondrial dysfunction, phospholipase C signaling, α-adrenergic signaling | 31 | 1.00 |
| 1 | MCD | Glioblastoma multiforme signaling | 6 | 1.00 |
| 1 | MGN | Xenobiotic metabolism signaling | 6 | 1.00 |
| 2 | IgAN, LN | Hepatic fibrosis hepatic stellate cell activation | 15 | 0.65 |
| 2 | DN, LN | Altered T cell and B cell signaling in rheumatoid arthritis, Cdc42 signaling, Fc-γ-receptor–mediated phagocytosis in macrophages and monocytes, neuroprotective role of THOP1 in Alzheimer's disease, Rac signaling | 20 | 0.59 |
| 2 | DN, IgAN | B cell development | 14 | 0.41 |
| 3 | DN, IgAN, LN | Allograft rejection signaling, autoimmune thyroid disease signaling, CD28 signaling in T-helper cells, complement system, graft-versus-host disease signaling, type 1 diabetes mellitus signaling, fMLP signaling in neutrophils, iCOS/iCOSL signaling in T-helper cells | 13 | 0.38 |
| 2 | DN, FSGS | Aryl hydrocarbon receptor signaling | 5 | 0.16 |
| 2 | DN, MCD | Virus entry through endocytic pathways | 4 | 0.12 |
| 3 | DN, FSGS, LN | Nur77 signaling in T lymphocytes | 4 | 0.11 |
| 4 | DN, FSGS, IgAN, LN | Antigen presentation pathway, dendritic cell maturation | 3 | 0.09 |
| 4 | DN, IgAN, LN, MGN | Calcium-induced T lymphocyte apoptosis, OX40 signaling pathway | 3 | 0.09 |
| 3 | DN, MCD, MGN | 1433-mediated signaling | 2 | 0.06 |
| 3 | IgAN, LN, MCD | PI3K signaling in B lymphocytes | 1 | 0.04 |
| 3 | DN, LN, MCD | PXR/RXR activation | 1 | 0.03 |
| 4 | DN, HTN, MCD, MGN | NRF2-mediated oxidative stress response | 1 | 0.03 |
| 5 | DN, FSGS, IgAN, LN, MGN | Cytotoxic T lymphocyte-mediated apoptosis of target cells | 1 | 0.03 |
Discussion
An Integrative Approach to GWAS Data Analyses
GWASs have identified several genetic loci with highly significant associations with CKD and eGFR. These genetic associations have yet to be translated into a molecular understanding of disease susceptibility (i.e., how the SNPs affect renal function mechanistically). SNPs in complex diseases have been shown to be able to influence protein function14 and mRNA expression.15 Various approaches have yielded insights into molecular mechanisms of CKD (reviews in refs. 16–18), but comprehensive functional integration of genetic CKD risk loci with the biology of CKD is still lacking.
Our study introduces an approach to extend GWAS results by transcriptomic analyses to uncover the rich information hidden in CKD GWAS. Several types of data were integrated to identify a hierarchy relating candidate genes to coexpressed transcripts to function in relationship to CKD (overview in Figure 1).19 The validity of this unbiased approach is supported by the identification of pathways with known CKD associations, providing a rationale to further evaluate the known and novel pathways as targets for therapeutic intervention.
The CKD Pathway Network: Crosstalk among Multiple Molecular Mechanisms
The concept of a single molecular pathway driving a disease process is a construct to facilitate our understanding of biology. In contrast to this concept, comprehensive studies in model organisms show genes and pathways in dense interrelationships, with multiple genes mapping onto multiple pathways.20 We exploit this property in our data by performing a pathway–crosstalk analysis of the gene sets linked to eGFR. Graphic presentation of the network of pathways reveals that the majority of the CKD pathways aggregates in either an inflammation- or a metabolism-related cluster, corresponding to two major hallmarks of CKD.21 Genes from pathways in those clusters display steady-state mRNA expression patterns consistent with previous findings in CKD (i.e., upregulation of inflammatory genes, such as HLA isoforms, TLR1, TLR3, and NFKB1) (review in ref. 22). Multiple eGFR-correlated genes, including RAC1, RAC2, TNFRSF1B, IL1R1, and RXRA, establish links between these two dominant CKD pathway groups.
