

Abstract
Autosomal dominant polycystic kidney disease is a genetic disorder associated with substantial variability in its natural course within and between affected families. Understanding predictors for rapid progression of this disease has become increasingly important with the emergence of potential new treatments. This systematic review of the literature since 1988 evaluates factors that may predict and/or effect autosomal dominant polycystic kidney disease progression. Predicting factors associated with early adverse structural and/or functional outcomes are considered. These factors include PKD1 mutation (particularly truncating mutation), men, early onset of hypertension, early and frequent gross hematuria, and among women, three or more pregnancies. Increases in total kidney volume and decreases in GFR and renal blood flow greater than expected for a given age also signify rapid disease progression. Concerning laboratory markers include overt proteinuria, macroalbuminuria, and perhaps, elevated serum copeptin levels in affected adults. These factors and others may help to identify patients with autosomal dominant polycystic kidney disease who are most likely to benefit from early intervention with novel treatments.
Keywords: autosomal dominant polycystic kidney disease, disease progression, PKD1/PKD2, hypertension, total kidney volume, renal blood flow
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder caused by mutations in the PKD1 gene located on chromosome 16p13.3 or the PKD2 gene located on chromosome 4q21.1 PKD1 and PKD2 encode the proteins polycystin-1 and -2, respectively; mutations disrupt the function of these proteins on the primary cilium, forming fluid-filled cysts that progressively increase in size, leading to gross enlargement of the kidneys and distortion of the renal architecture.2 Glomerular hyperfiltration compensates for the progressive loss of healthy glomeruli, and therefore, by the time GFR decline becomes detectable, as much as one half of the original functional glomeruli are irreversibly lost.3,4 The majority of patients with ADPKD ultimately progress to ESRD.3
The natural course of ADPKD varies significantly, with onset of ESRD reported from childhood to age >80 years, and a median age of 58 years was reported recently for PKD1, which is much more common than PKD2.5 Rapid ADPKD progression may be defined as onset of ESRD at age <55 years, development of stage 3 CKD at <40 years old, onset of hypertension at <18 years old, presence of total kidney volume (TKV) greater than that expected for a given age, or presence of multiple complications. Identification of patients at high risk for rapid progression has become increasingly important given the emergence of potential new treatments. These treatments are most likely to be beneficial when started early in the disease course. This systematic review evaluates markers of and factors contributing to ADPKD progression.
Methods
A comprehensive systematic review of the literature was undertaken using several structured steps (Table 1). All authors participated in each step of the review process and ranking of final articles for inclusion.
Table 1.
Summary of systematic review process
| Step | Procedure | Result |
|---|---|---|
| 1 | Systematic literature review through Medline, EMBASE, and Biosis for articles published from January of 1988 to May of 2013a | 2056 citations |
| 2 | Review abstracts; omit articles not dealing with factors affecting ADPKD severity or progression, reviews, case reports, editorials, commentaries, and letters | 1078 citations |
| 3 | Omit non-English language articles and articles with focus on transplantation, dialysis, or diabetes, with ESRD used as a cutoff for inclusion | 863 citations |
| 4 | Omit duplicate references; results separated into four categories: randomized clinical trials; other clinical trials; observational, retrospective, or epidemiologic studies; and preclinical studies | 666 articles ranked for inclusionb |
The searches comprised “ADPKD or autosomal dominant polycystic kidney disease” combined with the following terms: angiogenic growth factor; anxiety, depression, or quality of life; child or pediatric; cyst (number or area) or liver fibrosis index; cyst segmentation; depression and dialysis; diabetes mellitus; epidemiology, hospitalization, or cost; ESRD or renal disease; family history, genetic or allelic heterogeneity, genotype–phenotype correlation, mutation type or position, polycystin, vasopressin, or cAMP; glomerular hyperfiltration; HALT PKD; hematuria; hypertension; intracellular calcium, cell proliferation, or fluid secretion; kidney, liver, or renal disease progression; left ventricular mass hypertrophy; molecular diagnostics; proteinuria or microalbuminuria; psychonephrology; renal blood flow, urine osmolality, biomarker, proteome, copeptin, exosome, or serum uric acid; renal or kidney ultrasonography, computed tomography, or MRI; TKV or height-adjusted TKV; total liver volume; UTI; and vitamin D.
Additional references were identified by the authors on the basis of their personal knowledge of the literature that reported specific information important to ADPKD progression.
Renal Disease Progression in ADPKD
Genetic Factors
A summary of the main studies examining genetic predictors of rapid renal disease progression is provided in Table 2.
Table 2.
Genetic predictors of rapid ADPKD progression: phenotypic differences between PKD1 and PKD2 mutations
| Ref./Findings | PKD1 | PKD2 | P Value |
|---|---|---|---|
| 5 | |||
| Patients, n | 392 | 95 | |
| Mean age at hypertension diagnosis, yr | 39 | 49 | <0.001 |
| Median age at ESRD onset, yr | 58 | 79 | <0.001 |
| 6 | |||
| Patients, n | 146 | 20 | |
| Mean age at diagnosis of ADPKD, yr | 27 | 41 | <0.001 |
| Mean age at diagnosis of hypertension, yr | 35 | 50 | 0.001 |
| Mean age at onset of ESRD, yr | 54 | 73 | <0.001 |
| 7 | |||
| Patients, n | 287 | 34 | |
| Median age at onset of ESRD, yr | 53 | 68 | <0.001 |
| 8 | |||
| Patients, n | 333 | 291 | |
| Median age at diagnosis of ADPKD caused by symptoms, yr | 42 | 56 | NA |
| Median age at death/ESRD, yr | 53 | 69 | <0.001 |
| Median age at ESRD, yr | 54 | 74 | |
| 9 | |||
| Patients, n | 136 | 60 | |
| Median age at hypertension treatment, yr | 46 | 51 | NA |
| Median age at stage 3 CKD onset, yr | 50 | 66 | NA |
| Median age at ESRD onset, yr | 53 | Only 21% had ESRD by age 70 yr | NA |
| Median age at death, yr | 67 | 71 | NA |
| 10 | |||
| Patients, n | 185 | 34 | |
| Findings | Patients with PKD1 had significantly larger kidneys and more cysts versus patients with PKD2 mutations; they also had significantly higher frequency of hypertension/higher urinary albumin excretion | ||
| 11 | |||
| Patients, n | 50 | 10 | |
| Findings | Children with PKD1 had significantly larger kidneys with more and larger cysts than children with PKD2 (mean age of 8.6 versus 8.9 yr); they also had significantly higher daytime/nighttime systolic BP by ambulatory monitoring over 24 h | ||
NA, not available.
Locus Heterogeneity
Several large studies have shown that patients with mutations in PKD1 generally have a more severe form of ADPKD than patients with PKD2 mutations, with a younger age at diagnosis, a higher number of cysts, earlier onset of hypertension, and faster progression to ESRD.5–11 In the European PKD1-PKD2 study cohort of 624 patients, median ages at death or ESRD onset in patients with PKD1 and PKD2 were 53 and 69 years, respectively (P<0.001).8 Similarly, in the recent large Genkys study of 741 patients with ADPKD from northwestern France, median ages at onset of ESRD in PKD1 and PKD2 carriers were 58 and 79 years, respectively.5 Some PKD1 families, however, have mild disease that is indistinguishable from PKD2 families, whereas other PKD1 families have a particularly severe course.3,5 In contrast to different gene loci, this observation may be caused by allelic heterogeneity within the PKD1 locus.
Allele Heterogeneity: Mutation Type and Location
Results from the Genkys study showed that truncating PKD1 mutations (frameshift, nonsense, splice mutations, and large rearrangements), affecting approximately two thirds of PKD1 families, were associated with a significantly younger median age of ESRD onset than nontruncating mutations (in-frame and missense mutations; 55.6 versus 67.9 years [P<0.001]).5 In another study, mutations toward the 5′ end of PKD1 were associated with a vascular phenotype and ruptured intracranial aneurysms.12
Mutation location within the PKD2 gene has also been suggested to influence outcome in a study of 22 PKD2 families13; however, this result was not confirmed in a larger study of 461 subjects from 71 PKD2 families.14 In the latter study, after adjustment for sex, patients with splice mutations tended to have better renal survival than patients with other types of mutations.
Hypomorphic Alleles
Several families have been described with a mild or atypical disease presentation, in which ADPKD is not explained by the dominant inheritance of a single PKD1 or PKD2 mutation.15–18 In these families, sequence variants function as hypomorphic alleles, because they do not result in the full phenotype when occurring as the only abnormality in heterozygous subjects, but they do result in severe disease in homozygous (for this variant) individuals or when occurring together with a truncating mutation on the second allele.16–18 For example, a unique ADPKD family has been described in which 30 affected individuals had mild disease but only 14 individuals carried a PKD2 mutation, whereas the other 16 individuals did not have this mutation but rather a sequence (Y528C missense) variant in PKD1.15 Expression of this variant in cultured cells confirmed its pathogenicity. Two family members carried both the PKD2 mutation and PKD1 variant, resulting in more severe disease than observed in carriers of the PKD2 mutation alone. Mosaicism for a PKD mutation may also account for an atypical mild phenotype.19
Modifier Genes
Highly variable disease severity occurs even within families, which cannot be explained by locus or allelic heterogeneity.3,14,20 Intrafamilial variability may be caused by other genes that modify disease severity.20 Several gene polymorphisms (e.g., in the angiotensin-converting enzyme, endothelial nitric oxide synthase, and α8 integrin genes) have been associated with age at ESRD onset, although usually in men and not women.21–24 Many of these reports were, however, based on small study populations with lack of correction for population stratification. Additional insight regarding the identity of genes that modify the severity of ADPKD will be provided by the outcome of ongoing large genome-wide association studies. It is also significant that PKD1 mutations may modify the disease course in PKD2 carriers with resultant earlier onset of ESRD, which was described in a case report of a de novo PKD1 splice mutation on the background of a homozygous PKD2 variant.25
Demographic and Familial Factors
A summary of the main studies examining demographic and familial predictors of rapid renal disease progression is provided in Table 3.
Table 3.
Demographic, familial, clinical, and imaging predictors of rapid renal disease progression in ADPKD
| Predictor/Cohort Size (No. of Patients) | Type of Observational Study | Major Supporting Refs. |
|---|---|---|
| Sex | ||
| 1215 | Retrospective | 7 |
| 580 | Retrospective | 26 |
| 100 | Prospective | 40 |
| Race | ||
| 72 | Retrospective | 28 |
| Hyperfiltration in childhood | ||
| 180 | Retrospective | 32 |
| Gross hematuria | ||
| 1215 | Retrospective | 7 |
| 580 | Retrospective | 26 |
| 191 | Retrospective | 35 |
| 180 | Retrospective | 36 |
| 100 | Prospective | 40 |
| Pregnancy | ||
| 235 | Retrospective | 37 |
| Multiple UTIs | ||
| 256 | Retrospective | 39 |
| 180 | Retrospective | 36 |
| 94 | Retrospective | 38 |
| Cyst rupture | ||
| 100 | Prospective | 40 |
| Hypertension | ||
| 2085 | Retrospective | 70 |
| 1215 | Retrospective | 7 |
| 580 | Retrospective | 26 |
| 543 | Retrospective | 73 |
| 323 | Prospective | 48 |
| 235 | Retrospective | 37 |
| TKV | ||
| 312 | Prospective | 54 |
| 241 | Prospective | 4 |
| 229 | Prospective | 55 |
| RBF | ||
| 241 | Prospective | 58 |
| 59 |
P values for all studies were <0.01.
