

Abstract
The landmark discovery by Bayliss and Starling in 1902 of the first hormone, secretin, emerged from earlier observations that a response (pancreatic secretion) following a stimulus (intestinal acidification) occurred after section of the relevant afferent nerve pathway. Nearly 80 years elapsed before it became clear that visceral afferent neurons could themselves also be targets for gut and other hormones. The action of gut hormones on vagal afferent neurons is now recognised to be an early step in controlling nutrient delivery to the intestine by regulating food intake and gastric emptying. Interest in these mechanisms has grown rapidly in view of the alarming global increase in obesity. Several of the gut hormones (cholecystokinin (CCK); peptide YY3–36 (PYY3–36); glucagon-like peptide-1 (GLP-1)) excite vagal afferent neurons to activate an ascending pathway leading to inhibition of food intake. Conversely others, e.g. ghrelin, that are released in the inter-digestive period, inhibit vagal afferent neurons leading to increased food intake. Nutrient status determines the neurochemical phenotype of vagal afferent neurons by regulating a switch between states that promote orexigenic or anorexigenic signalling through mechanisms mediated, at least partly, by CCK. Gut–brain signalling is also influenced by leptin, by gut inflammation and by shifts in the gut microbiota including those that occur in obesity. Moreover, there is emerging evidence that diet-induced obesity locks the phenotype of vagal afferent neurons in a state similar to that normally occurring during fasting. Vagal afferent neurons are therefore early integrators of peripheral signals underling homeostatic mechanisms controlling nutrient intake. They may also provide new targets in developing treatments for obesity and feeding disorders.
 |
Graham J. Dockray is Emeritus Professor of Physiology at the University of Liverpool. He was a Fogarty International Fellow of the National Institutes of Health, USA and has held positions as Head of the Department of Physiology, Pro Vice-Chancellor and Deputy Vice-Chancellor. He works in the field of regulatory peptides of the gut and brain. He has delivered the G. L. Brown, G. W. Harris and Annual Review Prize Lectures of The Physiological Society and is a member of Academia Europaea, Fellow of the Academy of Medical Sciences, Honorary FRCP and Fellow of the Royal Society.
Introduction
I greatly appreciate the honour of being invited to give this lecture which was established by The Physiological Society as a memorial to Bayliss and Starling. I approach the task with considerable hesitation and humility. For one thing, there is the challenge of doing justice to two such formidable scientists but I'm mindful too of the high standing and enormous contributions of my predecessors in the role. The early lecturers in the series had all known Bayliss and Starling and were able to give very personal accounts of their work. Rod Gregory was the first not to have known them personally (Gregory, 1974). As it happens, it was Gregory who gave me my first job; I knew him for the last 20 years of his life and learnt a huge amount from him. He had been trained by the first lecturer in the series, Charles Lovatt Evans, who as a young man had worked with Bayliss and Starling and so there is a sort of personal link from them that runs through several generations to the present lecture.
From about 1898, the collaboration between Bayliss and Starling was focused mainly in the area of digestive physiology: together they published about 10 papers covering gut motility and enzyme activity but by far their most spectacular and enduring work was, of course, the discovery of hormonal reflex mechanisms. About 65 years later I was drawn to the field and still remember the impact their papers had on me as a young graduate student. Their key experiment was done in the afternoon of 16th January 1902. The events have been described in detail by Starling's friend Charles Martin who was present and while these are well known it is worth just reiterating the thought process. Bayliss and Starling knew that putting acid in the duodenum stimulates the flow of pancreatic juice and that this still occurs after section of the nerves to the intestine: they reasoned that the link must be via the circulation and they then showed that the active factor, which they named secretin, could be extracted from intestine (Bayliss & Starling, 1902a). The significance of the observation was immediately obvious to them and in a preliminary communication just one week later they pointed out that this was the first direct experimental proof of what, at the time, they called ‘chemical sympathy’ between different organs (Starling later introduced the word ‘hormone’) and with penetrating insight they speculated that this was just one of a general class of similar mechanisms (Bayliss & Starling, 1902b). It was to be nearly 80 years before it became clear that gut hormones might also signal to the CNS by acting on afferent neurons.
Gut hormones: a potted history
In the early 20th century, concepts of what I've called the dialogue between gut and brain were dominated by the work of Pavlov who had characterised nervous reflex mechanisms controlling gastrointestinal function. Understanding of the pathways by which the gut speaks to the brain were reasonably well developed and had been perceptively reviewed by Hurst in his Goulstonian Lectures where he drew attention to the role of vagal afferents in physiological control mechanisms and of spinal afferents in mediating painful sensations from the gut (Hertz, 1911). Nevertheless, this system was considered largely independent of the gut hormones. Over the 70 years or so following the discovery of secretin, many other gut hormones were reported. The main highlights are the discovery of gastrin in 1905 (Edkins, 1905), cholecystokinin (CCK) in 1928 and pancreozymin (PZ) in 1943 (Ivy & Oldberg, 1928; Harper & Raper, 1943). In the next generation, Gregory, Tracy and Kenner isolated, sequenced and synthesised gastrin (Gregory, 1968). At about the same time, Jorpes and Mutt isolated and sequenced secretin and CCK, and showed CCK and PZ were a single hormone (Jorpes & Mutt, 1966). Then, after Jorpes’ death, Viktor Mutt discovered many other regulatory peptides in the intestine including vasoactive intestinal polypeptide (VIP), neuropeptide Y (NPY), and peptide YY (PYY) (Mutt, 1982). By about the time that Rod Gregory gave his Bayliss and Starling Lecture in 1973 the standard model was that gut hormones were produced in specialised cells (now called enteroendocrine cells, EECs) scattered in the epithelium and responding to luminal nutrient; it was thought likely that some of the EEC products might also have local or paracrine effects, while the targets of hormonal action were recognised to include epithelial cells and smooth muscle. In the context of the gut–brain dialogue, however, it is striking that, with just the occasional exception, neurons had not been considered as targets for the gut hormones. In other words while both hormonal and nervous reflexes controlling gastrointestinal function were well known these were considered to be largely distinct mechanisms albeit converging, and potentially interacting, at the level of target organs.
Hormones and the gut–brain axis
Two developments changed things. First, Gibbs and Smith showed that CCK inhibited food intake in rats. They used doses that were within the range for stimulation of pancreatic secretion, and their design allowed them to exclude possible effects secondary to gastric motility (Gibbs et al. 1973): self-evidently, CCK could influence the brain to change behaviour. Second, it became clear that gut hormones, of which CCK was an early and good example, were also expressed in the brain frequently in concentrations higher than those in the intestine (Dockray, 1976; Rehfeld, 1978); the chemical identity of the neuronal CCK was shown to correspond to the minimal fragment of the intestinal hormone required for biological activity (Dockray et al. 1978). Shortly afterwards, came the demonstration that CCK receptors were also widely expressed by neurons raising, amongst other things, the prospect of cross-talk between gut hormones and CNS neurons (Innis & Snyder, 1980).
In principle, the inhibition of food intake by circulating CCK could be mediated by direct effects on CNS neurons after crossing or by-passing the blood–brain barrier, or it could be mediated by afferent neurons and in this context vagal afferents would be good candidates. Smith et al. showed by careful lesion experiments that the effect of CCK was likely mediated by an action on abdominal vagal afferent fibres (Smith et al. 1981). Many different lines of evidence have since supported this concept. Thus CCK1 receptors are expressed in nodose ganglia neurons and transported towards the gut (Moran et al. 1987; Moriarty et al. 1997; Fig. 1A), vagal afferent fibre discharge is increased by CCK (Davison, 1986; Schwartz et al. 1991) and similarly neurons in the nucleus tractus solitarii (NTS) that receive an input from vagal afferents are stimulated by CCK given by close arterial injection to the upper gastrointestinal tract (Raybould et al. 1988); the expression of fos in NTS neurons is also increased by peripheral CCK administration (Fraser & Davison, 1992; Luckman, 1992); finally the effects of lesioning afferent neurons with capsaicin support the conclusion that CCK acts via these neurons to change behaviour and autonomic outflow (Raybould & Tache, 1988; Forster et al. 1990).
