

Abstract
Smooth muscle contraction initiated by myosin regulatory light chain (RLC) phosphorylation is dependent on the relative activities of Ca2+–calmodulin-dependent myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). We have investigated the physiological role of the MLCP regulatory subunit MYPT1 in bladder smooth muscle containing a smooth muscle-specific deletion of MYPT1 in adult mice. Deep-sequencing analyses of mRNA and immunoblotting revealed that MYPT1 depletion reduced the amount of PP1cδ with no compensatory changes in expression of other MYPT1 family members. Phosphatase activity towards phosphorylated smooth muscle heavy meromyosin was proportional to the amount of PP1cδ in total homogenates from wild-type or MYPT1-deficient tissues. Isolated MYPT1-deficient tissues from MYPT1SM−/− mice contracted with moderate differences in response to KCl and carbachol treatments, and relaxed rapidly with comparable rates after carbachol removal and only 1.5-fold slower after KCl removal. Measurements of phosphorylated proteins in the RLC signalling and actin polymerization modules during contractions revealed moderate changes. Using a novel procedure to quantify total phosphorylation of MYPT1 at Thr696 and Thr853, we found substantial phosphorylation in wild-type tissues under resting conditions, predicting attenuation of MLCP activity. Reduced PP1cδ activity in MYPT1-deficient tissues may be similar to the attenuated MLCP activity in wild-type tissues resulting from constitutively phosphorylated MYPT1. Constitutive phosphorylation of MYPT1 Thr696 and Thr853 may thus represent a physiological mechanism acting in concert with agonist-induced MYPT1 phosphorylation to inhibit MLCP activity. In summary, MYPT1 deficiency may not cause significant derangement of smooth muscle contractility because the effective MLCP activity is not changed.
Key points
Smooth muscle myosin regulatory light chain (RLC) phosphorylation depends on the relative activities of myosin light chain kinase (MLCK), activated by Ca2+–calmodulin, and myosin light chain phosphatase (MLCP).
MYPT1 is a scaffolding protein subunit of MLCP that binds the catalytic subunit PP1cδ and myosin, affects the conformation of PP1cδ for effective inhibition by phosphorylated CPI-17, and is phosphorylated at two sites that inhibit PP1cδ activity.
A conditional knockout of MYPT1 in smooth muscles of adult mice resulted in modest changes in bladder smooth muscle contractile and relaxation responses to KCl or carbachol even though the amount of PP1cδ protein was reduced.
A new a procedure to quantify phosphorylation of MYPT1 showed substantial phosphorylation in wild-type tissues under resting conditions, predicting attenuation of MLCP activity. Reduced PP1cδ activity in MYPT1-deficient tissues may be similar to the attenuated MLCP activity in wild-type tissues resulting from constitutively phosphorylated MYPT1.
In contrast to results obtained with the conditional knockout of MLCK in smooth muscle, MYPT1 is not necessary for smooth muscle function because its loss may not change the effective MLCP activity.
Introduction
Smooth muscles contain the contractile protein myosin in thick filaments that track on actin thin filaments to initiate force development and shortening (Hartshorne, 1987; Conti & Adelstein, 2008; Lowey & Trybus, 2010). Smooth muscle myosin is activated by the short Ca2+–calmodulin-dependent myosin light chain kinase (MLCK) that phosphorylates the myosin regulatory light chain subunit (RLC), thereby displacing and activating the motor domain. Myosin light chain phosphatase (MLCP) dephosphorylates RLC to induce relaxation. The extent of RLC phosphorylation determines force development in smooth muscle tissues and is thus affected by the ratio of MLCK to MLCP activities (Kamm & Stull, 1985; Hartshorne, 1987; Somlyo & Somlyo, 2000; Lowey & Trybus, 2010).
Chemical messengers act on cell surface receptors with assorted signal transduction pathways and ion channels that converge to increase [Ca2+]i for contraction. Ca2+ binds to calmodulin which then activates MLCK to phosphorylate RLC and initiate smooth muscle contraction (Hartshorne, 1987; Kamm & Stull, 2001). MLCK appears to be the only kinase phosphorylating RLC physiologically in intestinal (He et al. 2008), airway (Zhang et al. 2010), mesenteric arterial (He et al. 2011), aortic and urinary bladder smooth muscles (Gao et al. 2013).