Sex
Several studies reported more severe disease in men than women, with earlier onset of hypertension, more severe hypertension, and earlier onset of ESRD.5–7,14,26,27 In one large retrospective cohort study of 580 patients with ADPKD who were followed for 25 years, men had worse renal function at a given age.26 In a second retrospective study of 1215 patients with ADPKD, median age at onset of ESRD was 4 years younger in men versus women (52 versus 56 years).7 Because none of these studies were population-based, the true sex effect remains unknown.
Race
ADPKD occurs in all races, but differences in severity between races have not been well studied. In one small study, African Americans with ADPKD had significantly earlier onset of ESRD than whites (age 43.2 versus 55.4 years), and African Americans with both ADPKD and sickle cell trait reached ESRD even earlier than African Americans with ADPKD alone (38 versus 48 years).28 More studies are needed to define the effect of race and ethnicity on ADPKD progression.
Parental History of Hypertension
Two studies have evaluated the effects of parental hypertension on ADPKD severity in offspring. In a European study of 162 subjects, hypertension in the non-ADPKD parent (versus normotension) was associated with ESRD at a significantly younger age in both male and female offspring (49 versus 54 years).29 Another study from the University of Colorado reported that hypertension in the parent with ADPKD was associated with a higher frequency of hypertension in affected children as well as younger age at diagnosis of hypertension, independent of kidney volume and renal function.30 Effects on ESRD onset in offspring were not examined.
Family History of Early ESRD
One study has explored whether family history of ESRD predicts the mutated gene and hence, the risk of disease progression. Barua et al.31 used a large population of 484 patients with ADPKD with known genotype and renal function data (including eGFR and age at ESRD) from 90 ADPKD pedigrees. The investigators found that the presence of one or more affected family members who developed ESRD at age ≤55 years was highly predictive of the PKD1 genotype (positive predictive value=100%; sensitivity=72%). In contrast, the presence of one or more affected family member who had adequate kidney function or developed ESRD at age ≥70 years was highly predictive of the PKD2 genotype (positive predictive value=100%; sensitivity=74%). The study provided a simple but effective prognostic tool by investigating carefully the history of renal function in the pedigree under analysis. Limitations of the study included the small population (only 90 pedigrees); the need to have a detailed family history, which limited the use of the approach for small pedigrees or de novo cases; and the impossibility of predicting age at ESRD in other family members given the overall high intrafamilial variability observed in ADPKD pedigrees.
Clinical Factors
The major studies reporting clinical and imaging characteristics predictive of rapid renal disease progression are summarized in Table 4.
Table 4.
Summary of the most relevant prospective and cross-sectional/retrospective studies where predictors and outcomes of rapid ADPKD progression were analyzed and validated
| Patients, N | Baseline or Major Patient Characteristics | Follow-Up Duration, Other Study Features, or Study Type/Summary | Predictors and Outcome (OM) | Refs. |
|---|---|---|---|---|
| Prospective observational studies | ||||
| 241 | Age 15–45 yr; CrCl>70 ml/min; other conditions affecting renal function (e.g., diabetes) excluded | Mean follow-up: 3,4,57 7.9,55 8.5,79 and 6 yr82 | TKV, RBF, serum copeptin, total cyst volume, genotype, percent cystic volume; OM: decline in eGFR (P<0.001) | 4,10,56–59,83,86 (CRISP) |
| 196 | Mean (SD) age (PKD1 and PKD2): 27±14 and 40±14 yr | Follow-up: 22 yr | PKD1 and PKD2; OM: age at onset of stage 3 CKD | 9 |
| 100 | Mean (SD) age 31.2±6.4 yr; mean (SD) estimated CrCl (Cockcroft–Gault): 109.8±25.5 ml/min | Follow-up: 6 mo | TKV and TCV change detectable at 6 mo | 40 |
| 140 | Children, mean age 11 yr (range=5 mo to 18 yr) | Follow-up: 2–5 yr | Increased cysts and KV; OM: disease severity measured by flank, back, or abdominal pain, HTN, inguinal hernia, palpitation, frequency, maximum urine osmolality, GFR (P<0.05) | 43 |
| 323 | Mean (SD) age 53±15 yr | Mean (SD) follow-up 100±38 mo (n=198) | Age, HTN, urinary stones, proteinuria, smoking; OM: disease progression, eGFR decline (P<0.05) | 48 |
| 312 | Children (<18 yr) | Mean follow-up 5.7 yr (range=3–15) for 115 of 185 affected children | Baseline and follow-up KV (mean of two kidneys), cysts; OM: disease severity (P<0.01) | 54 |
| 229 | Mean (SD) age 37±11 yr; mean (SD) eGFR=71±22 ml/min per 1.73 m2 (range=20–152) | Mean follow-up 7.8 yr (range=2.6–15.1) | Increased TKV; OM: eGFR decline (P<0.001) | 55 |
| 154 | Children; mean (SEM) age 9.6±0.5 yr (range=8 mo to 17 yr) | Mean 11 BP measurements, routine echocardiography | HTN; OM: LVMI (P<0.001) | 69 |
| 200 (ADPKD subset of MDRD) | Measured GFR=13–55 ml/min per 1.73 m2 | Secondary subgroup analysis of randomized controlled trial; mean follow-up 2.2 yr | Greater sCr, proteinuria, HTN, young age, low protein diet; OM: renal function decline | 78 |
| 209 (CRISP) | Mean age 32.1 yr; mean eGFR=89.9 ml/min per 1.73 m2 | Baseline and follow-up measurement at 1, 2, and 3 yr | NGAL, IL-8; OM: eGFR and TKV | 93 |
| 19 | Postmenopausal women; Cr<2.5 mg/dl; lack of other serious medical conditions | Postmenopausal estrogen use (n=11) versus no use (n=8); follow-up 1 yr (observational) | Postmenopausal estrogen use; OM: liver volume by CT scan (P<0.03) | 109 |
| Randomized interventional studies | ||||
| 72 | HTN and LVH; age 20–60 yr; CrCl>30 ml/min per 1.73 m2 | Prospective; strict versus standard BP control; follow-up 7 yr | HTN control; OM: LVM (P<0.01) | 49 |
| 24 | Mean age: 41 (amlodipine) to 43 yr (enalapril); CrCl>50 ml/min per 1.73 m2 | Follow-up 5 yr | HTN control; OM: decreased urinary albumin excretion (P<0.05) | 50 |
| 46 | ADPKD with HTN; age range=18–65 yr; sCr≤4 mg/dl | Double-blind 3-yr trial comparing ramipril with metoprolol | BP control; OM: renal function, LVM | 51 |
| 8 | Mean (SD) age 38±10 yr; mean (SD) sCr=0.7±0.1 mg/dl; mean (SD) eGFR=113±31 ml/min per 1.73 m2 | Conducted over a 2-wk interval | Water intake; OM: urine osmolality<285 mOsm/kg | 107 |
| Cross-sectional/retrospective studies | ||||
| 741 | Mean (SD) age 53.4±14.8 yr; stages 1–4 CKD; patients with no molecular data excluded | Genotype–phenotype correlation | PKD1 disease, PKD1 truncating mutation; OM: kidney function (P<0.001) | 5 |
| 336 (267 at risk) | Mean age: 35.4 (PKD1) and 44.5 yr (PKD2); 42 patients had CrCl<70 ml/min or ESRD | Genotype–phenotype correlation | PKD1; OM: kidney function (P<0.001) | 6 |
| 1215 | 622 alive without ESRD; 205 died before ESRD; patients with no specific event or younger than cutoff age excluded from analysis for specific predictor | Retrospective | Sex, PKD1, young age at diagnosis, HTN, gross hematuria; OM: renal survival (P<0.001) | 7 |
| 624 | 44% of PKD1 and 29% of PKD2 had reached ESRD | Genotype–phenotype correlation | PKD1; OM: kidney function (P<0.001) | 8 |
| 60 | Children with PKD1 (n=50) or PKD2 (n=10); patients without molecular data excluded | Genotype–phenotype correlation | PKD1; OM: bilateral renal cysts (P<0.001) | 11 |
| 51 PKD1 families | Families with evidence of vascular phenotype in unaffected relatives excluded | Genotype–phenotype correlation | PKD1 mutation position 5′ to 3′; OM: vascular phenotype (P<0.001) | 12 |
| 234 | PKD2 only | Genotype–phenotype correlation | PKD2 mutation location; OM: phenotypic score variability (P=0.002) | 13 |
| 461 | PKD2 only | Genotype–phenotype correlation | Sex, PKD2 mutation other than splice mutations; OM: renal function (P<0.001) | 14 |
| Cross-sectional/retrospective studies | ||||
| 704 | PKD1 only; 46% had ESRD | Genotype–phenotype correlation | PKD1, modifier genes; OM: phenotype heritability (P<0.05) | 20 |
| 155 | PKD1 only; mean (SD) age 37.4±17.29 yr; ESRD: 31.1%; patients with no molecular data excluded | Genotype–phenotype correlation | ACE DD polymorphism; OM: ESRD<50 yr (P=0.02) | 21 |
| 173 | PKD1 only; patients with linkage to PKD2 or no molecular data excluded | Genotype–phenotype correlation | ENOS Glu298Asp polymorphism in men; OM: age at ESRD (P=0.02) | 23 |
| 294 | PKD1 and PKD2; 41% reached ESRD | Genotype–phenotype correlation | ITGA8 polymorphism; OM: age at ESRD (P=0.03) | 24 |
| 580 | 71% alive without ESRD at age 50 yr, 53% at 58 yr, 22% at 70 yr | Retrospective | PKD1, younger age at diagnosis, men, HTN, increased LVM, hepatic cysts in women, three or more pregnancies, gross hematuria, UTIs in men, KV; OM: renal function (P<0.05) | 26 |
| 157 | Study A: from CrCl=30–50 ml/min per 1.73 m2 to ESRD; study B: CrCl=50–60 ml/min per 1.73 m2, followed for 4 yr | Retrospective | Sex, MAP, age; OM: renal function, age at ESRD (P<0.001) | 27 |