Figure 1. Overview of nutrient sensing mechanisms in the gut and signalling to the CNS.
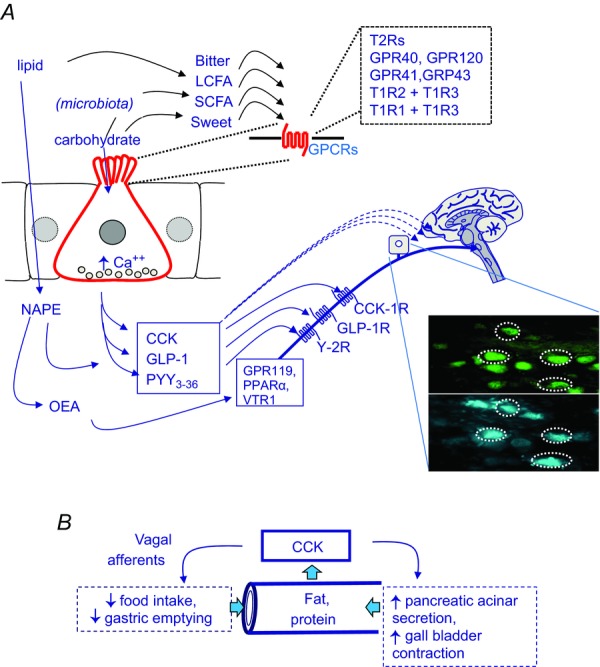
A, sensing of gut luminal chemicals is a property of enteroendocrine cells (red); examples of luminal sensing G-protein coupled receptors (GPCRs) on the apical membrane include those for bitter compounds (T2R), amino acids (extracellular calcium-sensing receptor, CaSR; T1R1 + T1R3), long chain fatty acids (LCFA: GPR40, GPR120), short chain fatty acids (SCFA: GPR41, GPR43) and sweet compounds (T1R2 + T1R3). Increased intracellular calcium controls exocytosis at the basolateral membrane of hormone-containing secretory vesicles. Examples of intestinal hormones that act on vagal afferent neurons include cholecystokinin (CCK) from I-cells predominantly found in the proximal small intestine, and acting at vagal CCK1 receptors, and GLP-1 and PYY3–36 from L-cells predominantly in the ileum and colon and acting at GLP-1 and Y2 receptors respectively (PYY3–36 is generated extracellularly from the primary secretory product PYY). Dietary oleate is converted by enterocytes to N-acylphosphatidylethanolamine (NAPE) which in turn gives rise to oleoylethanolamide (OEA) which may act on PPARα, GPR119 or TRPV1. The inset shows nodose ganglion cells labelled retrogradely from the stomach (blue) and expressing CCK1 receptors (green). B, CCK is a master-regulator of the luminal environment in the upper small intestine. It is released by fat and protein in the intestine and matches delivery of nutrient to the capacity for nutrient digestion: this is achieved by inhibition of nutrient delivery by slowing gastric emptying and inhibiting food intake (via vagal afferent stimulation) and by stimulating gall bladder contraction and pancreatic acinar cell secretion (both of which can be direct effects) thereby delivering deliver bile salt and digestive enzymes to the intestine.
The main consequences of vagal afferent stimulation by CCK (inhibition of food intake and gastric emptying) serve to restrict nutrient delivery to the intestine. CCK regulates, therefore, a balance between the delivery of pancreatic enzymes and bile salt to the intestine (which determine the capacity for fat and protein digestion) and the presence of substrate to be digested (Dockray, 1988; Fig. 1B). While CCK is one of the best-studied examples of the intestinal hormones that act on vagal afferent neurons to inhibit food intake, there are also others notably GLP-1 and the PYY product, PYY3–36, both originating from L-cells in the ileum and colon (Abbott et al. 2005; Burdyga et al. 2008; Bucinskaite et al. 2009; Fig. 1A). However, peptide hormones are not the only neurohumoral messengers involved in gut–brain signalling: lipid amides like oleoylethanolamide produced by enterocytes from dietary oleate also inhibit food intake through mechanisms depending on intact vagal afferent pathways with potential roles, in this case, for several different receptors including peroxisome proliferator–activator receptor (PPAR)-α, GPR119 and the vanilloid receptor (TRPV)-1 (Rodriguez et al. 2001; Wang et al. 2005; Overton et al. 2006; Schwartz et al. 2008; Fig. 1A).
Vagal afferent neurons: also targets for orexigenic hormones
Over the last decade or so it has become clear that in addition to gut signals that limit nutrient delivery, there are also signalling molecules released in the absence of food that tend to increase food intake and accelerate gastric emptying – examples include ghrelin (Kojima et al. 1999; Date et al. 2002; Burdyga et al. 2006b), orexin-A (Kirchgessner & Liu, 1999; Burdyga et al. 2003) and endocannabinoids such as anandamide (Gomez et al. 2002; Burdyga et al. 2004). In each of these cases, vagal afferent neurons express the appropriate receptors and there is extensive overlap in the pattern of expression of these receptors and those of factors that inhibit nutrient delivery (CCK, leptin, PYY3–36 and GLP-1; Fig. 2A and B). This clearly provides a basis for functional interactions and, for example, both orexin-A (Burdyga et al. 2003; Fig. 2C) and ghrelin inhibit vagal afferent activation in response CCK (Date et al. 2002). The interactions between orexigenic and satiety signals are likely to be particularly important in the early phase of digestion when the former are decreasing and the latter rising, as well as at the start of the interdigestive phase when this situation is reversed.
Figure 2. Vagal afferent neurons express receptors for both orexigenic and satiety factors.
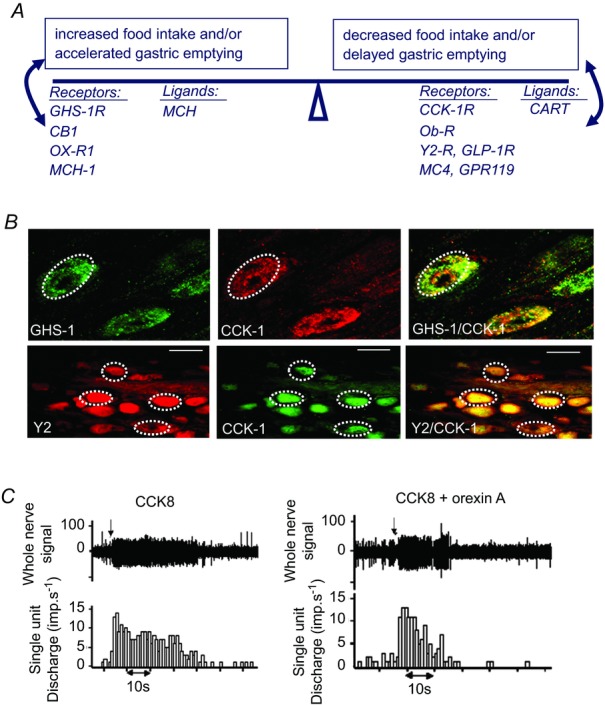
A, vagal afferent neurons express receptors for factors that stimulate food intake and/or gastric emptying including the GHS1, CB1, Ox-R1, MCH1 receptors, and for factors that inhibit food intake and/or gastric emptying including CCK1, Ob-R, GLP-1, Y2, MC4, GPR119. The same neurons also express two neuropeptide transmitters: MCH which stimulates food intake, and CART which inhibits it. B, the same neurons can express receptors for multiple orexigenic and satiety factors: examples are shown for GHS1 and CCK1 receptor expression and for Y2 and CCK1 receptor expression. C, orexigenic factors, in this case orexin-A, inhibit the stimulation of vagal afferent discharge by satiety factors, in this case CCK (Burdyga et al. 2003).