The structure of the network identifies three pathways with the most neighbors: the xenobiotic signaling pathway, the arylhydrocarbon-receptor signaling pathway, and the NRF2 oxidative stress pathway. The xenobiotic signaling pathway was originally identified as a major player in the elimination of drugs.23 Beyond this metabolic function, additional hypotheses for this pathway in the progression of CKD can be postulated. Transcriptional mechanisms mediating the coordinated downregulation of this pathway have been associated with disease progression in inflammatory bowel disease,24 and similar mechanisms could be involved in progression of CKD.25
The second most interconnected pathway is the arylhydrocarbon-receptor signaling pathway. Arylhydrocarbon receptors are members of the basic helix-loop-helix Per-Arnt-Sim receptor superfamily that are closely related to the xenobiotic signaling pathway, with receptor activation (i.e., by statins) leading to upregulation of xenobiotic pathway members.26,27 In CKD animal models, statins, through the arylhydrocarbon receptor, are involved in the elimination of uremic toxins and have been shown to decrease organ damage.27
The third most interconnected pathway, the NRF2 oxidative stress pathway, links to the relevance of oxidative stress in CKD, particularly in DN.28–30 In general, low concentrations of pro-oxidants have potentially protective effects, because they act as secondary messengers of cell survival.31 Excessive oxidative stress, however, is highly prevalent in patients with CKD and believed to contribute to more traditional cardiovascular risk factors in this patient cohort.28 Targeting NRF2, therefore, is an attractive strategy, and an NRF2 agonist has been studied in phase II and III trials in DN CKD stages 3 of 4. Studies were halted because of an increased cardiovascular mortality before conclusive evidence on the effect on CKD could be generated.32,33
The crosstalk between pathways indicates a plethora of connections between metabolism- and inflammation-related pathways. Because many genes enriched in one of these top three pathways are also enriched in other pathways, they form major hubs in the network and might be capable of affecting the entire regulatory network balance, establishing intriguing therapeutic targets.
Additional evidence for the CKD association for genes connecting pathways within the network is provided by their role as CKD candidate biomarkers (i.e., TIMP-1, FABP1, GGT1, and TNFRSF1B).6 These CKD coregulated genes also interact on a protein level (Supplemental Figure 1), supporting the concept of identifying shared functional concepts by coregulation analysis. Only 1 of the original 14 CKDGen candidates, ACSM5, was also a pathway-connecting gene, highlighting the importance of integrating additional molecular information from CKD to unravel information linked to GWAS candidate genes.
Pathways Specific to Individual Kidney Diseases
The bipartite disease–pathway network reveals relationships between individual renal diseases and CKD pathways (Figure 4), highlighting a key finding of this study: renal diseases show a substantial overlap in gene expression changes, irrespective of the initiating disease mechanism. The observation that the tubulointerstitial compartment shows robust correlation of CKDGen-associated transcripts across renal diseases is consistent with the longstanding knowledge in nephropathology that tubulointerstitial lesions correlate and predict renal function more robustly than glomerular alterations.34 The limited number of pathways common among diseases sharing a clinical pattern (i.e., nephrotic syndrome) might also be explained by the analysis of the tubulointerstitial versus glomerular compartment of renal biopsies. Additional mechanisms and pathways (e.g., for disease initiation and development) might be more similar in the glomerular compartment among nephrotic diseases.
Pathways Not Previously Associated with Kidney Disease
Although an in-depth analysis of 41 pathways not previously associated with CKD is beyond the scope of this work, a ranked list of these pathways on the basis of the number of candidate genes connected to the respective pathways is provided (Supplemental Table 2). This list could prioritize future research on mechanisms of CKD. Among 41 pathways not previously found to be associated with CKD is the hepatic fibrosis/hepatic stellate cell activation pathway, which might be activated in both CKD and hepatic fibrosis. Although the pathway was originally described for transcripts associated with liver disease, core components, like the identified transcripts CTGF, SMAD2, SMAD3, SMAD4, VEGFA, VEGFB, TGFBR1, TGFBR2, and MMP13, have also been found in renal fibrotic disease states.35–37 Elements of the renal fibrosis pathways replicating in independent organ systems can be considered core elements of chronic fibrotic mechanism of human disease.
Twenty-four of forty-one pathways with no previous association to CKD have been linked to specific renal diseases. As shown in Supplemental Figure 2, A–F between 9 and 18 of the pathways not previously implicated in CKD were enriched among transcripts differentially expressed in disease versus controls for specific diseases. Nine pathways (allograft rejection signaling, antigen presentation pathway, cytotoxic T lymphocyte-mediated apoptosis of target cells, graft-versus-host disease signaling, hepatic fibrosis/hepatic stellate cell activation, LPS/IL-1–mediated inhibition of RXR function, OX40 signaling pathway, starch and sucrose metabolism, and type 1 diabetes mellitus signaling) were enriched (P<0.05) among six of seven diseases, supporting the notion that these pathways or several members of these pathways do play a role in renal function decline for specific diseases.