| 58 | ESRD | Retrospective | Race; OM: ESRD (P<0.001) | 28 |
| 162 | ESRD (dialysis or renal transplant) | Retrospective | HTN in nonaffected parent; OM: age at renal death (P<0.03) | 29 |
| 475 | Patients’ mean (SD) age 40±11 yr | Retrospective | HTN in affected parent; OM: HTN in offspring (P<0.05) | 30 |
| 484 | Mean age (PKD1 and PKD2): 41 yr (range=2–67) and 56 yr (range=17–88); ESRD: PKD1, n=140 (mean age 48.9 yr) and PKD2, n=24 (mean age 70.2 yr) | Retrospective | Family history of ADPKD-related ESRD; OM: mutated gene | 31 |
| 180 | Children; age 4–18 yr with normal renal function | Retrospective | Glomerular hyperfiltration; OM: total renal volume, renal function (P<0.001) | 32 |
| 191 | Mean (SEM) age 41±1 yr (range=17–81) | Retrospective | Gross hematuria; OM: total renal volume, renal function (P<0.03) | 35 |
| 108 | Patients with history of two or more UTIs | Retrospective | Efficacy of antibiotic prophylaxis in reducing UTIs and gross hematuria and delaying disease progression, gross hematuria at age <30 yr; OM: renal function (P<0.001) | 36 |
| 235 | Mean (SEM) age 42±1 yr; 91 women excluded because of pregnancy at study visit, Cr excretion variability, different laboratory source for Cr data, or missing data | Retrospective | Preexisting HTN; OM: maternal complications in pregnancy (P<0.001) | 37 |
| Cross-sectional/retrospective studies | ||||
| 94 | Patients who doubled sCr in <36 mo (rapid progressors) or >36 mo (slow progressors) | Retrospective | HTN, UTI, hematuria, overt proteinuria; OM: renal function (P=0.01) | 38 |
| 256 | Stages 1–4 CKD; patients with <6 mo follow-up excluded | Retrospective | Recurrent, persistent pyuria; OM: renal function (P=0.01) | 39 |
| 32 | Children; mean (SD) age 12.3±4.7 yr | Cross-sectional | HTN onset in early-stage ADPKD; OM: renal volume, length (P<0.05) | 41 |
| 62 | Children; mean (SD) age 12.3±4.3 yr | Cross-sectional | HTN; OM: renal volume (P<0.01) | 42 |
| 199 | Children with ADPKD | Retrospective | Very early onset of ADPKD, ADPKD signs, symptoms; OM: KV, HTN, ESRD (P<0.005) | 44 |
| 147 | Age 16–45 yr; CrCl=75–150 ml/min per 1.73 m2 | Retrospective | HTN; OM: renal volume (P<0.005) | 45 |
| 134 | Mean (SD) age 42.3±13.1 yr | Retrospective | HTN; OM: clinical severity index (P<0.001) | 46 |
| 30 | Mean age 32 yr (range=19–45); CrCl>80 ml/min per 1.73 m2; patients with less than six sCr measurements or less than three sCr measurements in 1 yr excluded | Retrospective | HTN; OM: renal function (P<0.001) | 47 |
| 1044 | Studies A and B: age 15–49 and 18–64 yr; eGFR≥60 and 25–59 ml/min per 1.73 m2 | Cross-sectional | Albuminuria, TKV, RBF; OM: renal function (P<0.001) | 52 (HALT PKD baseline data) |
| 103 | Mean (SD) age 40±11 yr; mean (SD) GFR 92±36 ml/min | Cross-sectional comparison with 103 age- and sex-matched healthy controls (live kidney donors) | ADPKD status, age quartile; OM: MAP, total renal volume, measured GFR, effective renal plasma flow, renal vascular resistance, filtration fraction (P<0.001) | 60 |
| 116 | Adults; mean (SEM) age 40.7±1.1 yr | Cross-sectional | HTN; OM: LVH frequency (P<0.05) | 62 |
| 26 | Mean (SD) age 26.5±7.4 yr; normotensive by clinic BP; normal renal function | Cross-sectional | 24-h systolic BP by ambulatory monitoring; OM: LVMI (P=0.01) | 63 |
| 46 | Mean (SD) age 25.9±8.1 yr; normotensive by clinic BP; normal renal function | Cross-sectional comparison with 35 age- and sex-matched healthy controls | ADPKD; OM: LVMI, diastolic function (P<0.001) | 64 |
| 18 | Mean (SD) age 24.1±6 yr; mean (SD) sCr=74.1±10.6 µM/L | Cross-sectional | Exercise systolic and diastolic BP; OM: LVMI (P=0.001) | 65 |
| Cross-sectional/retrospective studies | ||||
| 20 | Mean (SD) age 46.7±13.3 yr; mean (SD) sCr=112±33 µM/L; patients with sCR>177 µM/L, cardiovascular complications, diabetes, pulmonary or liver disease, or pregnancy excluded | Cross-sectional | 24-h systolic and diastolic BP; OM: LVMI (P<0.001) | 66 |
| 24 | Age range 5.7–24.9 yr; normal renal function | Cross-sectional | HTN; OM: LVMI (P<0.002) | 67 |
| 85 | Children; mean (SEM) age 12±1 yr; normal renal function | Cross-sectional | HTN; OM: LVMI (P<0.001) | 68 |
| 2085 | Mean age 50 yr | Retrospective | HTN; OM: mortality (P<0.001) | 70 |
| 513 | Mean ages for women/men: 37/38 yr (1985–1992) and 39/36 yr (1992–2001) | Retrospective | HTN control; OM: time to ESRD, ADPKD man±HTN (P=0.02), ADPKD woman±HTN (P=0.02–0.03) | 71 |
| 693 | Mean age 55.9 yr (1990–1995) and 60.6 yr (2000–2007) | Retrospective | HTN control; OM: patient survival, age at ESRD (P<0.001) | 72 |
| 543 | Age 15–49 yr; GFR≥60 ml/min per 1.73 m2 | Cross-sectional | Systolic BP measured at office, sex; OM: LVMI (P<0.001) | 73 (HALT study A baseline data) |
| 31 | Mean age 35.8 yr (normotensive)–CrCl 39.6 yr (hypertensive), mean CrCl=91–106 ml/min per 1.73 m2; patients with diabetes, cardiovascular disease, and other conditions affecting endothelial function excluded | Cross-sectional | HTN; OM: endothelial-dependent dilation (P<0.05) | 75 |
| 15 | Mean (SD) age 40.1±10.3 yr; mean (SD) CrCl=87.8±13.7 ml/min per 1.73 m2; patients with diabetes, cardiovascular disease, hypercholesterolemia, or smokers excluded | Cross-sectional | HTN; OM: endothelial dysfunction (P<0.005) | 76 |
| 144 | n=52: HTN and eGFR<60 ml/min per 1.73 m2; n=50: HTN and eGFR≥60 ml/min per 1.73 m2; n=42: normotensive and eGFR≥60 ml/min per 1.73 m2; 51 healthy controls | Cross-sectional | ADPKD versus control; HTN, renal function; OM: peripheral augmentation index, circulating inflammation biomarkers (P<0.05) | 77 |
| 97 | Proteinuria<300 and >300 mg/d: mean age 39 and 48 yr, mean CrCl=82 and 37 ml/min per 1.73 m2 | Retrospective | Overt proteinuria (>300 mg/d), microalbuminuria; OM: renal function (P<0.001) | 79 |
| Cross-sectional/retrospective studies | ||||
| 102 | Mean (SD) age 40±11 yr; mean (SD) measured GFR=77±31 ml/min per 1.73 m2 | Cross-sectional | Plasma copeptin; OM: disease severity by measured GFR, effective RBF, total renal volume, albuminuria (P<0.001) | 82 |
| 102 | Mean (SD) age 40±11 yr; mean (SD) measured GFR=77±31 ml/min per 1.73 m2 | Cross-sectional | Urinary biomarkers; OM: disease severity by measured GFR, effective RBF, total renal volume (P<0.05) | 84 |
| 71 | Mean (SD) age 16±4 yr (range=8–26); mean (SD) CrCl=130.2±29.4 ml/min per 1.73 m2 | Cross-sectional | Serum angiogenic growth factors (particularly Ang-1); OM: renal function (P=0.01) | 85 |
| 680 | Consecutive adult ADPKD patients not in ESRD | Retrospective | Serum uric acid; OM: ESRD hazard ratio (P<0.001) | 87 |
| 91 | Adult normotensive ADPKD patients; normal renal function; no other chronic conditions | Retrospective | Uric acid, serum ADMA, eGFR; OM: endothelial dysfunction (P<0.001) | 88 |
| 55 | Age 22–79 yr; sCr=0.5–11.9 mg/dl (median=1.2); patients with clinical renal infections, on dialysis, or with transplant excluded | Retrospective | Urine excretion of MCP-1; OM: sCr | 89 |
| 144 | n=50: HTN and eGFR>60 ml/min per 1.73 m2; n=52: HTN and eGFR=25–60 ml/min per 1.73 m2; n=42: normotensive and eGFR>60 ml/min per 1.73 m2; patients with vascular disease, diabetes, and severe heart failure excluded | Cross-sectional | Disease severity defined by presence or absence of HTN and renal function (normal versus decreased); OM: levels of serum inflammatory and oxidative stress markers (P<0.001) | 90 |
| 100 | Mean (SD) age 31±6 yr; mean (SD) eGFR=94±18 ml/min per 1.73 m2; CKD stages 1/2 | Cross-sectional | FGF23; OM: increased renal phosphate excretion (P=0.07) | 91 |
| Test/validation sets: 134/158 | Test/validation sets: mean (SD) age 31.4±6.3/32.4±8.7 yr, mean (SD) eGFR=86.4±15.5/88.1±27.8 | Retrospective | Proteomic severity score made of 142 peptides; OM: hTKV (P<0.001) | 92 |
| 284 | Mean (SD) age at ESRD 54.1±9.9 yr | Retrospective | Low birth weight; OM: age at ESRD (P=0.01) | 95 |
| 102 | Mean (SD) age 39±12 yr | Cross-sectional | Coffee consumption; OM: caffeine in ADPKD patients versus controls (P<0.001) | 97 |
| Cross-sectional/retrospective studies | ||||
| 582 | ESRD; patients with systemic disease involving kidney, immunosuppressive therapy, and age at renal death <21 yr excluded | Retrospective | Tobacco consumption; OM: ESRD risk (P=0.002) | 98 |
| 239 | Mean (SEM) age 38.6±0.9 yr (range=2–80); patients with primary PLD diagnosis excluded | Retrospective | Sex; OM: massive hepatic cystic disease (P<0.04) | 108 |
OM, outcome; TCV, total cyst volume; KV, kidney volume; HTN, hypertension; sCr, serum creatinine; Cr, creatinine; CT, computed tomography; LVM, left ventricular mass; ACE DD, angiotensin converting enzyme DD genotype; ENOS, endothelial nitric oxide synthase; ITGA8, integrin α-8 gene; MAP, mean arterial pressure; Ang-1, angiotensin-1; ADMA, asymmetric dimethylarginine; MCP-1, monocyte chemoattractant protein-1; FGF23, fibroblast growth factor-23; hTKV, height-adjusted TKV; PLD, polycystic liver disease.
