

 |
Magdy Younes is Distinguished Professor Emeritus at the University of Manitoba, Canada. He obtained his medical degree from the University of Alexandria, Egypt. He subsequently trained in Internal and Pulmonary medicine at McGill University hospitals in Montreal and obtained a PhD in respiratory physiology from McGill University. He has held academic positions at McGill University, University of Texas Medical Branch, Dalhousie University in Halifax, Canada, and the University of Manitoba. He has been active in research in several areas of respiratory pathophysiology including control of breathing during exercise, pathogenesis of respiratory failure, haemodynamics of the pulmonary circulation, patient–ventilator interactions and pathogenesis of obstructive sleep apnoea. He has several inventions including the development of ‘proportional assist ventilation’ (PAV).
Before addressing this question it is necessary to define loop gain (LG) in the context of obstructive sleep apnoea (OSA). In a closed loop system, such as the breathing system, a perturbation in the controlled loop component (the respiratory apparatus, or ‘plant’) elicits changes in the feedback (blood gas tensions) received by the controller (respiratory centres), which in turn effects a compensatory response in the plant. The initial response may partially correct the blood gas changes, with the residual changes being gradually corrected later. This is a stable response. However, the initial response may result in over-correction of the gas changes (‘overshoot’) such that they are better than what existed before the perturbation. The ventilatory apparatus is then inhibited through the same changes in blood gas tensions that resulted in the initial response, and a second hypopnoea results. If the second hypopnoea is less severe than the original one, the disturbance in gas tensions will also be less severe, eliciting a lesser response and a less severe hypopnoea results, and so on. Ultimately, the system stabilizes. If the overshoot results in such improvement in gas tensions that the second hypopnoea is more severe than the initial one, the cycle can perpetuate indefinitely. LG is the ratio of the initial response to the initial perturbation. A LG of <1.0 is consistent with stable breathing while a LG >1.0 results in perpetual cycling.
LG is determined by: (a) how much blood gas tensions deteriorate before the controller can reverse the trend (maximal changes), and (b) how much the controller will respond to these maximal changes (‘controller gain’) (Khoo et al. 1982; Younes, 1989; Younes et al. 2001). Maximal changes are determined by the time course of changes in gas tensions after the onset of hypopnoea (‘plant gain’; related to lung volume, mixed venous gas tensions, and other factors) and the time it takes to elicit a ventilatory response (‘Delay’). The longer the Delay, the greater the maximal changes and the higher the plant gain. Controller gain is determined by the sensitivity of respiratory centres to blood gas changes, which is also time dependent; the longer the 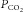 change lasts the greater the response to the same
change lasts the greater the response to the same 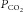 change (Cunningham et al. 1986). Thus, both components of LG increase as a function of ‘response delay’.
change (Cunningham et al. 1986). Thus, both components of LG increase as a function of ‘response delay’.
When respiratory mechanical impedance is fairly constant, such as in normal subjects or in patients with a stable upper airway (OSA on continuous positive airway pressure (CPAP)), Delay is strictly a function of the lung–carotid circulatory delay. Furthermore, because respiratory impedance is ‘constant’ it affects the initial perturbation and subsequent response equally. In this case, LG is a function of the relatively short circulatory delay (∼7 s), and the relatively low (because of the short Delay) plant and controller gains. Under such conditions recurrent apnoeas or hypopnoeas occur when circulatory delay is abnormally long (heart failure), plant gain is high (low lung volume, poor mixed venous blood gas tensions, etc.) and/or high ventilatory responses to CO2 and hypoxia. LG under such conditions predominantly reflects the behaviour of chemoreceptor-mediated feedback loops and I will refer to this as ‘Chemical LG’.
In OSA, the repetitive nature of the events is consistent with a LG that is >1.0. This high LG cannot be analysed in the same way as in central apnoeas because the delay between event onset and initiation of ventilatory response is not the lung–carotid delay, but the time taken for upper airway (UA) dilator muscles to open the airway (event duration). Furthermore, respiratory mechanics during and following the obstructive phase are vastly different. Event duration (up to 100 s) is invariably longer than circulation delay (∼7 s; Younes et al. 2007); progressive increase in effort occurs well before the airway opens (Remmers et al. 1978; Onal & Lopata, 1982; Berry & Gleeson, 1997), and the mechanisms that determine this duration are extremely complex (Younes, 2008; White & Younes, 2012; Younes et al. 2012). As the obstruction continues, plant gain increases and controller gain increases up to the level of respiratory pressure generation. Thus potential LG continues to increase, but actual LG is zero because there is no ventilatory response. At opening, potential LG may be very high, but it cannot be fully realized unless UA opens completely, which is not usually the case (snoring and flow limitation usually continue during the open phase). The net (actual) LG in OSA is determined by the potential LG at the end of obstruction (a function of event duration) as attenuated by the residual high impedance during the open phase. This net LG is what determines whether the cycle will repeat.