Limitations of the Approach
Our study analyzed genetic association and transcriptomic data from two independent cohorts by filtering candidate genes for significant correlation of their mRNA expression with eGFR in the European Renal cDNA Bank-Kroener-Fresenius Biopsy Bank (ERCB) and C-PROBE cohorts. All study participants in the ERCB cohort were of European descent. The majority (129; 87%) of all renal disease samples for this cohort was collected in study centers in southern Germany and northern Italy; the remaining 18 samples were procured in France, Czech Republic, Ireland, and Spain. No correction for ancestry was performed in our study. The association between gene expression levels and eGFR remained significant after correcting for age and sex in the 18 CKDGen candidate genes studied. The study reported here remains agnostic to how a specific SNP is affecting the mRNA levels found to be correlated with eGFR, and substantial additional work integrating multiple information sources and approaches, like cis-eQTL studies, will be needed to address this question (a recent study in DN genetics is in the work by Martini et al.38). With regard to the limited number of samples in each disease subgroup, our study analyzed the CKD cohort as a whole rather than facilitating adjustments for aforementioned potential confounders. The analysis of pathways shared among specific disease subgroups in the CKD cohort was not corrected for heterogeneity in sample size given the limited sample sizes in each disease subcohort. Diseases with larger sample sizes (i.e., LN) could, therefore, have a greater influence on the bipartite and the mode projection networks presented in Figures 4 and 5.
Of the original 40 CKDGen candidate genes, 29 genes were present on the Affymetrix Chip, excluding the missing genes from our analysis. Newer transcriptomic analysis techniques, such as RNA sequencing, could fill these gaps in the future. It should be noted that the decision to include correlated transcripts of candidate genes passing a cutoff of FDR<0.01 and |r|>0.50 was operationally derived. With this cutoff, each candidate gene generated a list of correlated transcripts of different length and thereby, potentially biased the influence of each candidate gene in identifying pathways. Finally, transcriptional data are a reflection of the regulatory mechanism activated in a given tissue and not a reflection of the functional state of the encoded proteins. Complementing the transcriptional approach with genome-scale proteomic analysis will be an important next step.
The CKD Pathway Network as a Resource for the Renal Research Community
To allow interrogation of the CKD pathway network and the underlying datasets for individual genes and pathways as well as pathway interaction, the data generated in this work are provided in Supplemental Material linked to a searchable interface through the open-source, platform-independent Cytoscape software (version 2.8.3). Using Cytoscape, all visualization and network analysis tools described in Concise Methods can be used. Information on individual pathways, genes that are shared among sets of pathways, and their eGFR correlation values from the entire cohort of 157 subjects can be retrieved through the dynamic interface. Unprocessed gene expression datasets are available in the Gene Expression Omnibus and have also been integrated into the renal search engine Nephromine (www.nephromine.org) for additional systems biology analysis by the renal research community.39
GWAS, intrarenal transcriptional profiles, and biologic knowledge together were able to define a tight pathway–crosstalk activated with impaired renal function. The activation of inflammatory signaling cascades and the loss of metabolite functions provide a wealth of information to be tested for causal relationships in additional experimental studies. Targeting the key regulatory hubs of the interlinked pathways will be a rational therapeutic approach to affect the CKD network at multiple levels.
Concise Methods
The goal of this study was to define CKD-related pathways and the functional connections among them by integrating CKD candidate gene information with transcriptomic and clinical data from 157 subjects with CKDs.
Strategy
In this study, previous observations from GWASs and transcriptomic studies in CKD were extended to integrate currently available GWAS results into their functional context. Three distinct types of data were used: GWAS candidates, renal biopsy-derived transcriptional profiles, and clinical information. Combining these diverse data for all CKDGen candidates allows the construction of a comprehensive network of CKD pathways. Correlation analyses were first used to test GWAS-derived CKD candidate genes for their association with renal function in a kidney biopsy gene expression dataset (Figure 1, step 2A). Pathway annotations were then used to identify functional categories for transcripts correlated with GWAS-derived candidate genes (Figure 1, step 2B). Relationships were captured among significantly enriched pathways and displayed through a network in which pathways are represented by single nodes and transcripts shared by pathway pairs form edges (connections between nodes) (Figure 1, steps 3 and 4). Individual pathways of this network were then interrogated for known associations with CKD. A disease-specific analysis defined the functional interplay of diseases on the basis of enriched pathways among differentially expressed genes of each glomerular disease studied (Figure 1, step 5). Genes forming the edges of the pathway network were tested for their association with CKD as well as their correlation with GFR (Figure 1, step 6).