Hyperfiltration in Childhood
The effect of glomerular hyperfiltration (creatinine clearance [CrCl]≥140 ml/min per 1.73 m2) was evaluated in a cohort of 140 children with ADPKD followed longitudinally for a median of 5.8 years.32 The subset of 32 children with glomerular hyperfiltration showed a higher rate of growth in TKV (corrected for body surface area) over 5 years and a faster decline in renal function than children without glomerular hyperfiltration. Because the final CrCl measures were similar in the two groups of children, it is unclear whether hyperfiltration translates into an earlier onset of ESRD.
Gross Hematuria
Early or frequent episodes of gross hematuria may be signs of more severe disease and contributors to renal function deterioration, perhaps by causing AKI and/or chronic iron toxicity. Cyst ruptures with gross hematuria may lead to the release and deposition of free iron and heme, promoting generation of reactive oxygen species and proinflammatory cytokines.33,34 An observational study of 191 patients with ADPKD found that patients who had one or more episode of gross hematuria had significantly larger kidneys and were more often hypertensive than patients without an episode.35 Patients who reported more episodes of gross hematuria had worse renal function than patients who had only one episode. Renal survival was significantly worse in 128 patients with ADPKD who had gross hematuria at age <30 years than in 448 patients without gross hematuria or whose first episode occurred at age >30 years (ESRD at 49 versus 59 years).7 Similar findings were reported in other studies.36 Although these observations are derived from retrospective studies, they indicate that gross hematuria is associated with more severe disease.
Pregnancy
No prospective study examining the effects of pregnancy on ADPKD renal outcome has been reported. An analysis of historical data from 235 women (605 pregnancies), however, showed that pregnancy did not seem to affect renal function in normotensive women, but hypertensive women with more than three pregnancies had significantly worse renal function than age-adjusted women with fewer pregnancies.37 Survival analysis of 236 women with ADPKD showed significantly shorter renal survival among women with three or more pregnancies.7 Because the latter analysis relied on historical data, it is unclear whether the shorter renal survival was because of complications of pregnancy, such as development or exacerbation of hypertension and preeclampsia, or other factors.
Multiple Urinary Tract Infections
Several small retrospective ADPKD studies have suggested that multiple urinary tract infections (UTIs) are associated with a more rapid decline in renal function. An uncontrolled study in 108 patients with a history of two or more UTIs reported that antibiotic prophylaxis significantly reduced the incidence of infection and limited the loss of renal function compared with no prophylaxis.36 In another study, patients classified as fast progressors (defined by doubling of serum creatinine in ≤36 months) had a higher incidence of single and recurrent UTIs than slow progressors.38 A third retrospective, single-center study suggested that asymptomatic pyuria and particularly, overt UTI were associated with faster decline in renal function, but it is unclear whether this result was independent of other factors, such as baseline GFR and TKV.39 In addition, overt UTI was uncommon in that study, occurring in only 33 of 256 patients during a mean observation period of 81 months. It remains, therefore, uncertain whether UTIs play a significant role in the loss of renal function in ADPKD.
Cyst Rupture
One study assessed changes in renal volume over a 6-month period by magnetic resonance imaging (MRI) in a cohort of 100 young patients with ADPKD and found direct evidence for cyst rupture, resulting in a decrease in renal volume in 6 patients.40 Compared with the rest of the study population, these six patients had higher baseline TKV and reported more symptoms (flank pain, macrohematuria, infection, and hypertension), suggesting that cyst rupture is a marker of more severe disease.
Hypertension and Kidney Function
Hypertension occurs in up to 80% of patients with ADPKD before significant loss of renal function and seems to be both a marker of more severe disease and a contributor to renal function loss.3 Hypertension is already present in a significant subset of children with ADPKD, and several cross-sectional studies have shown that the prevalence of hypertension in children correlates with kidney size and number of cysts.41–43 Another observational study of children with ADPKD found that those who were diagnosed by ultrasound at age <18 months had larger kidneys, more cysts, and more frequent hypertension on follow-up than children diagnosed at age >18 months, confirming the relationship between renal structural severity and prevalence of hypertension, even in children with ADPKD.44
Similarly, cross-sectional analysis of 147 adults with ADPKD revealed that renal volume was significantly greater in patients with hypertension versus normotension among both men and women, whereas serum creatinine and CrCl did not differ.45 Furthermore, TKV assessed by MRI was significantly higher in young (mean age of 31 years) ADPKD patients with hypertension (n=67) versus normotension (n=33).40 Another cross-sectional study of 134 patients found a strong and graded association between presence and severity of hypertension and decreased eGFR.46
Early onset of hypertension may lead to earlier onset of ESRD. A survival analysis using data from 506 adults with ADPKD revealed that patients with a diagnosis of hypertension at age <35 years developed ESRD 14 years earlier (at 51 versus 65 years old) than patients who were normotensive until >35 years old.7 Another small (n=30) retrospective study found that the decline in mean standardized CrCl slope over time was significantly greater in hypertensive versus normotensive patients.47 Similarly, in an earlier retrospective study of 157 patients with decreased renal function, patients with higher mean arterial pressure had a faster CrCl decline than patients with lower BP.27 In a prospective observational study of 198 patients with ADPKD followed for a mean of 100 months, hypertension was an independent risk factor of CKD progression.48 These data show that early-onset hypertension and severity of hypertension are associated with larger kidneys and progression to ESRD, and thus, they are markers of more severe disease. To date, no randomized clinical trial has been able to show an effect of BP control on progression to ESRD, but these trials were all limited by their small size and short duration.49–51 The effect of BP control on renal structural and functional disease progression is currently being examined in a large multicenter prospective trial (HALT PKD) involving 1044 patients over 5–8 years; results are expected by the end of 2014.52
Episodes of AKI
Although there is strong evidence in animal models that AKI accelerates the progression of PKD, it has not been studied in humans. Only one case-control study reported that patients with ADPKD hospitalized for pneumonia were significantly more likely to develop AKI (serum creatinine elevation≥0.3 mg/dl) than matched patients without ADPKD with similar severity of pneumonia.53
Renal Volume above Age-Corrected Mean
Two longitudinal observational studies in adults and one study in children showed that greater renal volume is predictive of more rapid ADPKD progression.4,54–56 In the children’s study (n=108), those patients with early renal enlargement had a significantly faster rate of renal growth (assessed over a mean follow-up of 5.7 years) than other children with ADPKD (26 versus 11 ml/kidney per year adjusted for age).54 In a study of 229 adults with ADPKD, sequential ultrasound examinations performed over a mean interval of 7.8 years showed that initial renal volume as well as renal volume growth rate were correlated with the rate of eGFR decline over time.55 The Consortium for Radiologic Imaging Studies in Polycystic Kidney Disease (CRISP) enrolled patients with ADPKD with CrCl>70 ml/min and used MRI to determine TKV.4,56 Age-adjusted renal volume was inversely related to GFR and albuminuria at baseline.56 During repeated assessments over 8 years, the strength of the correlation between increase in TKV and decrease in measured (iothalamate clearance) GFR increased.57 Receiver-operator characteristic curve analysis indicated that a baseline height-adjusted TKV≥600 cm3/m predicted progression to stage 3 CKD within 8 years with 74% sensitivity and 75% specificity.
Decreased Renal Blood Flow
In CRISP, mean renal blood flow (RBF) measured by MRI decreased progressively over a 3-year period and preceded the decline in GFR.58,59 Lower baseline RBF was associated with a larger increase in TKV and total cyst volume over 3 years and a decline in measured GFR. On multivariate analysis, RBF was an independent predictor of renal structural and functional disease progression.59 A different group of investigators using iothalamate and hippuran clearances in a cross-sectional analysis of 103 subjects also reported a marked decrease in effective renal plasma flow, whereas GFR was preserved in young adult patients with ADPKD compared with matched healthy control subjects.60 Although RBF measurements are currently not available for routine clinical practice, they may be useful as outcome parameters for future interventional studies. The early decrease in RBF is also important for elucidating the pathogenesis of renal function loss in ADPKD. It remains to be determined whether it is mainly because of mechanical compression of blood vessels by the cysts, intrinsic vascular abnormalities caused by PKD mutations, or a secondary effect of oxidative stress and inflammation causing vascular dysfunction.
In summary, the best evidence from carefully designed prospective studies and multiple cross-sectional and retrospective studies identifies PKD1 mutations (particularly if truncating), early onset of hypertension, early and multiple episodes of gross hematuria, large kidney volumes, and decrease in RBF or GFR at a young age as the most reliable predictors of rapid renal disease progression (Figure 1).
Figure 1.
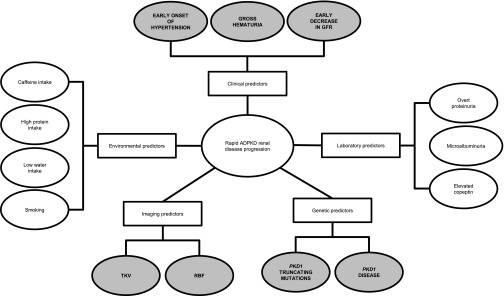
Diagram depicting the spectrum of predictors for rapid renal disease progression in ADPKD. Shaded ovals represent the most established predictors.
Cardiovascular Disease Progression in ADPKD
Hypertension and Left Ventricular Hypertrophy
Hypertension is the most commonly reported initial sign of ADPKD and a well recognized risk factor for left ventricular hypertrophy (LVH).61 In an earlier study, LVH was common in adults with ADPKD, which was shown in an echocardiographic study of 116 consecutive patients, where 41% had LVH at a mean age of only 44 years.62 Patients with ADPKD with versus without LVH had a significantly higher prevalence (83% versus 61%) and duration (7.5 versus 3.9 years) of hypertension and worse renal function (serum creatinine of 2.0 versus 1.4 mg/dl). Another study reported higher left ventricular mass index (LVMI) in young normotensive patients with ADPKD than healthy control subjects and found a correlation between 24-hour systolic BP on ambulatory monitoring and LVMI.63 These young patients also had signs of diastolic dysfunction by echocardiogram and higher BP during exercise stress testing than healthy control subjects.64,65 Levels of neurohormones were not correlated with LVMI, but 24-hour systolic and diastolic BPs were correlated.66 These observations highlight the importance of 24-hour BP load on the development of LVH in ADPKD.
Early Increase in LVMI
Even in children and young adults (ages<25 years) with ADPKD, a significantly higher LVMI than in healthy control subjects has been reported, although most LVMI values were in the normal range.67 These young adults had higher BP by ambulatory monitoring than control subjects but not children with ADPKD. In another study, children with ADPKD and hypertension or borderline hypertension had significantly higher LVMI than children with ADPKD and normotension.68 Similarly, children with ADPKD, but not their unaffected siblings, showed a significant correlation between systolic BP and LVMI; children with hypertension had significantly higher LVMI than children with normotension.69 Children with ADPKD also had a significantly higher incidence of mitral valve prolapse than their unaffected siblings.