In summary, there is no doubt that UA instability and the resulting lengthy Delay (event duration > circulatory delay) is the main mechanism of increased LG in OSA. Thus, the increased LG in OSA is, in large measure, the result of the obstructive event itself. What might be debated is whether Chemical LG is increased and, if so, whether this contributes to OSA severity and whether the increase was the result or cause of the disease. To evaluate Chemical LG in these patients it is necessary to measure it when UA is stable, for example by measuring ventilatory responses during wakefulness or while the patient is asleep on CPAP. Measurements of ventilatory responses during wakefulness yielded highly conflicting results (see Loewen et al. 2009 for review). These studies used chemical challenges that do not reflect the situation in OSA. In OSA the chemical challenge is brief stimulation by a mixture of hypoxia and hypercapnia (asphyxia), whereas ventilatory response studies during wakefulness used re-breathing or steady-state exposures. By contrast, when measured during sleep, directly by proportional assist ventilation (Younes et al. 2001; Wellman et al. 2004) or indirectly from dynamic ventilatory responses to brief changes in inspired gases (Younes et al. 2007; Loewen et al. 2009), Chemical LG was somewhat elevated in OSA patients, but the increase is insufficient to cause instability in the absence of unstable UA (Younes et al. 2001; Wellman et al. 2004). Hence, central apnoea develops infrequently in patients while on CPAP.
Does a somewhat increased Chemical LG aggravate the instability produced by abnormal UA mechanics? A higher Chemical LG would lead to a faster increase in respiratory effort during the obstruction. However, since UA opening via arousals (arousal threshold; Gleeson et al. 1990; Kimoff et al. 1994; Eckert & Younes 2014) or via reflex activation of pharyngeal dilators (‘effective recruitment threshold’; Younes, 2008; White & Younes, 2012; Younes et al. 2012) occurs when a certain chemical drive is reached, the increased Chemical LG would reduce event duration but not result in higher chemical stimulus at the time of opening. This, per se, would not aggravate the instability. However, because of circulation delay, chemical drive continues to increase for 1–2 breaths beyond event termination. An above normal ventilatory response might, therefore, result in greater overshoot, increasing the chance of recurrence. Whether the higher controller gain in OSA contributes to OSA severity is currently debatable. Chemical LG correlates with apnoea severity only in patients with mild UA mechanical abnormality (Wellman et al. 2004). Oxygen breathing (Martin et al. 1982; Smith et al. 1984; Gold et al. 1985, 1986; Wellman et al. 2008) and acetazolamide (Sharp et al. 1985; Tojima et al. 1988; Whyte et al. 1988; Edwards et al. 2012), both of which attenuate Chemical LG, partially reduce the apnoea–hypopnoea index in some but not all patients. This is understandable since Chemical LG is only modestly elevated and only in some patients (Younes et al. 2001; Wellman et al. 2004, 2008; Edwards et al. 2012).
With respect to whether the increased Chemical LG in OSA is inherent or acquired as a result of OSA, there is little to discuss. There is only one study that directly addressed this issue. We studied dynamic ventilatory responses to asphyxia mimicking obstructive events during sleep before and several weeks after therapy with CPAP. There was a dramatic reduction in these responses following treatment (Fig. 1; Loewen et al. 2009). Thus, the high Chemical LG did not pre-exist. Case closed!
Figure 1. Change in dynamic ventilatory response to asphyxia after several weeks of CPAP therapy.
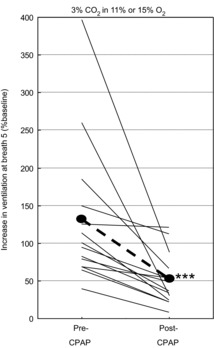
Increase in ventilation 5 breaths after changing inspired gas from air to 3% CO2 in 11% or 15% oxygen before and after several weeks of therapy with continuous positive airway pressure (CPAP). In both cases the patient was on optimal CPAP during the measurements. ***P < 0.005. Reproduced from Loewen et al. (2009).
In summary, the high LG in OSA is due to OSA and not to high Chemical LG. The contribution of Chemical LG to OSA severity, if any, is modest and limited to some patients in whom Chemical LG is high.
Call for comments
Readers are invited to give their views on this and the accompanying CrossTalk articles in this issue by submitting a brief comment. Comments may be posted up to 6 weeks after publication of the article, at which point the discussion will close and authors will be invited to submit a ‘final word’. To submit a comment, go to http://jp.physoc.org/letters/submit/jphysiol;592/14/2899
Additional information
Competing interests
None declared.