Selecting Subjects for Human Renal Biopsy
Forty CKD candidate genes were derived from a published meta-analysis that identified susceptibility loci for GFR.7 GWAS candidates were linked with gene expression profiles from the ERCB. Renal transcriptional expression profiles from renal biopsies of 157 subjects were selected representing the following nine forms of CKD: FSGS (n=16), MCD (n=12), MGN (n=18), DN (n=17), IgAN (n=24), LN (n=30), HN (n=20), thin-membrane disease (n=6), and tumor nephrectomy (n=4) as well as controls (LD; n=10). Biopsy specimens were obtained from patients after informed consent and with the approval of the respective local ethics committees. Clinical and histologic characteristics of the subjects are summarized in Table 2. Manual microdissection of glomeruli and tubulointerstitial compartments was performed as previously described.40
RNA Isolation, RNA Preparation, and Microarray Data Analyses
Total RNA from the microdissected glomerular and tubulointerstitial compartments was isolated, reverse transcribed, linearly amplified, and hybridized on Affymetrix HG-U133A and HG-U133Plus2 microarrays.40,41 Affymetrix CEL files were normalized using the robust multi-array average method and annotated at EntrezGene level (HumanEntrez Gene custom CDF annotation, version 10; http://brainarray.mbni.med.umich.edu/Brainarray/default.asp), corrected for batch effect,42 and log2 transformed through the GenePattern pipeline (http://www.GenePattern.org). Underexpressed genes were filtered by applying a cutoff of the median of the non-human Affymetrix control probe sets plus two times their SD.43 Gene expression datasets are available as CEL files in the Gene Expression Omnibus (submission no. GSE47183). In addition, expression datasets have been annotated with clinical information and uploaded into the renal systems biology search engine Nephromine39 to enable individual- and system-level analysis.
Correlation to eGFR
Candidate genes were prioritized by correlation of their expression profiles with log eGFR. In analogy of the analysis performed by CKDGen, eGFR was estimated using the Modification of Diet in Renal Disease equation. For 29 of 40 CKD candidate genes, renal gene expression profiles above background were available; the other 11 genes were not included on the Affymetrix 133 A expression platform. Pearson correlation of gene expression to log eGFR was calculated for these 29 candidate genes across all eight diseases as well as the LD controls to define the linear relationships between gene expression and log eGFR. Correlations with |r|≥0.25 and FDR≤0.01 were considered significant and calculated for both compartments, every disease individually, and all diseases together (Figure 2). Random association of transcripts with log eGFR was tested over 10 random gene sets of size identical to the CKD candidate gene set (n=40) generated using the above threshold parameters and statistically assessed by z score.
Gene Coexpression
A genome-wide expression correlation analysis detected transcripts with expression profiles that are similar to those of the CKDGen candidate genes (Figure 1, step 2). The goal of this step was to link specific transcripts by their coregulated gene set to defined molecular functions in CKD. This concept is on the basis of the observation that mRNA coexpression patterns can result from common upstream transcriptional regulatory events that determine the transcriptional response in the functional context of a disease.43,44 Pearson r correlation of tubulointerstitial compartment expression of each candidate gene was computed with the CKD gene expression dataset. Correlations with |r|≥0.5 and FDR≤0.01 were considered significant.
Analysis of Canonical Pathways
Candidate-correlated transcripts were analyzed for significant (P<0.05) enrichment of canonical pathways and functional groupings using the Ingenuity Pathway Analysis Software Suite. Pathway analysis was conducted separately for each list of correlated transcripts expanded from individual CKD candidate genes (Figure 1, step 3). FBXO22, ALMS1, PRUNE, and PIP5K1B were not included, because each had five or fewer correlated transcripts, limiting the ability to identify associated molecular pathways.45
Pathway Network, Generation, and Analyses
A network of all pathways significantly enriched in at least two sets of candidate gene-correlated transcripts was constructed (Figure 1, step 4). In this network, a node represents a pathway, and an edge between two pathways represents the occurrence of one or more transcripts both correlated with two or more candidate genes and shared between these two pathways. The network graph was visualized in Cytoscape (http://www.cytoscape.org/) using a spring-embedded layout (Figure 3).