BP Control
There is indirect evidence from three recent epidemiologic studies that BP control improves cardiovascular and renal outcomes. First, a population-based study using the United Kingdom General Practice Research Database (n=2085) found that antihypertensive drug use increased in frequency in patients with ADPKD over the period from 1991 to 2008, particularly for agents blocking the renin-angiotensin system (RAS).70 After adjusting for potential confounders, mortality decreased over the same time period. Second, a study that compared two ADPKD cohorts (n=513) over the 1985–1992 versus 1992–2001 periods found that patients in the later cohort had lower mean arterial pressure and increased use of angiotensin-converting enzyme inhibitors.71 Cardiovascular outcomes were not determined, but patients in the later cohort had longer renal and overall survival. Third, a study of patients with ADPKD in Denmark (n=693) found improved survival and an increase in age (by 4.7 years) at onset of ESRD between 1990 and 2007, coinciding with increased use of RAS-blocking drugs.72
Small randomized trials of hypertension control showed benefits of strict BP control for LVMI and LVH but not renal function.49,51
In contrast to earlier results,62 the prevalence of LVH was assessed by cardiac MRI in 543 hypertensive patients with ADPKD (mean age of 36 years) with normal renal function who were participating in the HALT PKD trial.73 Baseline data revealed that the prevalence of LVH was only 3.9% by nonindexed left ventricular mass and 0.9% by LVMI, despite a mean duration of diagnosed hypertension of 5.8 years. This low prevalence of LVH may be attributed to the fact that BP was well controlled (mean of approximately 124/82 mmHg at baseline) and that 61% of patients were using RAS blockers. There is, therefore, strong epidemiologic evidence that strict BP control using RAS-blocking drugs ameliorates cardiovascular disease progression in ADPKD.
Aneurysm Rupture
Intracranial aneurysm (ICA) rupture is one of the most dramatic cardiovascular disease manifestations of ADPKD. The only predictor that has been consistently identified is family history of ICA rupture, which was shown in a retrospective study of 608 adults from 199 ADPKD families.74 The frequency of ruptured ICAs was significantly higher in patients with ADPKD than unaffected family members, but ruptured ICAs were not randomly distributed; rather, clustering was evident within certain ADPKD families. As mentioned previously, mutations toward the 5′ end of PKD1 may be more likely to cause ICA and other vascular complications.12
Endothelial Dysfunction
Endothelial dysfunction is an early and common finding in patients with ADPKD. In one study, patients with ADPKD with and without hypertension exhibited greater endothelial dysfunction (measured by endothelium-dependent dilation of the brachial artery after transient ischemia) than healthy subjects with and without hypertension, respectively.75 Carotid intima media thickness was increased in patients with ADPKD and normotension compared with healthy subjects and further increased in patients with ADPKD and hypertension. Other studies have also shown evidence of endothelial dysfunction in early ADPKD before the onset of hypertension and renal dysfunction when measured in the cutaneous microcirculation by laser Doppler examination or peripheral artery tone recordings using finger plethysmography.76,77 Presence of endothelial dysfunction in early ADPKD likely confers increased atherosclerosis risk, consistent with the observed increase in carotid intima media thickness, and it may contribute to the early decline in RBF described previously. Endothelial dysfunction and carotid intima media thickness are not routinely assessed in clinical practice, but they can serve as markers or treatment targets in research settings.
Association of Laboratory Factors with Renal and Cardiovascular Disease Progression
A summary of the main studies examining laboratory predictors of rapid renal and cardiovascular disease progression is provided in Table 5.
Table 5.
Laboratory predictors associated with renal and cardiovascular disease progression in ADPKD
| Predictor | Association/Change | Refs. |
|---|---|---|
| Urine predictors | ||
| Urine-concentrating capacity | Reduced in children with >10 versus ≤10 cysts | 43 |
| MCP-1 | Increased in ADPKD; associated with TKV; predictor of stage 3 CKD | 57,89 |
| Increased in ADPKD; associated with TKV | 82 | |
| NGAL and IL-8 | NGAL increased in ADPKD; associated with GFR, TKV, effective RBF | 82 |
| Both increased in ADPKD | 93 | |
| Kidney injury molecule-1 | Increased in ADPKD; associated with GFR, TKV | 82 |
| Heart-type-fatty-acid binding protein | Increased in ADPKD; associated with GFR, RBF | 82 |
| Collagen-derived peptides and other biomarkers | Increased in ADPKD | 92 |
| Serum or other laboratory predictors | ||
| HDL cholesterol | Decreased HDL levels associated with GFR decline | 78,86 |
| Low levels associated with TKV increase | 86 | |
| Vascular endothelial growth factor | In children, correlated with TKV and LVMI; negatively correlated with CrCl | 85 |
| Angiopoietin 1 | In children, correlated with TKV and LVMI; negatively correlated with urinary protein excretion | 85 |
| Serum uric acid | Higher level associated with earlier hypertension onset, larger TKV, ESRD risk | 87 |
| Higher level associated with endothelial dysfunction | 88 | |
| FGF23 | Increased in ADPKD patients versus controls; associated with increased renal phosphate excretion | 91 |
| Inflammation-associated biomarkers (MCP-1 and TNF-α) | Overexpression of inflammation factors drives interstitial inflammation and fibrosis | 94 |
MCP-1, monocyte chemoattractant protein-1; FGF23, fibroblast growth factor-23.
Proteinuria and Microalbuminuria
The amount of proteinuria correlates with the risk for progression in many renal diseases, including ADPKD. Among 200 patients with ADPKD participating in the Modification of Diet in Renal Disease (MDRD) Study, higher levels of proteinuria were associated with a steeper decline in GFR.78 Another study of 270 consecutive adults with ADPKD identified overt proteinuria (>300 mg/d) in 48 patients (18%), which was associated with worse renal function, higher BP, and larger renal volume.79 Patients with versus without overt proteinuria reached a serum creatinine concentration of 1.5 mg/dl at a significantly younger age (44 versus 58 years old). The same study found microalbuminuria in 20 of 49 patients with hypertension and LVH but not overt proteinuria. BP, renal volume, and filtration fraction were significantly higher in patients with microalbuminuria. Similarly, in a study of 100 young patients (mean age of 31 years) with ADPKD and preserved renal function, the degree of albuminuria was correlated with TKV and kidney volume growth rate.40 Among 1044 adult participants in the HALT PKD trials at baseline, urine albumin excretion was positively correlated with TKV and negatively correlated with eGFR.52 Because TKV is usually not available in clinical practice, increased levels of urinary albumin excretion in adults may be a valuable marker of ADPKD severity before a decline in eGFR is evident. In children, microalbuminuria is not consistently correlated with disease severity.44,54,68
Serum Copeptin Levels
Copeptin is the carboxy-terminal portion of the precursor for arginine vasopressin and serves as a reliable and stable surrogate of circulating arginine vasopressin levels.80,81 Because vasopressin induces the production of cAMP, which is a potent stimulator of cyst formation and growth, elevated levels of copeptin may be a marker and/or contributor to rapid ADPKD progression. In a cross-sectional study of 102 patients with ADPKD, plasma copeptin concentrations were correlated with TKV, 24-hour urine albumin excretion, GFR, and effective RBF independent of age, sex, and diuretic use.82 More importantly, in the CRISP cohort of 241 patients with preserved renal function at baseline, higher baseline copeptin levels (after adjusting for age and sex) were associated with larger increases in TKV and decreases in GFR during a median follow-up of 8.5 years.83 Copeptin level was associated with plasma osmolality in the former study but not CRISP. More research studies to confirm the potential predictive value of serum copeptin level are necessary before it can be adopted for clinical use.
Additional Investigational Urinary and Serum Biomarkers
Levels of several urinary markers and other laboratory parameters are abnormal in ADPKD (Table 5). Many of these markers have been associated with renal structural and functional disease severity, and some have been associated with cardiac structure.43,84–88 In a cross-sectional analysis of 102 patients with ADPKD, neutrophil gelatinase-associated lipocalin (NGAL; a marker of proximal tubule damage), kidney injury molecule-1 (a marker of proximal tubule damage), heart-type-fatty-acid binding protein (a marker of distal tubule damage), and monocyte chemoattractant protein-1 (a marker of inflammation) were all significantly increased compared with healthy subjects.84 Monocyte chemoattractant protein-1 has been extensively investigated in other studies as well, suggesting that it may become an important predictive marker when further validated by larger population studies.57,89 Moreover, each of these markers was associated with renal structural or functional parameters. Among these markers, NGAL may be of most value for determining ADPKD severity, because it was associated with both renal structural and functional parameters. Validation of these markers in larger prospective cohorts is still required before their use can be recommended for clinical practice.
Patients with ADPKD, hypertension, and renal dysfunction have significantly higher levels of systemic markers of inflammation than patients with normal renal function (hypertensive or normotensive) or healthy subjects.90 In comparison, oxidative stress was apparent even in patients with ADPKD with normotension versus healthy subjects and likely precedes the appearance of hypertension, which may account for the increased levels of angiogenic growth factors described in young patients with ADPKD. Several other serum and laboratory factors have been associated with various aspects of ADPKD severity, including serum uric acid, HDL, fibroblast growth factor-23, collagen-derived peptides, NGAL, IL-8, and TNF-α.78,86–88,91–94 Although some of these factors may contribute to ADPKD progression, confirmation in additional studies is required as well as biomarker validation.
Association of Environmental Factors with Renal Disease Progression
A summary of the main studies examining environmental factors of rapid renal disease progression is provided in Table 6.
Table 6.
Environmental predictors of renal disease progression in ADPKD
| Environmental Predictor | No. of Patients/ Experimental Platform | Patient Characteristics/Experimental Platform | Study Features | Highest P Value | Major Refs. | Additional Supporting Refs. |
|---|---|---|---|---|---|---|
| High water intake | 8 | ADPKD patients; normal renal function; patients eating typical diets | Phase 1: baseline measurements; phase 2: water administration to reduce urine osmolality<285 mosM/kg | 0.02 | 106,107 | 99–105 |
| Caffeine | Cysts epithelial cells | Mural epithelial cell from human ADPKD cysts | cAMP levels determined after caffeine or receptor-mediated agonist stimulation | <0.001 | 96 | 97 |
| Low protein intake | 200 | ADPKD patients from MDRD; reduced renal function | 2.2 yr follow-up | 0.06 | 78 | NA |
| Smoking | 323 | Consecutive ADPKD patients at Istanbul University | Prospective observational (n=198); mean (SD) follow-up=100±38 mo | 0.01 | 48 | NA |
| 582 | Patients diagnosed with IgA nephropathy or ADPKD | Retrospective | 0.002 | 98 | NA | |
| Low birth weight | 284 | Retrospective cohort of ADPKD patients with ESRD from Denmark | Retrospective | 0.01 | 95 | NA |
NA, not applicable.