References
- Berry RB, Gleeson K. Respiratory arousal from sleep: mechanisms and significance. Sleep. 1997;20:654–675. doi: 10.1093/sleep/20.8.654. [DOI] [PubMed] [Google Scholar]
- Cunningham DJC, Robbins PA. Integration of respiratory responses to changes in alveolar partial pressures of CO2, O2 and in arterial pH. In: Widdicombe JG, Wolff CB, editors; Cherniack NS, editor. Handbook of Physiology, section 3, The Respiratory System, vol II, Control of Breathing. Bethesda, MD, USA: American Physiological Society; 1986. pp. 475–528. [Google Scholar]
- Eckert DJ, Younes M. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol (1985) 2014;116:302–313. doi: 10.1152/japplphysiol.00649.2013. [DOI] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Eckert DJ, White DP, Butler JP, Owens RL, Malhotra A, Wellman A. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012;590:1199–1211. doi: 10.1113/jphysiol.2011.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson K, Zwillich CW, White DP. The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis. 1990;142:295–300. doi: 10.1164/ajrccm/142.2.295. [DOI] [PubMed] [Google Scholar]
- Gold AR, Bleecker ER, Smith PL. A shift from central and mixed sleep apnea to obstructive sleep apnea resulting from low-flow oxygen. Am Rev Respir Dis. 1985;132:220–223. doi: 10.1164/arrd.1985.132.2.220. [DOI] [PubMed] [Google Scholar]
- Gold AR, Schwartz AR, Bleecker ER, Smith PL. The effect of chronic nocturnal oxygen administration upon sleep apnea. Am Rev Respir Dis. 1986;134:925–929. doi: 10.1164/arrd.1986.134.5.925. [DOI] [PubMed] [Google Scholar]
- Khoo MCK, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol. 1982;53:644–659. doi: 10.1152/jappl.1982.53.3.644. [DOI] [PubMed] [Google Scholar]
- Kimoff RJ, Cheong TH, Olha AE, Charbonneau M, Levy RD, Cosio MG, Gottfried SB. Mechanisms of apnea termination in obstructive sleep apnea. Role of chemoreceptor and mechanoreceptor stimuli. Am J Respir Crit Care Med. 1994;149:707–714. doi: 10.1164/ajrccm.149.3.8118640. [DOI] [PubMed] [Google Scholar]
- Loewen A, Ostrowski M, Laprairie J, Atkar R, Gnitecki J, Hanly P, Younes M. Determinants of ventilatory instability in obstructive sleep apnea: inherent or acquired? Sleep. 2009;32:1355–1365. doi: 10.1093/sleep/32.10.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RJ, Sanders MH, Gray BA, Pennock BE. Acute and long-term ventilatory effects of hyperoxia in the adult sleep apnea syndrome. Am Rev Respir Dis. 1982;125:175–180. doi: 10.1164/arrd.1982.125.2.175. [DOI] [PubMed] [Google Scholar]
- Onal E, Lopata M. Periodic breathing and the pathogenesis of occlusive sleep apnea. Am Rev Respir Dis. 1982;126:676–680. doi: 10.1164/arrd.1982.126.4.676. [DOI] [PubMed] [Google Scholar]
- Sharp JT, Druz WS, D'Souza V, Diamond E. Effect of metabolic acidosis upon sleep apnea. Chest. 1985;87:619–624. doi: 10.1378/chest.87.5.619. [DOI] [PubMed] [Google Scholar]
- Smith PL, Haponik EF, Bleecker ER. The effects of oxygen in patients with sleep apnea. Am Rev Respir Dis. 1984;130:958–963. doi: 10.1164/arrd.1984.130.6.958. [DOI] [PubMed] [Google Scholar]
- Remmers JE, Degroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol. 1978;44:931–938. doi: 10.1152/jappl.1978.44.6.931. [DOI] [PubMed] [Google Scholar]
- Tojima H, Kunitomo F, Kimura H, Tatsumi K, Kuriyama T, Honda Y. Effects of acetazolamide in patients with the sleep apnoea syndrome. Thorax. 1988;43:113–119. doi: 10.1136/thx.43.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Jordan AS, Malhotra A, Fogel RB, Katz ES, Schory K, Edwards JK, White DP. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. 2004;170:1225–1232. doi: 10.1164/rccm.200404-510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: Role of loop gain. Respir Physiol Neurobiol. 2008;162:144–151. doi: 10.1016/j.resp.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DP, Younes MK. Obstructive sleep apnea. Compr Physiol. 2012;2:2541–2594. doi: 10.1002/cphy.c110064. [DOI] [PubMed] [Google Scholar]
- Whyte KF, Gould GA, Airlie MA, Shapiro CM, Douglas NJ. Role of protriptyline and acetazolamide in the sleep apnea/hypopnea syndrome. Sleep. 1988;11:463–472. doi: 10.1093/sleep/11.5.463. [DOI] [PubMed] [Google Scholar]
- Younes M. The physiologic basis of central apnea and periodic breathing. Curr Pulmonol. 1989;10:265–326. [Google Scholar]
- Younes M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J Appl Physiol. 2008;105:1389–1405. doi: 10.1152/japplphysiol.90408.2008. [DOI] [PubMed] [Google Scholar]
- Younes M, Loewen AH, Ostrowski M, Laprairie J, Maturino F, Hanly PJ. Genioglossus activity available via non-arousal mechanisms vs. that required for opening the airway in obstructive apnea patients. J Appl Physiol. 2012;112:249–258. doi: 10.1152/japplphysiol.00312.2011. [DOI] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A, Hanly P. Mechanisms of ventilatory instability in individual patients with obstructive sleep apnea. J Appl Physiol. 2007;103:1929–1941. doi: 10.1152/japplphysiol.00561.2007. [DOI] [PubMed] [Google Scholar]
- Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163:1181–1190. doi: 10.1164/ajrccm.163.5.2007013. [DOI] [PubMed] [Google Scholar]