After the generation of the network, analysis focused on the pathways of the CKD network, the connecting genes of this network, and the network structure itself. First, the pathway network was evaluated for pathways with known CKD associations using the literature mining tool LitInspector (Genomatix) followed by verification in PubMed. For each result, an exemplar citation as well as a brief description of the proposed mechanism of action in CKD is given in Supplemental Table 1. Pathways that had not previously been implicated in CKD were ranked in descending order according to the number of original candidate genes that the pathway was associated with in Supplemental Table 2.
Second, we tested significantly differentially regulated genes between seven CKDs and LDs for their enrichment in any of 97 pathways in the network. This analysis identifies pathways likely to be involved in disease-specific processes among all CKD-associated pathways that are responsible for the development and progression of a particular CKD.
Third, we focused on the connecting genes of the CKD pathway network. These genes connect two or more pathways within the network, because they are correlated with original CKD candidate genes and also enriched in two or more pathways of the network. We tested the hypothesis that transcripts connecting pathways in the network play an important role in the pathogenesis of CKD. We calculated their correlation with log eGFR and tested whether pathway connecting transcripts were found among the CKD biomarkers as described in the review by Fassett et al.6
Fourth, we constructed a protein–protein interaction network using the Cytoscape MiMI plug-in from all 309 connecting genes (settings: all databases, human)46 and assessed the functional connections among the molecules of this network by comparing against a randomized version of the network (random networks generated and compared by Cytoscape Random Networks plug-in).
Fifth, we tested the hypothesis that biologically related pathways aggregate in the pathway network with an unbiased cluster analysis of the network using the Cytoscape plug-in Allegro mCode (default settings; analysis on the complete pathway network). This cluster analysis identifies strongly connected subnetworks, defining groups of possibly functionally related pathways. Gene expression levels of candidate-correlated transcripts that connect pathways within the resulting two subclusters were then compared separately between 153 subjects with CKDs and 32 healthy LD controls by SAM analysis in Tigr MeV to identify differential regulation of genes with the goal of detecting expression patterns reflecting established CKD pathomechanism.
The CKD pathway network and its detailed pathway and eGFR correlation information are available through a downloadable Cytoscape network file (Supplemental Material). Cytoscape is a multiplatform, open-source software, enabling researchers to scrutinize our data for their molecule(s) and/or pathway(s) of interest.47
CKD Pathway Validation
Pathway networks were validated in an independent CKD cohort of 43 subjects from the C-PROBE study cohort (Table 2). We followed the same procedure starting with the initial set of candidate genes. The overlap of pathways retrieved for each cohort was compared to assess reproducibility of our approach.
Disclosures
None.
Supplementary Material
Acknowledgments
We acknowledge the great efforts of all contributors of the CKDGen consortium. We also thank all participating centers of the European Renal cDNA Bank-Kroener-Fresenius Biopsy Bank (ERCB-KFB) and the Clinical Phenotyping Resource and Biobank Core (C-PROBE) and their participants for their cooperation. The expert technical assistance of Anna Henger is gratefully acknowledged. We thank Chrysta C. Lienczewski and Tennille S. Leak-Johnson for carefully proofreading the manuscript.
This study was supported, in part, by O’Brien Renal Center National Institutes of Health Grant P30-DK081943-01, National Institutes of Health Grant R01-DK079912-01, and National Center for Integrative Bioinformatics National Institutes of Health Grant U54-DA021519 01A1 (to M.K.). C.A.B. was supported by Else Kröner-Fresenius-Stiftung Grants Nr P48/08//A11/08 and 2012_A147.
Parts of this study were presented at the Keystone Meeting “Complex Traits: Genomics and Computational Approaches” in Beckenridge, CO on February 20–25, 2012.