Birth Weight
In a retrospective analysis of 284 patients with ADPKD, ESRD, and available birth weight, age at ESRD onset increased by 1.7 years for every 1-kg increase in birth weight after adjustment for BP, sex, birth decade, and type of antihypertensive treatment.95 This observation needs to be confirmed in other larger ADPKD cohorts, but it is consistent with the notion that people born with low birth weight have a reduced number of nephrons and therefore, are more likely to develop ESRD.
Caffeine
By inhibiting phosphodiesterase, caffeine increased cAMP levels in a concentration-dependent manner in cyst epithelial cell culture, resulting in activation of the extracellular signal-regulated kinase pathway and leading to increased cellular proliferation and transepithelial fluid secretion.96 High caffeine intake may, therefore, be a risk factor for cyst enlargement and ADPKD progression, although a small clinical study has suggested no correlation between mild caffeine intake and TKV by ultrasound.97 Nevertheless, it is recommended to counsel patients with ADPKD to limit their caffeine intake.
Smoking
In the prospective observational study by Ozkok et al.,48 93 patients with ADPKD were found to be fast progressors (defined as decline in eGFR>1 ml/min per year) and 78 patients were slow progressors (decline<1 ml/min per year) over a mean (SD) follow-up of 100±38 months. Fast progressors were significantly more likely to have a history of smoking than slow progressors, and the number of pack-years smoked was also significantly higher, adding to the increasing evidence that smoking adversely affects CKD progression. In support of this observation, smoking has been found to increase the risk of ESRD in men with inflammatory and noninflammatory renal diseases in a dose-dependent fashion.98
High Water Intake
The vasopressin pathway is upregulated in ADPKD because of an early loss of urine concentration capability.99,100 High water intake as well as vasopressin gene inactivation have been shown to effectively reduce cystogenesis in the polycystic kidney rat.101,102 On the basis of this rationale, it has been envisioned that high water intake may be effective in delaying disease progression in human ADPKD as well.103–106 In a small pilot study, Wang et al.107 showed the feasibility of safely achieving a target urine osmolality≤285 mOsm/kg in eight patients with ADPKD eating normal diets. Larger long-term studies are needed to assess the effect of such fluid intake on renal disease progression in ADPKD.
Low Protein Intake
In the subset of 200 patients with ADPKD in the MDRD study (mean follow-up of 2.2 years), patients in study B (GFR=13–24 ml/min per 1.73 m2) were on a low or very low protein diet.78 After controlling for actual protein intake, a trend was observed to a beneficial effect with the very low protein intake group. In study A (GFR=25–55 ml/min per 1.73 m2), however, this effect was not confirmed, questioning the use of a low protein diet in patients with ADPKD with moderately impaired renal function.
Liver Disease Progression in ADPKD
The factors associated with severity of hepatic cysts were evaluated in 239 patients with ADPKD (age range=2–80 years).108 The incidence and number of hepatic cysts increased with age. Although sex was not associated with hepatic cyst incidence, women were significantly more likely to have >15 hepatic cysts. Liver cyst size was larger in women, and number of cysts was correlated with number of pregnancies, suggesting that estrogens influence the severity of liver cystic disease. Patients with hepatic cysts also had larger TKV and lower CrCl. On logistic regression, increased age, higher renal structural severity index, and decreased CrCl were predictive of the presence of liver cysts, whereas increased age, higher renal severity index, women, and pregnancy were correlated with the number and size of hepatic cysts. Another study compared changes in liver cyst volume between women with ADPKD who took postmenopausal estrogen and women who did not (similar age and number of pregnancies), and it found that 1-year estrogen treatment was associated with a significant increase in total liver and liver cyst volume.109 At present, no guidelines related to use of hormone-based contraceptives or hormone replacement therapy and risk for liver cyst complications exist for women with ADPKD. Additional studies are needed to better assess this risk.
Conclusions
This systematic review discusses several factors associated with rapid ADPKD progression (Figure 1). PKD1 mutations confer greater risk of rapid progression than PKD2 mutations, with truncating PKD1 mutations having greater significance than nontruncating mutations. Other predictors include men, early onset of hypertension, early onset or repeated episodes of gross hematuria, and in women with hypertension, three or more pregnancies. Greater TKV and early decreases in GFR and RBF predict rapid disease progression. Similarly, the presence of overt proteinuria, microalbuminuria, and elevated serum copeptin levels has been associated with more rapid progression. Hypertension seems to be the main risk factor for early cardiovascular disease; small treatment trials have shown the benefit of strict BP control for prevention or regression of LVH, whereas large epidemiologic studies have indicated a mortality benefit. Increased age and hormonal environment are the major predictors for liver cyst progression. Interestingly, most data used to validate the predictors described in this review derive from retrospective or cross-sectional studies, suggesting that additional long-term prospective studies are needed to better understand the natural history of ADPKD at the population level. A better understanding of the risk predictors involved in rapid renal disease progression may be useful in selecting patients for treatment with new agents directed against mechanisms inherent to disease pathogenesis.
Disclosures
R.W.S. is a consultant with Ikaria, Janssen, Novartis, and Otsuka America Pharmaceutical, Inc. K.F. and S.R. are employees of Otsuka America Pharmaceutical, Inc. The other authors have nothing to disclose.
Acknowledgments
Editorial assistance was provided by Catherine Fontana, Geoff Marx, and Barry M. Weichman of BioScience Communications (New York, NY), which was funded by Otsuka America Pharmaceutical, Inc.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Takiar V, Caplan MJ: Polycystic kidney disease: Pathogenesis and potential therapies. Biochim Biophys Acta 1812: 1337–1343, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fick-Brosnahan GM, Ecder T, Schrier RW: Polycystic kidney disease. In: Diseases of the Kidney, 7th Ed., edited by Schrier R, Philadelphia, Lippincott Williams & Wilkins, 2001, pp 547–588 [Google Scholar]
- 4.Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF, Jr., Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP, CRISP Investigators : Volume progression in polycystic kidney disease. N Engl J Med 354: 2122–2130, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo M-P, Grall-Jezequel A, Saliou P, Férec C, Le Meur Y: Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 24: 1006–1013, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torra R, Badenas C, Darnell A, Nicolau C, Volpini V, Revert L, Estivill X: Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol 7: 2142–2151, 1996 [DOI] [PubMed] [Google Scholar]
- 7.Johnson AM, Gabow PA: Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol 8: 1560–1567, 1997 [DOI] [PubMed] [Google Scholar]
- 8.Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D, European PKD1-PKD2 Study Group : Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet 353: 103–107, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Dicks E, Ravani P, Langman D, Davidson WS, Pei Y, Parfrey PS: Incident renal events and risk factors in autosomal dominant polycystic kidney disease: A population and family-based cohort followed for 22 years. Clin J Am Soc Nephrol 1: 710–717, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, Guay-Woodford LM, King BF, Wetzel LH, Baumgarten DA, Kenney PJ, Consugar M, Klahr S, Bennett WM, Meyers CM, Zhang QJ, Thompson PA, Zhu F, Miller JP: Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 17: 3013–3019, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Fencl F, Janda J, Bláhová K, Hríbal Z, Stekrová J, Puchmajerová A, Seeman T: Genotype-phenotype correlation in children with autosomal dominant polycystic kidney disease. Pediatr Nephrol 24: 983–989, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Rossetti S, Chauveau D, Kubly V, Slezak JM, Saggar-Malik AK, Pei Y, Ong ACM, Stewart F, Watson ML, Bergstralh EJ, Winearls CG, Torres VE, Harris PC: Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 361: 2196–2201, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Hateboer N, Veldhuisen B, Peters D, Breuning MH, San-Millán JL, Bogdanova N, Coto E, van Dijk MA, Afzal AR, Jeffery S, Saggar-Malik AK, Torra R, Dimitrakov D, Martinez I, de Castro SS, Krawczak M, Ravine D: Location of mutations within the PKD2 gene influences clinical outcome. Kidney Int 57: 1444–1451, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Magistroni R, He N, Wang K, Andrew R, Johnson A, Gabow P, Dicks E, Parfrey P, Torra R, San-Millán JL, Coto E, Van Dijk M, Breuning M, Peters D, Bogdanova N, Ligabue G, Albertazzi A, Hateboer N, Demetriou K, Pierides A, Deltas C, St George-Hyslop P, Ravine D, Pei Y: Genotype-renal function correlation in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol 14: 1164–1174, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Pei Y, Lan Z, Wang K, Garcia-Gonzalez M, He N, Dicks E, Parfrey P, Germino G, Watnick T: A missense mutation in PKD1 attenuates the severity of renal disease. Kidney Int 81: 412–417, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, Chauveau D, Rees L, Barratt TM, van’t Hoff WG, Niaudet P, Torres VE, Harris PC: Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int 75: 848–855, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, Rudenhed B, Nasr SH, Torres VE, Torra R, Bogdanova N, Harris PC: Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 21: 1097–1102, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Losekoot M, Ruivenkamp CA, Tholens AP, Grimbergen JE, Vijfhuizen L, Vermeer S, Dijkman HB, Cornelissen EA, Bongers EM, Peters DJ: Neonatal onset autosomal dominant polycystic kidney disease (ADPKD) in a patient homozygous for a PKD2 missense mutation due to uniparental disomy. J Med Genet 49: 37–40, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Reiterová J, Štekrová J, Merta M, Kotlas J, Elišáková V, Lněnička P, Korabečná M, Kohoutová M, Tesař V: Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol 14: 59, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fain PR, McFann KK, Taylor MR, Tison M, Johnson AM, Reed B, Schrier RW: Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int 67: 1256–1267, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Pérez-Oller L, Torra R, Badenas C, Milà M, Darnell A: Influence of the ACE gene polymorphism in the progression of renal failure in autosomal dominant polycystic kidney disease. Am J Kidney Dis 34: 273–278, 1999 [DOI] [PubMed] [Google Scholar]