Cell signalling pathways also inhibit MLCP, thereby increasing RLC phosphorylation without changing elevated [Ca2+]i (Ca2+ sensitization; Somlyo & Somlyo, 2003; Hartshorne et al. 2004; Dimopoulos et al. 2007; Kitazawa, 2010; Grassie et al. 2011). MLCP is a holoenzyme composed of three distinct subunits: a 38 kDa catalytic subunit (PP1cδ), a large 110–130 kDa regulatory subunit (MYPT1), and a small 20 kDa subunit of unknown function (Matsumura & Hartshorne, 2008; Grassie et al. 2011). Biochemically, MYPT1 binds PP1cδ and myosin, and thus functions as a scaffolding protein. Agonist-mediated Ca2+ sensitization is due to two major signalling pathways, including phosphorylation of MYPT1 by a Rho kinase pathway (ROCK), and phosphorylation of a small inhibitor protein CPI-17 by protein kinase C (PKC; Kitazawa et al. 2000; Hirano et al. 2003; Murthy, 2006; Mizuno et al. 2008). The extent of RLC phosphorylation is thus balanced by the regulated MLCK and MLCP activities involving temporally dynamic and distinct cellular processes.
MYPT1, like MLCK, is thus proposed as a central player in signalling to RLC (Hartshorne et al. 1998; Somlyo & Somlyo, 2000; Grassie et al. 2011). In spite of an abundance of pharmacological studies with inhibitors of PKC and ROCK on Ca2+ sensitization in intact tissues (Somlyo & Somlyo, 2003; Hartshorne et al. 2004; Grassie et al. 2011), genetic approaches for investigating physiological functions of MYPT1 in vivo and in vitro are just emerging, with a recent report on a conditional knockout of MYPT1 in smooth muscles of adult animals (He et al. 2013). It was anticipated that the knockout of MYPT1 would lead to marked phenotypic responses, perhaps similar to results obtained with the conditional knockout of MLCK that includes smooth muscle contractile failure in vivo and in vitro, and death. However, MYPT1 knockout in smooth muscle tissues of adult mice resulted in no marked phenotypic responses in intact animals and small changes in contractile responses of isolated intestinal tissues (He et al. 2013). We have explored the reasons for these surprising results in the urinary bladder. We developed new approaches for quantifying phosphorylation of MYPT1 Thr696 and Thr853 in addition to using established quantitative measurements for CPI-17 as well as RLC phosphorylation. Results from these studies suggest that the constitutive phosphorylation of MYPT1 may play a significant role in attenuating the activity of MLCP in normal tissues. Further, knockout of MYPT1 results in a reduced amount of PP1cδ activity that may be similar to that predicted with constitutively inhibited MLCP from MYPT1 phosphorylation in wild-type tissues. Thus, the overall balance of MLCK to MLCP activities appears not to be greatly perturbed in MYPT1-deficient smooth muscle tissues.
Methods
Ethical approval
Experiments were performed in accordance with NIH and Institutional Animal Care and Use Guidelines. The Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center approved all procedures and protocols. All protocols and procedures are in compliance with The Journal of Physiology guidelines (Drummond, 2009). Animals were killed by the intraperitoneal administration of a lethal dose of Avertin (250 mg kg−1) for tissue collection.
Generation of genetically modified mice
Mice containing floxed Mypt1 alleles (Mypt1f/f; He et al. 2013) were crossed with a SMMHC-CreERT2 transgenic mouse line expressing a fusion protein of the Cre recombinase with the modified oestrogen receptor binding domain (CreERT2) under the control of the smooth muscle myosin heavy chain (SMMHC) promoter (Wirth et al. 2008). Cre-mediated recombination occurred robustly and exclusively in smooth muscle cells in tissues, but only after tamoxifen treatment (Wirth et al. 2008). Mice were bred and screened as described previously (Wirth et al. 2008; He et al. 2013).
Mice were injected intraperitoneally with tamoxifen for five consecutive days each week for two weeks at a dose of 1 mg day−1. The tamoxifen (200 mg, Sigma, St. Louis, MO, USA) was dissolved in 2 ml of ethanol followed by 18 ml of sunflower oil at a concentration 10 mg ml−1 and stored at −20°C for up to 1 month. Bladders were harvested from transgenic mice containing Mypt1f/f, Cre+ alleles (denoted as MYPT1SM−/− mice). Vehicle-treated age-matched male littermate mice (denoted as MYPT1SM+/+ mice) were used as controls. Experiments were conducted 6–7 weeks post tamoxifen injections.