The contribution of V.N. in this study is, in part, to fulfill the thesis requirement at the Ludwig Maximilian University of Munich. M.K. is the guarantor of this work and as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Active members of the ERCB-KFB at the time of the study: Clemens David Cohen, Holger Schmid, Michael Fischereder, Lutz Weber, Walter Samtleben, Matthias Kretzler, and Detlef Schlöndorff (Ludwig-Maximilians University, Munich, Germany); Jean Daniel Sraer and Pierre Ronco (Tenon Hospital, Paris, France); Maria Pia Rastaldi and Giuseppe D'Amico (San Carlo Borromeo Hospital, Milan, Italy); Peter Doran and Hugh Brady (University College Dublin, Dublin, Ireland); Detlev Mönks and Christoph Wanner (University of Würzburg, Germany); Andrew Rees (University of Aberdeen, UK); Frank Strutz and Gerhard Anton Müller (University of Göttingen, Germany); Peter Mertens and Jürgen Floege (Rheinisch-Westfälische Technische Hochschule Aachen University, Germany); Norbert Braun and Teut Risler (University of Tübingen, Germany); Loreto Gesualdo and Francesco Paolo Schena (University of Bari, Italy); Jens Gerth and Gunter Wolf (University of Jena, Germany); Rainer Oberbauer and Dontscho Kerjaschki (University of Vienna, Austria); Bernhard Banas and Bernhard Krämer (University of Regensburg, Germany); Moin Saleem (University of Bristol, UK); Rudolf Wüthrich (University of Zürich, Switzerland); Harm Peters and Hans-Hellmut Neumayer (Charite, Berlin, Germany); Mohamed Daha (University of Leiden, Netherlands); Katrin Ivens and Bernd Grabensee (University of Düsseldorf, Germany); Francisco Mampaso (University of Madrid, Spain); Jun Oh, Franz Schaefer, and Martin Zeier (University of Heidelberg, Germany); Hermann-Joseph Gröne (German Cancer Research Center [DKFZ], Heidelberg, Germany); Peter Gross (University of Dresden, Germany); Giancarlo Tonolo (University of Sassari, Italy); Vladimir Tesar (University of Prag, Czech Republic); Harald Rupprecht (Klinikum Bayreuth, Germany); Hermann Pavenstädt (University of Münster, Germany); and Hans-Peter Marti (University of Bern, Switzerland).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013080906/-/DCSupplemental.
References
- 1.US Renal Data System : USRDS 2010 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, Bethesda, MD, US Renal Data System, 2010 [Google Scholar]
- 2.Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D: Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 24: 1506–1523, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Sharma S, Sirin Y, Susztak K: The story of Notch and chronic kidney disease. Curr Opin Nephrol Hypertens 20: 56–61, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ly J, Alexander M, Quaggin SE: A podocentric view of nephrology. Curr Opin Nephrol Hypertens 13: 299–305, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Zoja C, Abbate M, Remuzzi G: Progression of chronic kidney disease: Insights from animal models. Curr Opin Nephrol Hypertens 15: 250–257, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Fassett RG, Venuthurupalli SK, Gobe GC, Coombes JS, Cooper MA, Hoy WE: Biomarkers in chronic kidney disease: A review. Kidney Int 80: 806–821, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Köttgen A, Glazer NL, Dehghan A, Hwang SJ, Katz R, Li M, Yang Q, Gudnason V, Launer LJ, Harris TB, Smith AV, Arking DE, Astor BC, Boerwinkle E, Ehret GB, Ruczinski I, Scharpf RB, Chen YD, de Boer IH, Haritunians T, Lumley T, Sarnak M, Siscovick D, Benjamin EJ, Levy D, Upadhyay A, Aulchenko YS, Hofman A, Rivadeneira F, Uitterlinden AG, van Duijn CM, Chasman DI, Paré G, Ridker PM, Kao WH, Witteman JC, Coresh J, Shlipak MG, Fox CS: Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet 41: 712–717, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Köttgen A, Pattaro C, Böger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, O’Connell JR, Li M, Schmidt H, Tanaka T, Isaacs A, Ketkar S, Hwang SJ, Johnson AD, Dehghan A, Teumer A, Paré G, Atkinson EJ, Zeller T, Lohman K, Cornelis MC, Probst-Hensch NM, Kronenberg F, Tönjes A, Hayward C, Aspelund T, Eiriksdottir G, Launer LJ, Harris TB, Rampersaud E, Mitchell BD, Arking DE, Boerwinkle E, Struchalin M, Cavalieri M, Singleton A, Giallauria F, Metter J, de Boer IH, Haritunians T, Lumley T, Siscovick D, Psaty BM, Zillikens MC, Oostra BA, Feitosa M, Province M, de Andrade M, Turner ST, Schillert A, Ziegler A, Wild PS, Schnabel RB, Wilde S, Munzel TF, Leak TS, Illig T, Klopp N, Meisinger C, Wichmann HE, Koenig W, Zgaga L, Zemunik T, Kolcic I, Minelli C, Hu FB, Johansson A, Igl W, Zaboli G, Wild SH, Wright AF, Campbell