Gut–brain signalling in humans
The worldwide increase in obesity has inevitably focused attention on these control systems in humans. It has been clear for over a decade that the different forms of bariatric surgery produce long term weight loss in obese patients (Buchwald et al. 2004); there may also be rapid improvement in glycaemic control (Schauer et al. 2012). These clinical observations give rise to the idea that disruption of gut–brain signalling can be therapeutically beneficial and consequently that the development of non-invasive approaches that mimic the effect of bariatric surgery would be advantageous. However, developing novel therapeutic approaches clearly depends on an understanding of the mechanisms of gut–brain signalling in humans and this presents challenges.
Functional magnetic resonance imaging (fMRI) has been used by several groups to define regions of the human brain activated by exogenous gut hormones administered intravenously, notably ghrelin (Malik et al. 2008) and PYY3–36 (Batterham et al. 2007), as well as leptin (Farooqi et al. 2007). Moreover, this approach identifies human CNS responses to ingested glucose with some evidence that the effects are mediated by insulin (Liu et al. 2000). Together with David Thompson, Shane McKie and John McLaughlin in Manchester, we have applied fMRI to studies of gut–brain signalling mediated by endogenous CCK in normal humans. In developing robust test meals for these studies we made use of an earlier observation that ingested dodecanoic acid (C12) provides a chemically defined, readily administered stimulus to CCK release producing (in the appropriate dose) plasma concentrations in the physiological range and that are associated with inhibition of gastric emptying and stimulation of gall-bladder contraction (McLaughlin et al. 1999). In fMRI studies, ingested C12 produced a robust increase in the blood oxygen level-dependent (BOLD) signal in the brain stem and hypothalamus of normal subjects and a CCK1 receptor antagonist completely inhibited these response (Lassman et al. 2010). Interestingly, ghrelin depressed the BOLD signal in brain stem and hypothalamus in response to C12, consistent with suppression of a satiety signalling pathway (Jones et al. 2012; Fig. 3).
Figure 3. fMRI reveals increased hypothalamic and brain stem blood flow in response to ingestion of fatty acid, and inhibition by ghrelin.
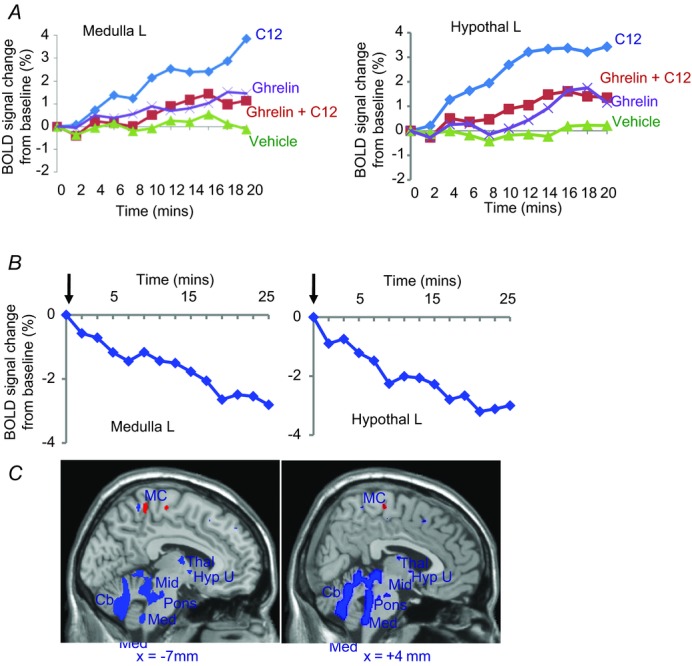
Blood oxygen-level dependent (BOLD) signals detected by functional magnetic resonance imaging in the hypothalamus and brain stem of healthy individuals are inhibited by ghrelin. A, after intragastric C12 to stimulate CCK release, there is increased hypothalamic and brain stem BOLD signals that are depressed by infusion of ghrelin in a dose (1.25 pmol kg−1 min−1) that increased plasma concentrations in the physiological range. B, after a mixed meal in healthy subjects, a bolus injection of ghrelin (0.3 nmol kg−1) decreased BOLD signal in hypothalamus and brain stem. C, brain images showing areas exhibiting increased BOLD signal (red) and decreased (blue) after administration of i.v. ghrelin compared with saline 2 h after a meal at P < 0.005 for display purposes (Jones et al. 2012).
Recent work suggests this sort of imaging approach may also help elucidate interactions between homeostatic and hedonic mechanisms controlling food intake. Van Oudenhove et al. recently reported that the C12 protocol I've just described attenuated BOLD signals in response to sadness induced by auditory and visual cues, and including those in areas such as the brain stem and hypothalamus (Van Oudenhove et al. 2011). About the same time, stress in the form of social defeat in mice was shown to release plasma ghrelin and promote a preference for dietary fat (Chuang et al. 2011). Together these studies start to provide a mechanistic framework for understanding the way that homeostatic and hedonic systems controlling food intake might interact, as well as providing a basis for understanding the mechanisms of so called ‘comfort feeding’ (Cizza & Rother, 2011).
Neurochemical plasticity of vagal afferent neurons
Vagal afferent neurons are more than simple reporters of changes at their periphery. For one thing their neurochemical phenotype reflects nutrient status. In the rat, fasting for just a few hours depresses expression of some receptors, e.g. the Y2 receptor which responds to the intestinal satiety peptide PYY3–36 (Burdyga et al. 2008), while there is increased expression of others, e.g. the cannabinoid CB1 receptor (Burdyga et al. 2004) which is associated with increased food intake (Gomez et al. 2002; Fig. 4). This switch in neurochemical phenotype can occur when as little as 10% of daily food intake is withdrawn, for example by fasting during the light phase (Burdyga et al. 2010). Refeeding fasted animals rapidly reverses the phenotype by increasing Y2 expression and inhibiting CB1 expression. The switch in neurochemical phenotype is mediated by CCK since, importantly, the effect of refeeding is blocked by a CCK1 receptor antagonist and is mimicked by exogenous CCK.
Figure 4. Nutrient-dependent expression of CB1 and Y2 receptors by vagal afferent neurons.
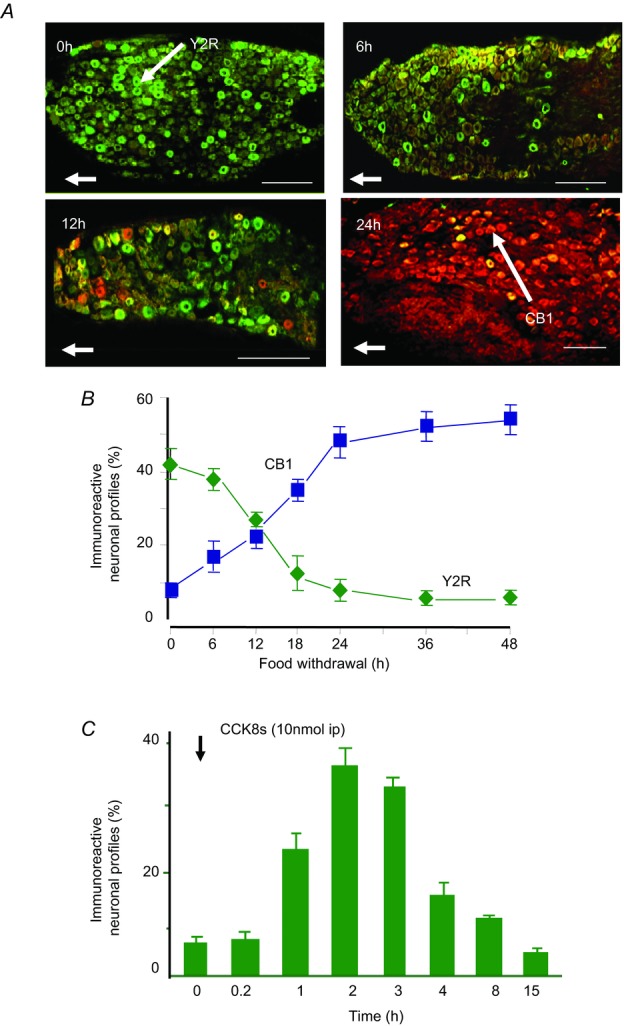
A, immunohistochemical localisation of CB1 receptors (red) and Y2 receptors (green) in nodose ganglia from rats fed ad libitum (0 h) or fasted for 6, 12 or 24 h. B, quantification of neurons expressing CB1 and Y2R in nodose ganglia of fasted rats. C, CCK8s (10 nmol kg−1 i.p.) stimulates expression of Y2R in nodose ganglia of rats fasted for 24 h (Burdyga et al. 2008).