Messenger RNA measurement by RNA sequencing
Total RNA was purified from urothelium-denuded bladder with RNeasy Protect Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. The quality of RNA used in cDNA library preparation was measured with an Agilent Bioanalyzer RNA 6000 Nano kit by the Microarray Core at UT Southwestern Medical Center. Library preparation, RNA sequencing quality control and measurement of expression levels were performed by the Genomics Core at the Immunology Department in UT Southwestern Medical Center using an Illumina HiSeqH2000 RNA sequencer. Over 30 million reads were generated, corresponding to greater than 30,000 transcripts measured for each sample; RPKM values great than 5 were used for comparison of expression levels.
MLCP activity assays
MLCP activity in bladder homogenates was assayed using published methods (Etter et al. 2001) with minor modifications. Dissected bladder strips were weighed and homogenized in a glass homogenizer on ice in phosphatase assay buffer (50 mm Mops, pH 7.4, 50 mm NaCl, 0.1 mm EDTA, 1 mm DTT, 1 nm okadaic acid, protease inhibitor cocktail (Pierce; EDTA-Free, used at concentration of 1 tablet per 10 ml of buffer) with 1% Nonidet P-40), in the proportion of 1 mg of tissue (wet weight) per 1000 μl of buffer. Okadaic acid (1 nm) was added to the buffer for eliminating endogenous PP2A activity.
For the substrate, 22 μm recombinant smooth muscle heavy meromyosin (Kuang et al. 2012) containing RLC was phosphorylated by purified MLCK in 10 mm Mops, pH 7.4, 5 mm MgCl2, 100 mm NaCl, 1.5 μm calmodulin, 0.3 mm CaCl2 with 0.2 mm [γ-32P]ATP (500–800 c.p.m. pmol−1; Chang et al. 2010). After 30 min at 30°C, the reaction was terminated with the addition of 10% volume of 440 mm Mops, pH 7.4, 66 mm EDTA, 5.5 mm EGTA, and immediately used in phosphatase assays. These conditions established full monophosphorylation of RLC (data not shown).
Bladder homogenates were incubated at 30°C for 10 min to allow MYPT1 to be dephosphorylated at both Thr696 and Thr853. Dephosphorylation of MYPT1 prior to assay was confirmed by immunoblot with phospho-specific antibodies to phospho-MYPT1 Thr696 or Thr853 (data not shown). Bladder homogenate was added at a 1:14,000 final dilution in 21 μl total reaction volume containing 2 μm 32P-labelled smooth muscle heavy meromyosin. After 6 min incubation in 30°C, 15 μl of the reaction mixture was added to Whatman 3MM chromatography paper, and washed in 10% trichloroacetic acid (Blumenthal & Stull, 1982). Radioactivity was measured by liquid scintillation spectroscopy and phosphatase activity was calculated as pmoles 32P hydrolysed per minute per microgram of bladder homogenate protein. A duplicate set of reaction mixtures containing 50 nm calyculin A to inhibit MLCP activity was used to obtain control values which were identical to reaction mixtures containing no added homogenate. Reaction time and concentrations of homogenate and substrate were optimized for linear dephosphorylation rates which were normalized to homogenate protein concentration measured by the Bradford assay. For normalization to total PP1cδ protein, an aliquot of the bladder homogenate used in the phosphatase assay was precipitated with an equal volume of 20% trichloroacetic acid. Precipitated protein was washed by three 5 min washes in ethyl ether to remove trichloroacetic acid, and dried protein was solubilized in 8 m urea sample buffer. Protein concentration was determined by the Bradford assay prior to boiling in 2× sample buffer for SDS–PAGE. The relative amount of PP1cδ protein used to calculate rates was measured by immunoblot and normalized to tubulin loading control for bladder tissues from MYPT1SM+/+ and MYPT1SM−/− mice.
Protein solubility measurements
Frozen bladder smooth muscle strips were homogenized in 50 × (vol./wet weight) of a buffer containing 50 mm Mops, pH 6.8, 25 mm KCl, 5 mm EDTA, 1 mm DTT, 0.5 mm E64 (a cysteine protease inhibitor), and 0.5% Triton X-100. After incubating the homogenates on ice for 15 min, myofilaments were pelleted in an aliquot by centrifugation at 100,000 × g for 30 min at 4°C. Both supernatant fractions and the total homogenates were diluted with Laemmli sample buffer for immunoblotting after SDS–PAGE. Primary antibodies included: mouse monoclonal anti-smooth muscle RLC (1:10,000, Sigma), mouse monoclonal anti-MYPT1 (1:5000, BD Transduction Laboratories, Lexington, KY, USA), rabbit polyclonal anti-MBS85 (1:1000, Abcam Inc., Cambridge, MA, USA), rabbit polyclonal anti-GAPDH (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), chicken polyclonal anti-pan PP1c (1:40,000, Millipore, Billerica, MA, USA), rabbit polyclonal anti-PP1cα (1:5000, Millipore), goat polyclonal anti-PP1cγ (1:5000, Santa Cruz), rabbit anti-PP1cδ (1:3000, Millipore). Amounts loaded and antibody dilutions were optimized for each protein.