H, Ellinghaus D, Schreiber S, Aulchenko YS, Felix JF, Rivadeneira F, Uitterlinden AG, Hofman A, Imboden M, Nitsch D, Brandstätter A, Kollerits B, Kedenko L, Mägi R, Stumvoll M, Kovacs P, Boban M, Campbell S, Endlich K, Völzke H, Kroemer HK, Nauck M, Völker U, Polasek O, Vitart V, Badola S, Parker AN, Ridker PM, Kardia SL, Blankenberg S, Liu Y, Curhan GC, Franke A, Rochat T, Paulweber B, Prokopenko I, Wang W, Gudnason V, Shuldiner AR, Coresh J, Schmidt R, Ferrucci L, Shlipak MG, van Duijn CM, Borecki I, Krämer BK, Rudan I, Gyllensten U, Wilson JF, Witteman JC, Pramstaller PP, Rettig R, Hastie N, Chasman DI, Kao WH, Heid IM, Fox CS: New loci associated with kidney function and chronic kidney disease. Nat Genet 42: 376–384, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodríguez-Iturbe B, García García G: The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin Pract 116: c81–c88, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Leblond FA, Giroux L, Villeneuve JP, Pichette V: Decreased in vivo metabolism of drugs in chronic renal failure. Drug Metab Dispos 28: 1317–1320, 2000 [PubMed] [Google Scholar]
- 11.Naud J, Michaud J, Beauchemin S, Hébert MJ, Roger M, Lefrancois S, Leblond FA, Pichette V: Effects of chronic renal failure on kidney drug transporters and cytochrome P450 in rats. Drug Metab Dispos 39: 1363–1369, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Vaziri ND: Dyslipidemia of chronic renal failure: The nature, mechanisms, and potential consequences. Am J Physiol Renal Physiol 290: F262–F272, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Song J, Singh M: How and when should interactome-derived clusters be used to predict functional modules and protein function? Bioinformatics 25: 3143–3150, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sauna ZE, Kimchi-Sarfaty C: Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet 12: 683–691, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, Lee MP: Allelic variation in gene expression is common in the human genome. Genome Res 13: 1855–1862, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohtat D, Susztak K: Fine tuning gene expression: The epigenome. Semin Nephrol 30: 468–476, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perco P, Oberbauer R: Integrative analysis of -omics data and histologic scoring in renal disease and transplantation: Renal histogenomics. Semin Nephrol 30: 520–530, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spasovski G, Ortiz A, Vanholder R, El Nahas M: Proteomics in chronic kidney disease: The issues clinical nephrologists need an answer for. Proteomics Clin Appl 5: 233–240, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Keller BJ, Martini S, Sedor JR, Kretzler M: A systems view of genetics in chronic kidney disease. Kidney Int 81: 14–21, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Li M, Bucan M: Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet 81: 1278–1283, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mak RH, Cheung W, Cone RD, Marks DL: Mechanisms of disease: Cytokine and adipokine signaling in uremic cachexia. Nat Clin Pract Nephrol 2: 527–534, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Slee AD: Exploring metabolic dysfunction in chronic kidney disease. Nutr Metab (Lond) 9: 36, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vondrácek J, Umannová L, Machala M: Interactions of the aryl hydrocarbon receptor with inflammatory mediators: Beyond CYP1A regulation. Curr Drug Metab 12: 89–103, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Reiff C, Delday M, Rucklidge G, Reid M, Duncan G, Wohlgemuth S, Hörmannsperger G, Loh G, Blaut M, Collie-Duguid E, Haller D, Kelly D: Balancing inflammatory, lipid, and xenobiotic signaling pathways by VSL#3, a biotherapeutic agent, in the treatment of inflammatory bowel disease. Inflamm Bowel Dis 15: 1721–1736, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Handschin C, Podvinec M, Stöckli J, Hoffmann K, Meyer UA: Conservation of signaling pathways of xenobiotic-sensing orphan nuclear receptors, chicken xenobiotic receptor, constitutive androstane receptor, and pregnane X receptor, from birds to humans. Mol Endocrinol 15: 1571–1585, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA: Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A 93: 6731–6736, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki T, Toyohara T, Akiyama Y, Takeuchi Y, Mishima E, Suzuki C, Ito S, Soga T, Abe T: Transcriptional regulation of organic anion transporting polypeptide SLCO4C1 as a new therapeutic modality to prevent chronic kidney disease. J Pharm Sci 100: 