What we call the ‘CCK switch’ extends not only to some receptors, but also to two neuropeptide transmitters synthesized by vagal afferent neurons (Fig. 5). One of these, CART (cocaine and amphetamine regulated transcript) was shown some time ago to be co-expressed in vagal afferent neurons with CCK1 receptors (Broberger et al. 1999), and is associated with inhibition of food intake; the other, MCH (melanin concentrating hormone) is expressed in the same neurons and is associated with stimulation of food intake (Burdyga et al. 2006a). There is decreased CART expression and increased MCH expression in nodose neurons in fasted rats, and this is reversed by treatment with CCK or refeeding. The CCK-switch can also be observed in cultured neurons. Thus, when rat nodose ganglion neurons are cultured in serum-free conditions, which equates to the fasted state, there is expression of MCH but not CART, while addition of CCK leads to rapid (60–90 min) stimulation of CART and decreased MCH expression (de Lartigue et al. 2007); the actions of CCK are inhibited by ghrelin.
Figure 5. Nutrient-dependent neurochemical switching in vagal afferent neurons.
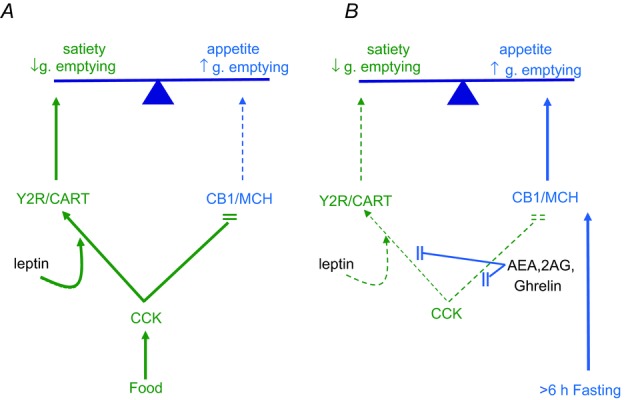
A, release of CCK in the post-prandial state stimulates expression in vagal afferent neurons of Y2R and the satiety peptide CART, while expression of CB1 and the orexigenic peptide MCH is suppressed. Leptin potentiates the action of CCK. These effects promote satiety signalling and inhibition of gastric emptying. B, in the fasted state (starting in rats approximately 6 h after a meal), plasma CCK is depressed while there is increased ghrelin secretion and endocannabinoid synthesis. The latter are associated with increased CB1 and MCH expression and depressed Y2R and CART expression, leading to enhanced appetite and stimulation of gastric emptying.
Co-operative interactions between CCK and leptin on vagal afferent neurons
In cultured neurons the stimulation of CART or Y2-receptor expression occurs in response to relatively high concentrations of CCK (10 nm) – approximately 1000 times those in plasma after a meal. However, the response to CCK is strongly potentiated by leptin (Fig. 6A). It has been known for many years that leptin stimulates the discharge of vagal afferent neurons (Wang et al. 1997), potentiates stimulation by CCK (Gaige et al. 2002; Peters et al. 2004) and acts locally in the gut to potentiate the satiety effect of CCK (Peters et al. 2006). In the present context, it is significant that in the presence of leptin the dose–response curve for CCK stimulation of CART or Y2 expression is moved to the left and concentrations of CCK approaching those in the physiological range (10 pm) are biologically effective (de Lartigue et al. 2010; Fig. 6A). In addition to these interactions between leptin and CCK there are also longer term interactions that enhance the satiety effect of CCK (de Lartigue et al. 2010). The neurochemical phenotype of vagal afferent neurons is therefore dependent on integration of signals representing both adipocyte status and recent nutrient ingestion.
Figure 6. Co-operative interactions between leptin and CCK to regulate CART expression in vagal afferent neurons.
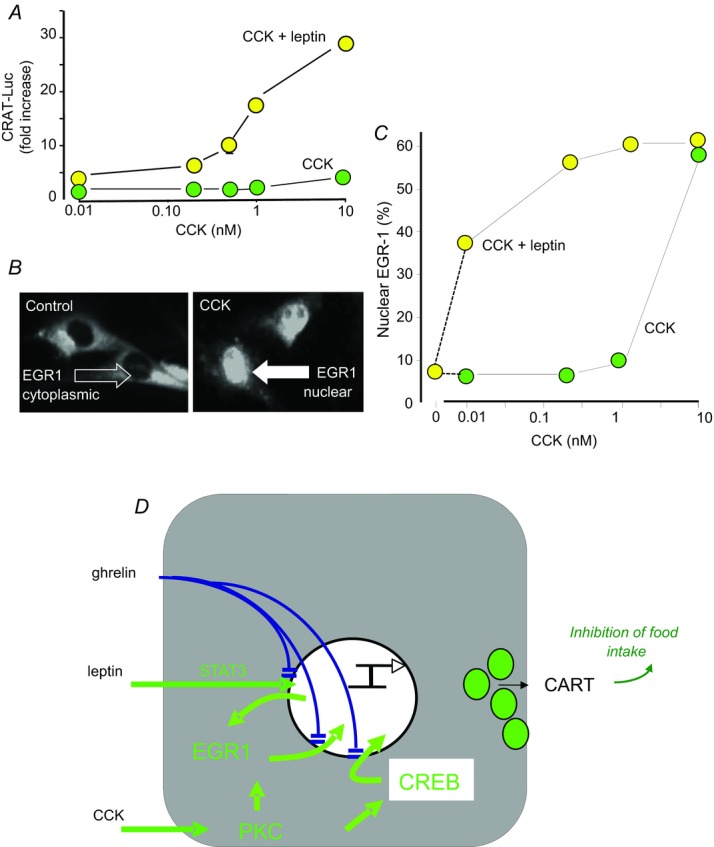
A, in cultured rat nodose ganglion neurons transfected with a CART promoter–luciferase reporter construct there is modest stimulation of CART expression by high concentrations of CCK, and there is strong potentiation by leptin. B, the immediate early gene EGR1, exhibits cytosolic localisation in unstimulated rat nodose ganglion neurons, but after CCK there is rapid nuclear translocation. C, stimulation of EGR1 nuclear translocation is strongly potentiated by leptin; leptin alone does not stimulate translocation. D, however, leptin stimulates EGR1 expression via STAT3. CCK does not stimulate EGR1 expression but acts via PKC and p42/44 MAP kinase to stimulate nuclear translocation which, together with activation of CREB, stimulates CART expression. Ghrelin inhibits nuclear translocation of STAT3, EGR1 and CREB thereby suppressing the effects of both leptin and CCK (de Lartigue et al. 2010).