Isolation of detrusor smooth muscle for isometric force measurements
Maintained MYPT1SM+/+ or MYPT1SM−/− mice were killed with a lethal-dose injection of Avertin. The urinary bladder was removed and placed in physiological salt solution (PSS, in 118.5 mm NaCl, 4.75 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 24.9 mm NaHCO3, 1.6 mm CaCl2, and 10 mm d-glucose, aerated with 95% O2–5% CO2 to maintain pH at 7.4 at 37°C). After removing the urothelium by dissection, three or four longitudinal strips of the detrusor muscle were prepared per bladder and mounted in a water-jacketed 8 ml organ bath for isometric force recording (Tsai et al. 2012). Muscle strips were adjusted to the length for optimal tension development, and allowed to equilibrate for at least 45 min followed by two alternating 5 min exposures to 65 mm KCl-PSS (equimolar replacement of NaCl with KCl) every 10 min in order to establish viability. Subsequently, muscle strips were stimulated with either a single dose of 10 μm carbachol for 5 min or cumulative doses (10 nm to 30 μm) of carbachol to establish a concentration–response curve. For measurement of 65 mm KCl-induced force, muscle strips were pre-treated with 1 μm atropine to block potential muscarinic receptor activation caused by release of acetylcholine from parasympathetic nerve varicosities during membrane depolarization. Contractions in response to KCl and carbachol stimulation were measured using Grass FT03 force transducers, with the outputs recorded on a Powerlab 8/SP data acquisition unit (AD Instruments, Colorado Springs, CO, USA). Force measurements were normalized as grams of developed force per milligram tissue wet weight.
Bladder strips were stimulated with either 10 μm carbachol or 65 mm KCl (pretreated with 1 μm atropine for 10 min) and then snap-frozen with a clamp pre-chilled in liquid nitrogen at different times. Strips were also pretreated with the Rho kinase inhibitor H-1152 (1 μm) or protein kinase C inhibitor GF 109203X (3 μm) (Kitazawa & Kitazawa, 2012). After 20 min preincubation, 10 μm carbachol was applied and muscle strips were snap-frozen at 1 min. Frozen tissues were stored at −80°C until they were added to frozen slurry of acetone with 10% trichloroacetic acid, slowly thawed, and homogenized in 10% trichloroacetic acid and 10 mm DTT. Samples were centrifuged to process for protein phosphorylation measurements as described below. Protein content was determined by a bicinchoninic acid (BCA) protein assay kit (Pierce) with bovine serum albumin as the standard.
Measurement of RLC phosphorylation
RLC phosphorylation was measured by urea–glycerol PAGE as previously described (Colburn et al. 1988). Briefly, muscle proteins precipitated in 10% trichloroacetic acid and 10 mm DTT were washed in ethyl ether and solubilized in 8 m urea sample buffer and subjected to urea–glycerol PAGE at 400 V for 90 min to separate non-phosphorylated and monophosphorylated RLC. Following electrophoresis, proteins were transferred to nitrocellulose membranes and probed with rabbit polyclonal antibodies against smooth muscle RLC. The ratio of mono-phosphorylated RLC to total RLC (non-phosphorylated plus mono-phosphorylated) was quantified by densitometry. The images were acquired with a STORM 840 scanner (Amersham Pharmacia Biotech) and the signal intensity was quantified by densitometry using ImageQuant 5.2 software (Molecular Dynamics).