- 22.Persu A, El-Khattabi O, Messiaen T, Pirson Y, Chauveau D, Devuyst O: Influence of ACE (I/D) and G460W polymorphism of α-adducin in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 18: 2032–2038, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Persu A, Stoenoiu MS, Messiaen T, Davila S, Robino C, El-Khattabi O, Mourad M, Horie S, Feron O, Balligand JL, Wattiez R, Pirson Y, Chauveau D, Lens XM, Devuyst O: Modifier effect of ENOS in autosomal dominant polycystic kidney disease. Hum Mol Genet 11: 229–241, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Zeltner R, Hilgers KF, Schmieder RE, Porst M, Schulze BD, Hartner A: A promoter polymorphism of the alpha 8 integrin gene and the progression of autosomal-dominant polycystic kidney disease. Nephron Clin Pract 108: c169–c175, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Dedoussis GVZ, Luo Y, Starremans P, Rossetti S, Ramos AJ, Cantiello HF, Katsareli E, Ziroyannis P, Lamnissou K, Harris PC, Zhou J: Co-inheritance of a PKD1 mutation and homozygous PKD2 variant: A potential modifier in autosomal dominant polycystic kidney disease. Eur J Clin Invest 38: 180–190, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Gabow PA, Johnson AM, Kaehny WD, Kimberling WJ, Lezotte DC, Duley IT, Jones RH: Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 41: 1311–1319, 1992 [DOI] [PubMed] [Google Scholar]
- 27.Choukroun G, Itakura Y, Albouze G, Christophe JL, Man NK, Grünfeld JP, Jungers P: Factors influencing progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 6: 1634–1642, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Yium J, Gabow P, Johnson A, Kimberling W, Martinez-Maldonado M: Autosomal dominant polycystic kidney disease in blacks: Clinical course and effects of sickle-cell hemoglobin. J Am Soc Nephrol 4: 1670–1674, 1994 [DOI] [PubMed] [Google Scholar]
- 29.Geberth S, Stier E, Zeier M, Mayer G, Rambausek M, Ritz E: More adverse renal prognosis of autosomal dominant polycystic kidney disease in families with primary hypertension. J Am Soc Nephrol 6: 1643–1648, 1995 [DOI] [PubMed] [Google Scholar]
- 30.Schrier RW, Johnson AM, McFann K, Chapman AB: The role of parental hypertension in the frequency and age of diagnosis of hypertension in offspring with autosomal-dominant polycystic kidney disease. Kidney Int 64: 1792–1799, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Barua M, Cil O, Paterson AD, Wang K, He N, Dicks E, Parfrey P, Pei Y: Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol 20: 1833–1838, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helal I, Reed B, McFann K, Yan XD, Fick-Brosnahan GM, Cadnapaphornchai M, Schrier RW: Glomerular hyperfiltration and renal progression in children with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 2439–2443, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah SV, Baliga R, Rajapurkar M, Fonseca VA: Oxidants in chronic kidney disease. J Am Soc Nephrol 18: 16–28, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Tracz MJ, Alam J, Nath KA: Physiology and pathophysiology of heme: Implications for kidney disease. J Am Soc Nephrol 18: 414–420, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Gabow PA, Duley I, Johnson AM: Clinical profiles of gross hematuria in autosomal dominant polycystic kidney disease. Am J Kidney Dis 20: 140–143, 1992 [DOI] [PubMed] [Google Scholar]
- 36.Idrizi A, Barbullushi M, Petrela E, Kodra S, Koroshi A, Thereska N: The influence of renal manifestations to the progression of autosomal dominant polycystic kidney disease. Hippokratia 13: 161–164, 2009 [PMC free article] [PubMed] [Google Scholar]
- 37.Chapman AB, Johnson AM, Gabow PA: Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 5: 1178–1185, 1994 [DOI] [PubMed] [Google Scholar]
- 38.Ahmed ER, Tashkandi MA, Nahrir S, Maulana A: Retrospective analysis of factors affecting the progression of chronic renal failure in adult polycystic kidney disease. Saudi J Kidney Dis Transpl 17: 511–515, 2006 [PubMed] [Google Scholar]
- 39.Hwang JH, Park HC, Jeong JC, Ha Baek S, Han MY, Bang K, Cho JY, Yu SH, Yang J, Oh KH, Hwang YH, Ahn C: Chronic asymptomatic pyuria precedes overt urinary tract infection and deterioration of renal function in autosomal dominant polycystic kidney disease. BMC Nephrol 14: 1, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kistler AD, Poster D, Krauer F, Weishaupt D, Raina S, Senn O, Binet I, Spanaus K, Wüthrich RP, Serra AL: Increases in kidney volume in autosomal dominant polycystic kidney disease can be detected within 6 months. Kidney Int 75: 235–241, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Seeman T, Sikut M, Konrad M, Vondrichová H, Janda J, Schärer K: Blood pressure and renal function in autosomal dominant polycystic kidney disease. Pediatr Nephrol 11: 592–596, 1997 [DOI] [PubMed] [Google Scholar]
- 42.Seeman T, Dusek J, Vondrichová H, Kyncl M, John U, Misselwitz J, Janda J: Ambulatory blood pressure correlates with renal volume and number of renal cysts in children with autosomal dominant polycystic kidney disease. Blood Press Monit 8: 107–110, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Fick GM, Duley IT, Johnson AM, Strain JD, Manco-Johnson ML, Gabow PA: The spectrum of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol 4: 1654–1660, 1994 [DOI] [PubMed] [Google Scholar]
- 44.Shamshirsaz AA, Reza Bekheirnia M, Kamgar M, Johnson AM, McFann K, Cadnapaphornchai M, Nobakhthaghighi N, Schrier RW: Autosomal-dominant polycystic kidney disease in infancy and childhood: Progression and outcome. Kidney Int 68: 2218–2224, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Gabow PA, Chapman AB, Johnson AM, Tangel DJ, Duley IT, Kaehny WD, Manco-Johnson M, Schrier RW: Renal structure and hypertension in autosomal dominant polycystic kidney disease. Kidney Int 38: 1177–1180, 1990 [DOI] [PubMed] [Google Scholar]
- 46.Augustyniak-Bartosik H, Krajewska M, Weyde W, Mazanowska O, Rurek M, Klinger M: The phenotypic characteristics of adult polycystic kidney disease have greater impact on the course of progressive disease than the type of mutation of the polycystin 1 gene. Adv Clin Exp Med 17: 155–159, 2008 [Google Scholar]
- 47.Gonzalo A, Gallego A, Rivera M, Orte L, Ortuño J: Influence of hypertension on early renal insufficiency in autosomal dominant polycystic kidney disease. Nephron 72: 225–230, 1996 [DOI] [PubMed] [Google Scholar]
- 48.Ozkok A, Akpinar TS, Tufan F, Kanitez NA, Uysal M, Guzel M, Caliskan Y, Alisir S, Yazici H, Ecder T: Clinical characteristics and predictors of progression of chronic kidney disease in autosomal dominant polycystic kidney disease: A single center experience. Clin Exp Nephrol 17: 345–351, 2013 [DOI] [PubMed] [Google Scholar]
- 49.Schrier R, McFann K, Johnson A, Chapman A, Edelstein C, Brosnahan G, Ecder T, Tison L: Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: Results of a seven-year prospective randomized study. J Am Soc Nephrol 13: 1733–1739, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Ecder T, Chapman AB, Brosnahan GM, Edelstein CL, Johnson AM, Schrier RW: Effect of antihypertensive therapy on renal function and urinary albumin excretion in hypertensive patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis 35: 427–432, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD: Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 23: 573–579, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Torres VE, Chapman AB, Perrone RD, Bae KT, Abebe KZ, Bost JE, Miskulin DC, Steinman TI, Braun WE, Winklhofer FT, Hogan MC, Oskoui FR, Kelleher C, Masoumi A, Glockner J, Halin NJ, Martin DR, Remer E, Patel N, Pedrosa I, Wetzel LH, Thompson PA, Miller JP, Meyers CM, Schrier RW, HALT PKD Study Group : Analysis of baseline parameters in the HALT polycystic kidney disease trials. Kidney Int 81: 577–585, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franco Palacios C, Keddis MT, Qin D, Zand L, Li G, Wang X, Cartin-Ceba R, Hartman RP, Qian Q: Acute kidney injury in ADPKD patients with pneumonia. Int J Nephrol 2011: 617904, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fick-Brosnahan GM, Tran ZV, Johnson AM, Strain JD, Gabow PA: Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int 59: 1654–1662, 2001 [DOI] [PubMed] [Google Scholar]
- 55.Fick-Brosnahan GM, Belz MM, McFann KK, Johnson AM, Schrier RW: Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: A longitudinal study. Am J Kidney Dis 39: 1127–1134, 2002 [DOI] [PubMed] [Google Scholar]
- 56.Chapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Bae KT, Baumgarten DA, Kenney PJ, King BF, Jr., Glockner JF, Wetzel LH, Brummer ME, O’Neill WC, Robbin ML, Bennett WM, Klahr S, Hirschman GH, Kimmel PL, Thompson PA, Miller JP, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort : Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 64: 1035–1045, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Chapman AB, Bost JE, Torres VE, Guay-Woodford L, Bae KT, Landsittel D, Li J, King BF, Martin D, Wetzel LH, Lockhart ME, Harris PC, Moxey-Mims M, Flessner M, Bennett WM, Grantham JJ: Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 7: 479–486, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.King BF, Torres VE, Brummer ME, Chapman AB, Bae KT, Glockner JF, Arya K, Felmlee JP, Grantham JJ, Guay-Woodford LM, Bennett WM, Klahr S, Hirschman GH, Kimmel PL, Thompson PA, Miller JP, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) : Magnetic resonance measurements of renal blood flow as a marker of disease severity in autosomal-dominant polycystic kidney disease. Kidney Int 64: 2214–2221, 2003 [DOI] [PubMed] [Google Scholar]
- 59.Torres VE, King BF, Chapman AB, Brummer ME, Bae KT, Glockner JF, Arya K, Risk D, Felmlee JP, Grantham JJ, Guay-Woodford LM, Bennett WM, Klahr S, Meyers CM, Zhang X, Thompson PA, Miller JP, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) : Magnetic resonance measurements of renal blood flow and disease progression in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2: 112–120, 2007 [DOI] [PubMed] [Google Scholar]
- 60.Meijer E, Rook M, Tent H, Navis G, van der Jagt EJ, de Jong PE, Gansevoort RT: Early renal abnormalities in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 5: 1091–1098, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taylor M, Johnson AM, Tison M, Fain P, Schrier RW: Earlier diagnosis of autosomal dominant polycystic kidney disease: Importance of family history and implications for cardiovascular and renal complications. Am J Kidney Dis 46: 415–423, 2005 [DOI] [PubMed] [Google Scholar]
- 62.Chapman AB, Johnson AM, Rainguet S, Hossack K, Gabow P, Schrier RW: Left ventricular hypertrophy in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 8: 1292–1297, 1997 [DOI] [PubMed] [Google Scholar]