The stimulation of CART expression by CCK involves activation of both the immediate early gene EGR1 (early growth response-1) and phosphorylation of CREB (cAMP response element binding protein). In the case of EGR1, studies with promoter–luciferase reporter constructs transfected in rat nodose ganglion cells indicate that deletion of the cis-acting element reduces responses to CCK by approximately 60%; over-expression of EGR1 has a modest stimulatory effect on its own, but with CCK stimulation CART expression is increased more than 30-fold and this is attributable to stimulation of nuclear translocation of EGR1 via PKC and p42/44 MAPkinase (de Lartigue et al. 2010). Interestingly, CCK seems not to stimulate EGR1 expression, but there is strong induction of EGR1 gene expression by leptin and this is mediated by STAT3. Moreover, leptin strongly potentiates the action of CCK in stimulating EGR1 nuclear translocation (Fig. 6C). The co-operative effects of CCK and leptin to control CART expression in nodose ganglion neurons are therefore based on the ability of leptin to determine the capacity of the system for CART synthesis by regulating availability of a key transcription factor, while on its own having little effect on CART expression. CCK is a primary regulator of CART expression but its effects depend on leptin to set the gain in the system (Fig. 6D). Ghrelin inhibits the effect of both leptin and CCK by restricting translocation to the nucleus of EGR1 and of STAT3 (Fig. 6D; de Lartigue et al. 2010).
Obesity locks the vagal afferent phenotype in the ‘fasting mode’
Recently, several different groups have drawn attention to the fact that in diet-induced obesity vagal afferent neurons exhibit decreased sensitivity to stimulation. Thus Daly et al. showed reduced sensitivity to CCK, 5HT and distension in diet-induced obesity in mice (Daly et al. 2011); in a rat model of diet-induced obesity, de Lartigue et al. showed insensitivity to leptin that was associated with a shift in expression of Y2 and CB1 receptors towards that normally seen in fasted animals (de Lartigue et al. 2011a, 2012); moreover, Page's group showed increased sensitivity to ghrelin as well as loss of leptin sensitivity, and a pattern of discharge of vagal afferent tension receptors in diet-induced obesity in mice similar to that in control fasted animals (Kentish et al. 2012, 2013). Collectively these observations indicate that the normal switching of vagal afferent phenotype in the feeding cycle is lost in obesity and that vagal afferent neurons are locked in a phenotype characteristic of that in fasted animals.
Microbiota, metabolism and gut–brain signalling
To understand what might be happening we have to consider the microbiota. It is a slightly disconcerting fact that 90% of the cells in our bodies are not human – they are bacterial. There are trillions of them in the gut and probably in excess of 2000 species. A stream of recent papers indicates the microbiome participates in two-way gut–brain signalling with relevance to brain development, stress responses, pain and behaviour (Sudo et al. 2004; Rousseaux et al. 2007; Rhee et al. 2009; Bravo et al. 2011; Diaz Heijtz et al. 2011). Moreover, many studies have identified potential links between the gut microbiota, immunity and energy metabolism; microbiota-related dysfunction is also linked to a range of disorders, including inflammatory bowel disease, irritable bowel syndrome, the metabolic syndrome, neurodevelopmental disorders, autoimmune diseases and allergic diseases (Shanahan, 2012; Tremaroli & Backhed, 2012).
The absence of a gut microbiota is associated with resistance to adiposity in mice (Backhed et al. 2004); moreover, both in human obesity and in animal models of obesity there are shifts in the composition of the microbiota, notably decreases in Bacteriodetes and increases in Firmicutes species (Ley et al. 2005, 2006; Kau et al. 2011). This shift is associated with increased energy harvesting in the form of extraction of short chain fatty acids from fibre and there may also be links to EEC signalling, since short chain fatty acids can act at the G-protein coupled receptor, GPR-41, which is expressed by L-cells (Turnbaugh et al. 2006; Samuel et al. 2008).
Microbiota and vagal afferent signalling
The relationships between gut hormones and vagal afferent neurons are known to be influenced by infection and inflammation in several different ways including increased CCK release in intestinal infection, for example by Giardia (Leslie et al. 2003) and by Trichinella spiralis (McDermott et al. 2006), as well potentiation of CCK stimulation of vagal afferent neurons by proinflammatory cytokines such as IL-1β (Bucinskaite et al. 1997; Gaige et al. 2004). In addition, recent work has identified other mechanisms that link changes in the microbiota in obesity to alterations in vagal afferent sensitivity to CCK.
Obesity-related changes in the microbiome produce a mild inflammatory state that is associated with increased intestinal permeability to the bacterial product, lipopolysaccharide (LPS). The latter acts via toll-like receptor (TLR)4 expressed by vagal afferent neurons to stimulate expression of the suppressor of cytokine signalling (SOCS)-3 which leads to leptin resistance (de Lartigue et al. 2011a; Raybould, 2012). Loss of sensitivity to leptin in turn reduces sensitivity to CCK. Although insensitivity to leptin is a well-known feature of obesity, it is important to note that in an animal model of diet-induced obesity the leptin resistance of vagal afferent neurons occurred well before that in hypothalamic neurons, suggesting that it may be of particular importance in the development of obesity (de Lartigue et al. 2011a,b).
A second example involves plasminogen activator inhibitor (PAI)-1 which is widely expressed, associated with inhibition of thrombolysis, and which increases in plasma in obesity (Landin et al. 1990; Shimomura et al. 1996). In the gut, expression of PAI-1 increases dramatically with infection, for example Helicobacter infection in the stomach (Keates et al. 2008; Kenny et al. 2008), as well as in radiation damage (Abderrahmani et al. 2012) and experimental colitis (Hyland et al. 2009). Wild-type mice infected with Helicobacter felis are insensitive to the satiety action of CCK whereas infected PAI-1 null mice retain CCK sensitivity (Kenny et al. 2013a). The implication is that induction of PAI-1 inhibits the effect of CCK, and more generally that with gastric infection there is an adjustment of the normal satiety signals that maintain food intake (Fig. 7A).
Figure 7. Gastric PAI-1 acts to restrain the satiety effects of CCK.
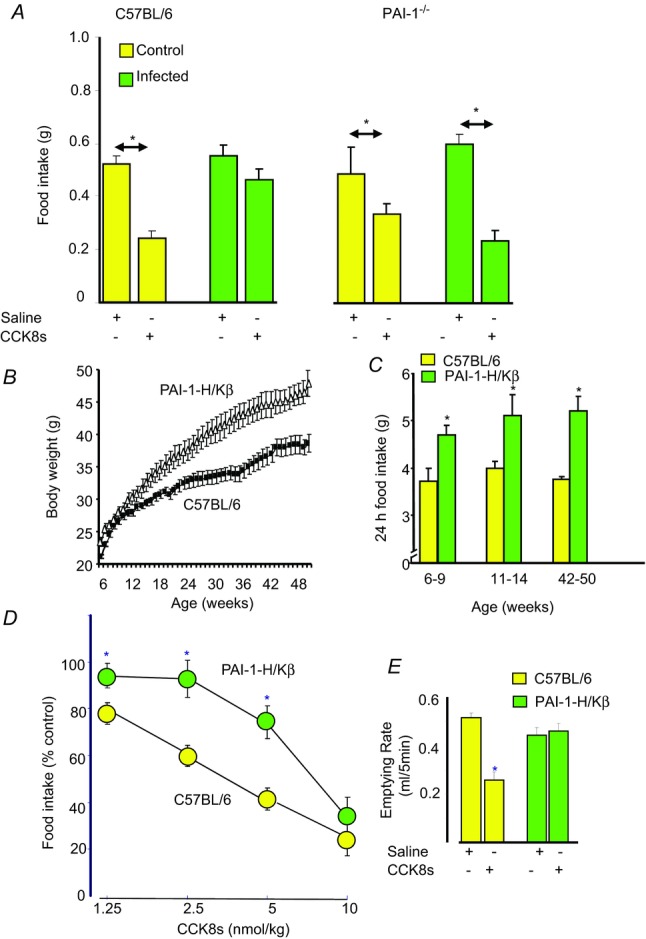
A, in control wild-type mice (C57BL/6) CCK inhibits food intake, but this effect is attenuated in mice infected with Helicobacter felis, which increases gastric PAI-1. In mice null for PAI-1, the inhibitory effect of CCK on food intake is preserved in Helicobacter infection. B, mice transgenic for the coding sequence of PAI-1 under the control of a proximal portion of the promoter of the β-subunit of the H+–K+-ATPase (PAI-1-H/Kβ mice) exhibit increased PAI-1 expression in gastric parietal cells and moderate obesity. C, they are also hyperphagic compared with wild-type mice. D, the dose–response curve for inhibition of food intake by CCK is moved to the right in PAI-1-H/Kβ mice. E, the latter are also insensitive to the action of CCK in inhibiting gastric emptying (Gamble et al. 2013; Kenny et al. 2013a).