Measurement of MYPT1, MLCK, FAK and paxillin phosphorylation
Tissue extracts prepared in urea sample buffer were boiled in Laemmli sample buffer and subjected to SDS–PAGE. Proteins were then transferred electrically onto nitrocellulose membranes. After being blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 h at room temperature, membranes were incubated overnight at 4°C with phosphospecific anti-MYPT1 Thr696 antibody (1:6000, Millipore), anti-MYPT1 Thr853 antibody (1:4000, Millipore), anti-MLCK Ser1760 antibody (1:7500, ProSci, Poway, CA, USA), anti-FAK Tyr397(1:1000, Santa Cruz) and anti-paxillin Tyr118 (1:1000, Cell, Signalling, Danvers, MA, USA). For MLCP subunit isoform detection, the following antibodies were used: MYPT1 (1:7500, BD Transduction Laboratories), MYPT2 (1:10,000), MYPT3 (1:1000, Santa Cruz), pan-MYPTs (1:5000, from Dr Masumi Eto), PP1cα (1:1000, Upstate), PP1cδ (1:3000, Millipore), PP1cγ (1:1000, Santa Cruz). The pan-MYPTs antibody recognizes the conserved autoinhibitory sequence in MYPT1, MYPT2 and MBS85. The blots were washed and incubated with horseradish peroxidase-conjugated secondary antibodies for 1.5 h at room temperature. Immunoreactive bands were visualized by ECL Plus Western Blotting detection reagents (GE Healthcare, Piscataway, NJ, USA). Membranes were then stripped of bound antibodies by incubation in a buffer containing 62.5 mm Tris-HCl (pH 6.8), 2% SDS, and 10 mm DTT for 30–35 min at 55°C with gentle agitation. The blots were re-probed with anti-MYPT1 (1:7500, BD Transduction Laboratories), anti-MLCK (1:10,000), anti-FAK (1:1000, Santa Cruz), anti-paxillin (1:2000, BD Transduction Laboratories) and GAPDH (1:3000, Santa Cruz) for loading controls. The images were acquired with a STORM 840 scanner (Amersham Pharmacia Biotech) and the signal intensity was quantified by densitometry using ImageQuant 5.2 software (Molecular Dynamics). Quantifications for all proteins were established to be in the linear range in relation to protein loaded. With the gradient gel system used for these analyses, the different isoforms for CPI-17 were resolved (Yamawaki et al. 2001; Tsai et al. 2012; Gao et al. 2013).
Quantitation of CPI-17 phosphorylation
Bladder strips were treated with 5 μm phorbol 12,13-dibutyrate (PDBu) for 30 min and snap-frozen with clamps pre-chilled in liquid nitrogen. The tissues were homogenized as described for RLC phosphorylation in urea sample buffer and then boiled in equal volume of 2× SDS–PAGE sample buffer. Non-phosphorylated and phosphorylated CPI-17 were separated by 10% Phos-tag PAGE as previously described, with slight modifications (Takeya et al. 2008; Johnson et al. 2009). The resolving gel (10% acrylamide (29:1), 375 mm Tris, pH 8.8, 0.1% SDS, 60 mm MnCl2, 30 μm Phos-tag acrylamide, 0.05% ammonium persulfate, 0.1% TEMED) was overlaid with water-saturated isobutanol and polymerized for 30 min. Isobutanol was removed by washing and standard stacking gel (3.9% acrylamide (29:1), 125 mm Tris, pH 6.8, 0.1% SDS, 0.05% ammonium persulfate, 0.1% TEMED) was polymerized for at least 30 min. Samples (15 μg bladder homogenate) were electrophoresed for approximately 2 h at 20 mA in 25 mm Tris, 192 mm glycine, and 0.1% SDS until tracking dye migrated out of gel. The gel was transferred directly onto PVDF membrane for 2 h at 0.4A in a chilled modified Phos-tag transfer buffer (25 mm Tris, 192 mm glycine, 0.1% SDS, 10% methanol and 1 mm EDTA). Post-transfer membrane was fixed in 0.4% glutaraldehyde–PBS for 15 min at room temperature, rinsed with PBS, blocked with 5% non-fat dry milk–TBST for 30 min, and then probed with pan CPI-17 antibody in 3% BSA–TBST overnight at 4°C. Quantification was performed as described for RLC phosphorylation. (Johnson et al. 2009; El-Yazbi et al. 2010; Moreno-Dominguez et al. 2013). The PDBu-treated samples were run in parallel with tissues treated with carbachol and KCl on regular SDS–PAGE and probed with phospho-specific anti-CPI-17Thr38 antibody (1:500, Santa Cruz) as described above, the ratio of the density of the bands corresponding to the phosphorylated proteins to the bands corresponding to total proteins was determined. The value 72% was then used as a factor to calculate total phosphorylation of CPI-17 in tissues treated with carbachol and KCl.