- 63.Valero FA, Martinez-Vea A, Bardají A, Gutierrez C, Garcia C, Richart C, Oliver JA: Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 10: 1020–1026, 1999 [DOI] [PubMed] [Google Scholar]
- 64.Bardají A, Vea AM, Gutierrez C, Ridao C, Richart C, Oliver JA: Left ventricular mass and diastolic function in normotensive young adults with autosomal dominant polycystic kidney disease. Am J Kidney Dis 32: 970–975, 1998 [DOI] [PubMed] [Google Scholar]
- 65.Martinez-Vea A, Bardají A, Gutierrez C, Garca C, Peralta C, Marcas L, Oliver JA: Exercise blood pressure, cardiac structure, and diastolic function in young normotensive patients with polycystic kidney disease: A prehypertensive state. Am J Kidney Dis 44: 216–223, 2004 [DOI] [PubMed] [Google Scholar]
- 66.Martinez-Vea A, Valero FA, Bardají A, Gutierrez C, Broch M, Garcia C, Richart C, Oliver JA: Left ventricular hypertrophy in hypertensive patients with autosomal dominant polycystic kidney disease: Influence of blood pressure and humoral and neurohormonal factors. Am J Nephrol 20: 193–200, 2000 [DOI] [PubMed] [Google Scholar]
- 67.Zeier M, Geberth S, Schmidt KG, Mandelbaum A, Ritz E: Elevated blood pressure profile and left ventricular mass in children and young adults with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 3: 1451–1457, 1993 [DOI] [PubMed] [Google Scholar]
- 68.Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW: Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int 74: 1192–1196, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ivy DD, Shaffer EM, Johnson AM, Kimberling WJ, Dobin A, Gabow PA: Cardiovascular abnormalities in children with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 5: 2032–2036, 1995 [DOI] [PubMed] [Google Scholar]
- 70.Patch C, Charlton J, Roderick PJ, Gulliford MC: Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: A population-based study. Am J Kidney Dis 57: 856–862, 2011 [DOI] [PubMed] [Google Scholar]
- 71.Schrier RW, McFann KK, Johnson AM: Epidemiological study of kidney survival in autosomal dominant polycystic kidney disease. Kidney Int 63: 678–685, 2003 [DOI] [PubMed] [Google Scholar]
- 72.Orskov B, Rømming Sørensen V, Feldt-Rasmussen B, Strandgaard S: Improved prognosis in patients with autosomal dominant polycystic kidney disease in Denmark. Clin J Am Soc Nephrol 5: 2034–2039, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perrone RD, Abebe KZ, Schrier RW, Chapman AB, Torres VE, Bost J, Kaya D, Miskulin DC, Steinman TI, Braun W, Winklhofer FT, Hogan MC, Rahbari-Oskoui F, Kelleher C, Masoumi A, Glockner J, Halin NJ, Martin D, Remer E, Patel N, Pedrosa I, Wetzel LH, Thompson PA, Miller JP, Meyers CM, Bae KT, HALT PKD Study Group : Cardiac magnetic resonance assessment of left ventricular mass in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 2508–2515, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Belz MM, Hughes RL, Kaehny WD, Johnson AM, Fick-Brosnahan GM, Earnest MP, Gabow PA: Familial clustering of ruptured intracranial aneurysms in autosomal dominant polycystic kidney disease. Am J Kidney Dis 38: 770–776, 2001 [DOI] [PubMed] [Google Scholar]
- 75.Kocaman O, Oflaz H, Yekeler E, Dursun M, Erdogan D, Demirel S, Alisir S, Turgut F, Mercanoglu F, Ecder T: Endothelial dysfunction and increased carotid intima-media thickness in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis 43: 854–860, 2004 [DOI] [PubMed] [Google Scholar]
- 76.Ramunni A, Brescia P, Quaranta D, Bianco MS, Ranieri P, Dolce E, Coratelli P: Cutaneous microcirculation is impaired in early autosomal dominant polycystic kidney disease. Nephron Clin Pract 113: c71–c75, 2009 [DOI] [PubMed] [Google Scholar]
- 77.Heffernan KS, Kuvin JT, Sarnak MJ, Perrone RD, Miskulin DC, Rudym D, Chandra P, Karas RH, Menon V: Peripheral augmentation index and vascular inflammation in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 26: 2515–2521, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klahr S, Breyer JA, Beck GJ, Dennis VW, Hartman JA, Roth D, Steinman TI, Wang SR, Yamamoto ME, Modification of Diet in Renal Disease Study Group : Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. J Am Soc Nephrol 5: 2037–2047, 1995 [DOI] [PubMed] [Google Scholar]
- 79.Chapman AB, Johnson AM, Gabow PA, Schrier RW: Overt proteinuria and microalbuminuria in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 5: 1349–1354, 1994 [DOI] [PubMed] [Google Scholar]
- 80.Morgenthaler NG, Struck J, Alonso C, Bergmann A: Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem 52: 112–119, 2006 [DOI] [PubMed] [Google Scholar]
- 81.Morgenthaler NG, Struck J, Jochberger S, Dünser MW: Copeptin: Clinical use of a new biomarker. Trends Endocrinol Metab 19: 43–49, 2008 [DOI] [PubMed] [Google Scholar]
- 82.Meijer E, Bakker SJL, van der Jagt EJ, Navis G, de Jong PE, Struck J, Gansevoort RT: Copeptin, a surrogate marker of vasopressin, is associated with disease severity in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 361–368, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boertien WE, Meijer E, Li J, Bost JE, Struck J, Flessner MF, Gansevoort RT, Torres VE, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease CRISP : Relationship of copeptin, a surrogate marker for arginine vasopressin, with change in total kidney volume and GFR decline in autosomal dominant polycystic kidney disease: Results from the CRISP cohort. Am J Kidney Dis 61: 420–429, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meijer E, Boertien WE, Nauta FL, Bakker SJL, van Oeveren W, Rook M, van der Jagt EJ, van Goor H, Peters DJM, Navis G, de Jong PE, Gansevoort RT: Association of urinary biomarkers with disease severity in patients with autosomal dominant polycystic kidney disease: A cross-sectional analysis. Am J Kidney Dis 56: 883–895, 2010 [DOI] [PubMed] [Google Scholar]
- 85.Reed BY, Masoumi A, Elhassan E, McFann K, Cadnapaphornchai MA, Maahs DM, Snell-Bergeon JK, Schrier RW: Angiogenic growth factors correlate with disease severity in young patients with autosomal dominant polycystic kidney disease. Kidney Int 79: 128–134, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Torres VE, Grantham JJ, Chapman AB, Mrug M, Bae KT, King BF, Jr., Wetzel LH, Martin D, Lockhart ME, Bennett WM, Moxey-Mims M, Abebe KZ, Lin Y, Bost JE, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) : Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 640–647, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Helal I, McFann K, Reed B, Yan X-D, Schrier RW, Fick-Brosnahan GM: Serum uric acid, kidney volume and progression in autosomal-dominant polycystic kidney disease. Nephrol Dial Transplant 28: 380–385, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kocyigit I, Yilmaz MI, Orscelik O, Sipahioglu MH, Unal A, Eroglu E, Kalay N, Tokgoz B, Axelsson J, Oymak O: Serum uric acid levels and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Nephron Clin Pract 123: 157–164, 2013 [DOI] [PubMed] [Google Scholar]
- 89.Zheng D, Wolfe M, Cowley BD, Jr., Wallace DP, Yamaguchi T, Grantham JJ: Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 14: 2588–2595, 2003 [DOI] [PubMed] [Google Scholar]
- 90.Menon V, Rudym D, Chandra P, Miskulin D, Perrone R, Sarnak M: Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin J Am Soc Nephrol 6: 7–13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pavik I, Jaeger P, Kistler AD, Poster D, Krauer F, Cavelti-Weder C, Rentsch KM, Wüthrich RP, Serra AL: Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int 79: 234–240, 2011 [DOI] [PubMed] [Google Scholar]
- 92.Kistler AD, Serra AL, Siwy J, Poster D, Krauer F, Torres VE, Mrug M, Grantham JJ, Bae KT, Bost JE, Mullen W, Wüthrich RP, Mischak H, Chapman AB: Urinary proteomic biomarkers for diagnosis and risk stratification of autosomal dominant polycystic kidney disease: A multicentric study. PLoS ONE 8: e53016, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parikh CR, Dahl NK, Chapman AB, Bost JE, Edelstein CL, Comer DM, Zeltner R, Tian X, Grantham JJ, Somlo S: Evaluation of urine biomarkers of kidney injury in polycystic kidney disease. Kidney Int 81: 784–790, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ta MH, Harris DC, Rangan GK: Role of interstitial inflammation in the pathogenesis of polycystic kidney disease. Nephrology (Carlton) 18: 317–330, 2013 [DOI] [PubMed] [Google Scholar]
- 95.Orskov B, Christensen KB, Feldt-Rasmussen B, Strandgaard S: Low birth weight is associated with earlier onset of end-stage renal disease in Danish patients with autosomal dominant polycystic kidney disease. Kidney Int 81: 919–924, 2012 [DOI] [PubMed] [Google Scholar]
- 96.Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ: The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 13: 2723–2729, 2002 [DOI] [PubMed] [Google Scholar]
- 97.Vendramini LC, Nishiura JL, Baxmann AC, Heilberg IP: Caffeine intake by patients with autosomal dominant polycystic kidney disease. Braz J Med Biol Res 45: 834–840, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Orth SR, Stöckmann A, Conradt C, Ritz E, Ferro M, Kreusser W, Piccoli G, Rambausek M, Roccatello D, Schäfer K, Sieberth HG, Wanner C, Watschinger B, Zucchelli P: Smoking as a risk factor for end-stage renal failure in men with primary renal disease. Kidney Int 54: 926–931, 1998 [DOI] [PubMed] [Google Scholar]
- 99.Gattone VH, 2nd, Wang X, Harris PC, Torres VE: Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9: 1323–1326, 2003 [DOI] [PubMed] [Google Scholar]
- 100.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH, 2nd: Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10: 363–364, 2004 [DOI] [PubMed] [Google Scholar]
- 101.Nagao S, Nishii K, Katsuyama M, Kurahashi H, Marunouchi T, Takahashi H, Wallace DP: Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 17: 2220–2227, 2006 [DOI] [PubMed] [Google Scholar]
- 102.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE: Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19: 102–108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grantham JJ: Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 19: 1–7, 2008 [DOI] [PubMed] [Google Scholar]
- 104.Torres VE, Bankir L, Grantham JJ: A case for water in the treatment of polycystic kidney disease. Clin J Am Soc Nephrol 4: 1140–1150, 2009 [DOI] [PubMed] [Google Scholar]
- 105.Bankir L, Bouby N, Ritz E: Vasopressin: A novel target for the prevention and retardation of kidney disease? Nat Rev Nephrol 9: 223–239, 2013 [DOI] [PubMed] [Google Scholar]
- 106.Wang CJ, Grantham JJ, Wetmore JB: The medicinal use of water in renal disease. Kidney Int 84: 45–53, 2013 [DOI] [PubMed] [Google Scholar]
- 107.Wang CJ, Creed C, Winklhofer FT, Grantham JJ: Water prescription in autosomal dominant polycystic kidney disease: A pilot study. Clin J Am Soc Nephrol 6: 192–197, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT: Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology 11: 1033–1037, 1990 [DOI] [PubMed] [Google Scholar]
- 109.Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, Everson GT: Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology 26: 1282–1286, 1997 [DOI] [PubMed] [Google Scholar]