To gain insight into these mechanisms we generated mice moderately over-expressing a PAI-1 transgene in the stomach using a promoter sequence of the β-subunit of H+–K+-ATPase which directs expression to gastric parietal cells (Lorenz & Gordon, 1993). These animals have moderately increased gastric PAI-1 mRNA abundance but crucially they exhibit obesity and life-long hyperphagia (Fig. 7B and C), and are also insensitive to the actions of CCK in inhibiting food intake and gastric emptying (Gamble et al. 2013; Kenny et al. 2013a; Fig. 7D and E). These observations support the idea that PAI-1 attenuates vagal responses to CCK and direct evidence of this is provided by studies of vagal afferent neurons in vitro. Thus, PAI-1 inhibits the effect of CCK on EGR1 nuclear translocation and induction of Y2R in nodose neurons in culture (Kenny et al. 2013a). It seems then, that PAI-1 is a new sort of modulator of gut–brain signalling. It is induced in response to infection and inflammation and as well as having protective effects involving inhibition of thrombolysis (Kenny et al. 2013b) it acts to preserve nutrient intake, probably by countering the action of proinflammatory cytokines that enhance the normal satiety effects of CCK.
Overview
To conclude, the idea of chemical sympathy between organs originally proposed by Bayliss and Starling is fundamental to understanding how the gut speaks to the brain, both in feeding and fasting, and in health and disease. The gut hormones exert both acute effects on afferent neurons but also longer term effects that determine neuronal phenotype in a way that reflects nutrient status. They provide the capacity for integration of nutrient signals external to the CNS and, importantly, this mechanism appears to be subverted in obesity, infection and inflammation. Understanding of the interactions involved in this system should pave the way for the development of novel therapies for the treatment of feeding disorders and metabolic disease.
Acknowledgments
It is a pleasure to acknowledge the many graduate students, post-doctoral researchers and collaborators with whom I have enjoyed countless hours of stimulating discussion over the years.
Glossary
- CART
cocaine and amphetamine regulated transcript
- CCK
cholecystokinin
- EEC
enteroendocrine cells
- EGR1
early growth response-1
- fMRI
functional magnetic resonance imaging
- GLP-1
glucagon-like peptide-1
- MCH
melanin concentrating hormone
- PYY3–36
peptide YY3–36
Additional information
Competing interests
None declared.
References
- Abbott CR, Monteiro M, Small CJ, Sajedi A, Smith KL, Parkinson JR, Ghatei MA, Bloom SR. The inhibitory effects of peripheral administration of peptide YY3–36 and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal–brainstem–hypothalamic pathway. Brain Res. 2005;1044:127–131. doi: 10.1016/j.brainres.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Abderrahmani R, Francois A, Buard V, Tarlet G, Blirando K, Hneino M, Vaurijoux A, Benderitter M, Sabourin JC, Milliat F. PAI-1-dependent endothelial cell death determines severity of radiation-induced intestinal injury. PloS One. 2012;7:e35740. doi: 10.1371/journal.pone.0035740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham RL, ffytche DH, Rosenthal JM, Zelaya FO, Barker GJ, Withers DJ, Williams SC. PYY modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature. 2007;450:106–109. doi: 10.1038/nature06212. [DOI] [PubMed] [Google Scholar]
- Bayliss WM, Starling EH. The mechanism of pancreatic secretion. J Physiol. 1902a;28:325–353. doi: 10.1113/jphysiol.1902.sp000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss WM, Starling EH. On the causation of the so-called ‘peripheral reflex secretion’ of the pancreas. Proc Roy Soc Lond. 1902b;69:352–353. [Google Scholar]
- Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, Bienenstock J, Cryan JF. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A. 2011;108:16050–16055. doi: 10.1073/pnas.1102999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, Holmberg K, Kuhar MJ, Hokfelt T. Cocaine- and amphetamine-regulated transcript in the rat vagus nerve: A putative mediator of cholecystokinin-induced satiety. Proc Natl Acad Sci U S A. 1999;96:13506–13511. doi: 10.1073/pnas.96.23.13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, Schoelles K. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292:1724–1737. doi: 10.1001/jama.292.14.1724. [DOI] [PubMed] [Google Scholar]
- Bucinskaite V, Kurosawa M, Miyasaka K, Funakoshi A, Lundeberg T. Interleukin-1beta sensitizes the response of the gastric vagal afferent to cholecystokinin in rat. Neurosci Lett. 1997;229:33–36. doi: 10.1016/s0304-3940(97)00406-0. [DOI] [PubMed] [Google Scholar]
- Bucinskaite V, Tolessa T, Pedersen J, Rydqvist B, Zerihun L, Holst JJ, Hellstrom PM. Receptor-mediated activation of gastric vagal afferents by glucagon-like peptide-1 in the rat. Neurogastroenterol Motil. 2009;21:978–e978. doi: 10.1111/j.1365-2982.2009.01317.x. [DOI] [PubMed] [Google Scholar]
- Burdyga G, de Lartigue G, Raybould HE, Morris R, Dimaline R, Varro A, Thompson DG, Dockray GJ. Cholecystokinin regulates expression of Y2 receptors in vagal afferent neurons serving the stomach. J Neurosci. 2008;28:11583–11592. doi: 10.1523/JNEUROSCI.2493-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Lal S, Spiller D, Jiang W, Thompson DG, Attwood S, Saeed S, Grundy D, Varro A, Dimaline R, Dockray GJ. Localization of orexin-1 receptors to vagal afferent neurons in the rat and humans. Gastroenterology. 2003;124:129–139. doi: 10.1053/gast.2003.50020. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Lal S, Varro A, Dimaline R, Thompson DG, Dockray GJ. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J Neurosci. 2004;24:2708–2715. doi: 10.1523/JNEUROSCI.5404-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Feeding-dependent depression of melanin-concentrating hormone and melanin-concentrating hormone receptor-1 expression in vagal afferent neurones. Neuroscience. 2006a;137:1405–1415. doi: 10.1016/j.neuroscience.2005.10.057. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Ghrelin receptors in rat and human nodose ganglia: putative role in regulating CB-1 and MCH receptor abundance. Am J Physiol Gastrointest Liver Physiol. 2006b;290:G1289–G1297. doi: 10.1152/ajpgi.00543.2005. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Expression of cannabinoid CB1 receptors by vagal afferent neurons: kinetics, and role in influencing neurochemical phenotype. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1514–G1524. doi: 10.1152/ajpgi.00059.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang JC, Perello M, Sakata I, Osborne-Lawrence S, Savitt JM, Lutter M, Zigman JM. Ghrelin mediates stress-induced food-reward behavior in mice. J Clin Invest. 2011;121:2684–2692. doi: 10.1172/JCI57660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizza G, Rother KI. Was Feuerbach right: are we what we eat? J Clin Invest. 2011;121:2969–2971. doi: 10.1172/JCI58595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly DM, Park SJ, Valinsky WC, Beyak MJ. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol. 2011;589:2857–2870. doi: 10.1113/jphysiol.2010.204594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123:1120–1128. doi: 10.1053/gast.2002.35954. [DOI] [PubMed] [Google Scholar]