3696–3707, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Del Vecchio L, Locatelli F, Carini M: What we know about oxidative stress in patients with chronic kidney disease on dialysis—clinical effects, potential treatment, and prevention. Semin Dial 24: 56–64, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Granata S, Zaza G, Simone S, Villani G, Latorre D, Pontrelli P, Carella M, Schena FP, Grandaliano G, Pertosa G: Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics 10: 388, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuchta A, Pacanis A, Kortas-Stempak B, Cwiklińska A, Ziętkiewicz M, Renke M, Rutkowski B: Estimation of oxidative stress markers in chronic kidney disease. Kidney Blood Press Res 34: 12–19, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Khan HY, Zubair H, Ullah MF, Ahmad A, Hadi SM: A prooxidant mechanism for the anticancer and chemopreventive properties of plant polyphenols. Curr Drug Targets 13: 1738–1749, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG, BEAM Study Investigators : Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365: 327–336, 2011 [DOI] [PubMed] [Google Scholar]
- 33.Zoja C, Benigni A, Remuzzi G: The Nrf2 pathway in the progression of renal disease. Nephrol Dial Transplant 29[Suppl 1]: S19–S24, 2014 [DOI] [PubMed] [Google Scholar]
- 34.Bohle A, Mackensen-Haen S, von Gise H, Grund KE, Wehrmann M, Batz C, Bogenschütz O, Schmitt H, Nagy J, Müller C: The consequences of tubulo-interstitial changes for renal function in glomerulopathies. A morphometric and cytological analysis. Pathol Res Pract 186: 135–144, 1990 [DOI] [PubMed] [Google Scholar]
- 35.Samarakoon R, Dobberfuhl AD, Cooley C, Overstreet JM, Patel S, Goldschmeding R, Meldrum KK, Higgins PJ: Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal 25: 2198–2209, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Berthier CC, Bethunaickan R, Gonzalez-Rivera T, Nair V, Ramanujam M, Zhang W, Bottinger EP, Segerer S, Lindenmeyer M, Cohen CD, Davidson A, Kretzler M: Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol 189: 988–1001, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodgin JB, Nair V, Zhang H, Randolph A, Harris RC, Nelson RG, Weil EJ, Cavalcoli JD, Patel JM, Brosius FC, 3rd, Kretzler M: Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 62: 299–308, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martini S, Nair V, Patel SR, Eichinger F, Nelson RG, Weil EJ, Pezzolesi MG, Krolewski AS, Randolph A, Keller BJ, Werner T, Kretzler M: From single nucleotide polymorphism to transcriptional mechanism: A model for FRMD3 in diabetic nephropathy. Diabetes 62: 2605–2612, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martini S, Eichinger F, Nair V, Kretzler M: Defining human diabetic nephropathy on the molecular level: Integration of transcriptomic profiles with biological knowledge. Rev Endocr Metab Disord 9: 267–274, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen CD, Frach K, Schlöndorff D, Kretzler M: Quantitative gene expression analysis in renal biopsies: A novel protocol for a high-throughput multicenter application. Kidney Int 61: 133–140, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Schmid H, Boucherot A, Yasuda Y, Henger A, Brunner B, Eichinger F, Nitsche A, Kiss E, Bleich M, Gröne HJ, Nelson PJ, Schlöndorff D, Cohen CD, Kretzler M, European Renal cDNA Bank (ERCB) Consortium : Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 55: 2993–3003, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Johnson WE, Li C, Rabinovic A: Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8: 118–127, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, Kaplan MJ: The detrimental effects of IFN-α on vasculogenesis in lupus are mediated by repression of IL-1 pathways: Potential role in atherogenesis and renal vascular rarefaction. J Immunol 185: 4457–4469, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fessele S, Maier H, Zischek C, Nelson PJ, Werner T: Regulatory context is a crucial part of gene function. Trends Genet 18: 60–63, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Huang W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tarcea VG, Weymouth T, Ade A, Bookvich A, Gao J, Mahavisno V, Wright Z, Chapman A, Jayapandian M, Ozgür A, Tian Y, Cavalcoli J, Mirel B, Patel J, Radev D, Athey B, States D, Jagadish HV: Michigan molecular interactions r2: From interacting proteins to pathways. Nucleic Acids Res 37: D642–D646, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang PL, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ, Ideker T, Bader GD: Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.