- Davison JS. Activation of vagal gastric mechanoreceptors by cholecystokinin. Proc West Pharmacol Soc. 1986;29:363–366. [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab. 2011a;301:E187–E195. doi: 10.1152/ajpendo.00056.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PloS One. 2012;7:e32967. doi: 10.1371/journal.pone.0032967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, de La Serre CB, Raybould HE. Vagal afferent neurons in high fat diet-induced obesity; intestinal microflora, gut inflammation and cholecystokinin. Physiol Behav. 2011b;105:100–105. doi: 10.1016/j.physbeh.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Dimaline R, Varro A, Dockray GJ. Cocaine- and amphetamine-regulated transcript: stimulation of expression in rat vagal afferent neurons by cholecystokinin and suppression by ghrelin. J Neurosci. 2007;27:2876–2882. doi: 10.1523/JNEUROSCI.5508-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Lur G, Dimaline R, Varro A, Raybould H, Dockray GJ. EGR1 is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology. 2010;151:3589–3599. doi: 10.1210/en.2010-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray GJ. Immunochemical evidence of cholecystokinin-like peptides in brain. Nature. 1976;264:568–570. doi: 10.1038/264568a0. [DOI] [PubMed] [Google Scholar]
- Dockray GJ. Regulatory peptides and the neuroendocrinology of gut–brain relations. Q J Exp Physiol. 1988;73:703–727. doi: 10.1113/expphysiol.1988.sp003191. [DOI] [PubMed] [Google Scholar]
- Dockray GJ, Gregory RA, Hutchison JB, Harris JI, Runswick MJ. Isolation, structure and biological activity of two cholecystokinin octapeptides from sheep brain. Nature. 1978;274:711–713. doi: 10.1038/274711a0. [DOI] [PubMed] [Google Scholar]
- Edkins JS. On the chemical mechanism of gastric secretion. Proc Roy Soc Lond B. 1905;76:376–376. [Google Scholar]
- Farooqi IS, Bullmore E, Keogh J, Gillard J, O'Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster ER, Green T, Elliot M, Bremner A, Dockray GJ. Gastric emptying in rats: role of afferent neurons and cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 1990;258:G552–G556. doi: 10.1152/ajpgi.1990.258.4.G552. [DOI] [PubMed] [Google Scholar]
- Fraser KA, Davison JS. Cholecystokinin-induced c-fos expression in the rat brain stem is influenced by vagal nerve integrity. Exp Physiol. 1992;77:225–228. doi: 10.1113/expphysiol.1992.sp003579. [DOI] [PubMed] [Google Scholar]
- Gaige S, Abou E, Abysique A, Bouvier M. Effects of interactions between interleukin-1ß and leptin on cat intestinal vagal mechanoreceptors. J Physiol. 2004;555:297–310. doi: 10.1113/jphysiol.2003.054379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaige S, Abysique A, Bouvier M. Effects of leptin on cat intestinal vagal mechanoreceptors. J Physiol. 2002;543:679–689. doi: 10.1113/jphysiol.2002.021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble J, Kenny S, Dockray GJ. Plasminogen activator inhibitor (PAI)-1 suppresses inhibition of gastric emptying by cholecystokinin (CCK) in mice. Regul Pept. 2013;185:9–13. doi: 10.1016/j.regpep.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J, Young RC, Smith GP. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature. 1973;245:323–325. doi: 10.1038/245323a0. [DOI] [PubMed] [Google Scholar]
- Gomez R, Navarro M, Ferrer B, Trigo JM, Bilbao A, Del AI, Cippitelli A, Nava F, Piomelli D, Rodriguez de Fonseca F. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci. 2002;22:9612–9617. doi: 10.1523/JNEUROSCI.22-21-09612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RA. Gastrin-the natural history of a peptide hormone. Harvey Lect. 1968;64:121–155. [PubMed] [Google Scholar]
- Gregory RA. The Bayliss–Starling lecture 1973. The gastrointestinal hormones: a review of recent advances. J Physiol. 1974;241:1–32. doi: 10.1113/jphysiol.1974.sp010637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AA, Raper HS. Pancreozymin, a stimulant of the secretion of pancreatic enzymes in extracts of the small intestine. J Physiol. 1943;102:115–125. doi: 10.1113/jphysiol.1943.sp004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz AF. The Sensibility of the Alimentary Tract. London: Hodder & Stoughton; 1911. [Google Scholar]
- Hyland NP, Chambers AP, Keenan CM, Pittman QJ, Sharkey KA. Differential adipokine response in genetically predisposed lean and obese rats during inflammation: a role in modulating experimental colitis? Am J Physiol Gastrointest Liver Physiol. 2009;297:G869–G877. doi: 10.1152/ajpgi.00164.2009. [DOI] [PubMed] [Google Scholar]
- Innis RB, Snyder SH. Distinct cholecystokinin receptors in brain and pancreas. Proc Natl Acad Sci U S A. 1980;77:6917–6921. doi: 10.1073/pnas.77.11.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy AC, Oldberg E. A hormone mechanism for gallbladder contraction and evacuation. Am J Physiol. 1928;86:599–613. [Google Scholar]
- Jones RB, McKie S, Astbury N, Little TJ, Tivey S, Lassman DJ, McLaughlin J, Luckman S, Williams SR, Dockray GJ, Thompson DG. Functional neuroimaging demonstrates that ghrelin inhibits the central nervous system response to ingested lipid. Gut. 2012;61:1543–1551. doi: 10.1136/gutjnl-2011-301323. [DOI] [PubMed] [Google Scholar]
- Jorpes E, Mutt V. Cholecystokinin and pancreozymin, one single hormone. Acta Physiol Scand. 1966;66:196–202. doi: 10.1111/j.1748-1716.1966.tb03185.x. [DOI] [PubMed] [Google Scholar]
- Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keates AC, Tummala S, Peek RM, Jr, Csizmadia E, Kunzli B, Becker K, Correa P, Romero-Gallo J, Piazuelo MB, Sheth S, Kelly CP, Robson SC, Keates S. Helicobacter pylori infection stimulates plasminogen activator inhibitor 1 production by gastric epithelial cells. Infect Immun. 2008;76:3992–3999. doi: 10.1128/IAI.00584-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny S, Duval C, Sammut SJ, Steele I, Pritchard DM, Atherton JC, Argent RH, Dimaline R, Dockray GJ, Varro A. Increased expression of the urokinase plasminogen activator system by Helicobacter pylori in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;295:G431–G441. doi: 10.1152/ajpgi.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny S, Gamble J, Lyons S, Vlatkovic N, Dimaline R, Varro A, Dockray GJ. Gastric expression of plasminogen activator inhibitor (PAI)-1 is associated with hyperphagia and obesity in mice. Endocrinology. 2013a;154:718–726. doi: 10.1210/en.2012-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny S, Steele I, Lyons S, Moore AR, Murugesan SV, Tiszlavicz L, Dimaline R, Pritchard DM, Varro A, Dockray GJ. The role of plasminogen activator inhibitor-1 in gastric mucosal protection. Am J Physiol Gastrointest Liver Physiol. 2013b;304:G814–G822. doi: 10.1152/ajpgi.00017.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentish S, Li H, Philp LK, O'Donnell TA, Isaacs NJ, Young RL, Wittert GA, Blackshaw LA, Page AJ. Diet-induced adaptation of vagal afferent function. J Physiol. 2012;590:209–221. doi: 10.1113/jphysiol.2011.222158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentish SJ, O'Donnell TA, Isaacs NJ, Young RL, Li H, Harrington AM, Brierley SM, Wittert GA, Blackshaw LA, Page AJ. Gastric vagal afferent modulation by leptin is influenced by food intake status. J Physiol. 2013;591:1921–1934. doi: 10.1113/jphysiol.2012.247577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchgessner AL, Liu M. Orexin synthesis and response in the gut. Neuron. 1999;24:941–951. doi: 10.1016/s0896-6273(00)81041-7. [DOI] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Landin K, Stigendal L, Eriksson E, Krotkiewski M, Risberg B, Tengborn L, Smith U. Abdominal obesity is associated with an impaired fibrinolytic activity and elevated plasminogen activator inhibitor-1. Metabolism. 1990;39:1044–1048. doi: 10.1016/0026-0495(90)90164-8. [DOI] [PubMed] [Google Scholar]
- Lassman DJ, McKie S, Gregory LJ, Lal S, D'Amato M, Steele I, Varro A, Dockray GJ, Williams SC, Thompson DG. Defining the role of cholecystokinin in the lipid-induced human brain activation matrix. Gastroenterology. 2010;138:1514–1524. doi: 10.1053/j.gastro.2009.12.060. [DOI] [PubMed] [Google Scholar]
- Leslie FC, Thompson DG, McLaughlin JT, Varro A, Dockray GJ, Mandal BK. Plasma cholecystokinin concentrations are elevated in acute upper gastrointestinal infections. QJM. 2003;96:870–871. doi: 10.1093/qjmed/hcg140. [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gao JH, Liu HL, Fox PT. The temporal response of the brain after eating revealed by functional MRI. Nature. 2000;405:1058–1062. doi: 10.1038/35016590. [DOI] [PubMed] [Google Scholar]
- Lorenz RG, Gordon JI. Use of transgenic mice to study regulation of gene expression in the parietal cell lineage of gastric units. J Biol Chem. 1993;268:26559–26570. [PubMed] [Google Scholar]
- Luckman SM. Fos-like immunoreactivity in the brainstem of the rat following peripheral administraiton of cholecystokinin. J Neuroendocrinol. 1992;4:149–151. doi: 10.1111/j.1365-2826.1992.tb00152.x. [DOI] [PubMed] [Google Scholar]
- McDermott JR, Leslie FC, D'Amato M, Thompson DG, Grencis RK, McLaughlin JT. Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut. 2006;55:492–497. doi: 10.1136/gut.2005.081752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, Grazia LM, Jones MN, D'Amato M, Dockray GJ, Thompson DG. Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology. 1999;116:46–53. doi: 10.1016/s0016-5085(99)70227-1. [DOI] [PubMed] [Google Scholar]
- Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008;7:400–409. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Moran TH, Smith GP, Hostetler AM, McHugh PR. Transport of cholecystokinin (CCK) binding sites in subdiaphragmatic vagal branches. Brain Res. 1987;415:149–152. doi: 10.1016/0006-8993(87)90278-2. [DOI] [PubMed] [Google Scholar]
- Moriarty P, Dimaline R, Thompson DG, Dockray GJ. Characterization of cholecystokininA and cholecystokininB receptors expressed by vagal afferent neurons. Neuroscience. 1997;79:905–913. doi: 10.1016/s0306-4522(96)00675-6. [DOI] [PubMed] [Google Scholar]
- Mutt V. Chemistry of the gastrointestinal hormones and hormone-like peptides and a sketch of their physiology and pharmacology. Vitam Horm. 1982;39:231–427. doi: 10.1016/s0083-6729(08)61138-3. [DOI] [PubMed] [Google Scholar]
- Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167–175. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–3657. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- Peters JH, Simasko SM, Ritter RC. Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav. 2006;89:477–485. doi: 10.1016/j.physbeh.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Raybould HE. Gut microbiota, epithelial function and derangements in obesity. J Physiol. 2012;590:441–446. doi: 10.1113/jphysiol.2011.222133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould HE, Gayton RJ, Dockray GJ. Mechanisms of action of peripherally administered cholecystokinin octapeptide on brain stem neurons in the rat. J Neurosci. 1988;8:3018–3024. doi: 10.1523/JNEUROSCI.08-08-03018.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould HE, Tache Y. Cholecystokinin inhibits gastric motility and emptying via a capsaicin-sensitive vagal pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1988;255:G242–G246. doi: 10.1152/ajpgi.1988.255.2.G242. [DOI] [PubMed] [Google Scholar]
- Rehfeld JF. Immunochemical studies on cholecystokinin. II. Distribution and molecular heterogeneity in the central nervous system and small intestine of man and hog. J Biol Chem. 1978;253:4022–4030. [PubMed] [Google Scholar]
- Rhee SH, Pothoulakis C, Mayer EA. Principles and clinical implications of the brain–gut–enteric microbiota axis. Nature Rev Gastroenterol Hepatol. 2009;6:306–314. doi: 10.1038/nrgastro.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- Rousseaux C, Thuru X, Gelot A, Barnich N, Neut C, Dubuquoy L, Dubuquoy C, Merour E, Geboes K, Chamaillard M, Ouwehand A, Leyer G, Carcano D, Colombel JF, Ardid D, Desreumaux P. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nature Med. 2007;13:35–37. doi: 10.1038/nm1521. [DOI] [PubMed] [Google Scholar]
- Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, Gordon JI. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. 2008;105:16767–16772. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, Thomas S, Abood B, Nissen SE, Bhatt DL. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med. 2012;366:1567–1576. doi: 10.1056/NEJMoa1200225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ, Fu J, Astarita G, Li X, Gaetani S, Campolongo P, Cuomo V, Piomelli D. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 2008;8:281–288. doi: 10.1016/j.cmet.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GJ, McHugh PR, Moran TH. Integration of vagal afferent responses to gastric loads and cholecystokinin in rats. Am J Physiol Regul Integr Comp Physiol. 1991;261:R64–R69. doi: 10.1152/ajpregu.1991.261.1.R64. [DOI] [PubMed] [Google Scholar]
- Shanahan F. The gut microbiota – a clinical perspective on lessons learned. Nature Rev Gastroenterol Hepatol. 2012;9:609–614. doi: 10.1038/nrgastro.2012.145. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, Yamashita S, Miura M, Fukuda Y, Takemura K, Tokunaga K, Matsuzawa Y. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. 1996;2:800–803. doi: 10.1038/nm0796-800. [DOI] [PubMed] [Google Scholar]
- Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ. Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science. 1981;213:1036–1037. doi: 10.1126/science.7268408. [DOI] [PubMed] [Google Scholar]
- Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, Kubo C, Koga Y. Postnatal microbial colonization programs the hypothalamic–pituitary–adrenal system for stress response in mice. J Physiol. 2004;558:263–275. doi: 10.1113/jphysiol.2004.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Van Oudenhove L, McKie S, Lassman D, Uddin B, Paine P, Coen S, Gregory L, Tack J, Aziz Q. Fatty acid-induced gut–brain signaling attenuates neural and behavioral effects of sad emotion in humans. J Clin Invest. 2011;121:3094–3099. doi: 10.1172/JCI46380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Miyares RL, Ahern GP. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J Physiol. 2005;564:541–547. doi: 10.1113/jphysiol.2004.081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, Tache Y, Sheibel AB, Go VL, Wei JY. Two types of leptin-responsive gastric vagal afferent terminals: an in vitro single-unit study in rats. Am J Physiol Regul Integr Comp Physiol. 1997;273:R833–R837. doi: 10.1152/ajpregu.1997.273.2.R833. [DOI] [PubMed] [Google Scholar]