Quantitation of MYPT1 phosphorylation
Total phosphorylation of MYPT1 Thr656 and Thr853 in calyculin A-treated bladder strips was measured by comparison with purified GST-MYPT1 peptide (654–880; Khromov et al. 2009) phosphorylated by ROCK1 (Life Technologies, Gaithersburg, MD, USA, PV3691) using previously published procedures (Khromov et al. 2009). Complete diphosphorylation of the GST-MYPT1 fragment was confirmed after separation of non-phosphorylated and phosphorylated forms by 6% Phos-tag PAGE (6% acrylamide (29:1), 375 mm Tris, pH 8.8, 0.1% SDS, 60 mm MnCl2, 30 μm Phos-tag acrylamide, 0.05% ammonium persulfate, 0.1% TEMED) using the same procedures as described above for CPI-17 and immunoblotting for total MYPT1 (BD Transduction Laboratories). This antibody was raised to residues 723–840 of rat MYPT1, which is contained within and 100% conserved in respect to the human MYPT1 residues 654–880 in the GST-MYPT1 fragment. As the residues in the GST-MYPT1 fragment are 100% conserved in respect to mouse MYPT1 sequence, antibodies to phosphorylated Thr696 and Thr853 are assumed to bind with equal affinity.
Statistical analyses
Data were expressed as means ± SEM. For multiple comparisons, one-way ANOVA followed by Dunnett's post hoc test or two-way ANOVA with Tukey's post hoc test was used (Prism 6.0; GraphPad Software, San Diego, CA, USA). Student's unpaired, two-tailed t test was used for comparison between control and treatment group. P values less than 0.05 were considered statistically significant.
Results
Profiles of mRNA and protein expression for signalling proteins relevant to RLC phosphorylation in MYPT1-deficient smooth muscle
We measured the mRNA expression profile of different proteins relevant to the regulation of RLC phosphorylation in wild-type and MYPT1-deficient smooth muscle by mRNA deep sequencing in the urothelium-denuded bladder. As indicated in Table 1 and Fig. 1A, MYPT1 mRNA expression was dramatically decreased in MYPT1SM−/− tissue compared to that in MYPT1SM+/+ mice. The mRNA for MLCP catalytic subunit PP1cδ partially decreased. No compensatory increases in other relevant mRNAs were observed, including MYPT1 family members (MYPT2, MYPT3 and MBS85) and PP1c isoforms, α and γ. The protein expression for MYPT1 decreased 96% while PP1cδ decreased about 70% (Fig. 1B). Using an antibody raised to a common sequence in MYPT1 and MBS85, the measured amount of MBS85 was 16 ± 3% compared to MYPT1 in wild-type tissue. The amount of expression of MBS85 was similar in tissues from both MYPT1SM+/+ and MYPT1SM−/− mice. The amounts of expression of other relevant proteins in the RLC signalling module, including MLCK, Rho-kinase (ROCK I), RLC and CPI-17 were comparable in MYPT1SM+/+ and MYPT1SM−/− bladder smooth muscle (data not shown). Thus, MYPT1 is deficient in detrusor smooth muscle from MYPT1SM−/− mice and was associated with a partial reduction of MLCP catalytic subunit PP1cδ.
Table 1.
mRNA expression levels of MLCP subunits MYPT1 and PP1c isoform in bladder smooth muscle from MYPT1SM+/+ (+/+) and MYPT1SM−/− (−/−) mice
| +/+ | −/− | |
|---|---|---|
| MHC | 2989.8 ± 80.8 | 2507.7 ± 171.9 |
| MYPT1 | 248.7 ± 47.0 | 16.9 ± 3.3** |
| MYPT2 | 37.0 ± 3.8 | 26.8 ± 2.6 |
| MYPT3 | 9.0 ± 0.7 | 9.9 ± 0.4 |
| MBS85 | 160.3 ± 5.2 | 194.3 ± 16.1 |
| PP1cα | 82.5 ± 1.1 | 86.7 ± 0.8* |
| PP1cγ | 59.7 ± 3.2 | 61.4 ± 3.6 |
| PP1cδ | 313.9 ± 14.0 | 240.4 ± 5.8** |
RNA purified from UBSM was processed for deep sequencing using HiSeq H2000 (Illumina). The read counts of each known transcript were normalized to the length of the individual transcript and to the total mapped read counts in each sample and expressed as RPKM (reads per kilobase of exon per million mapped reads). Data represent means ± SEM (n = 3).
P < 0.05,
P < 0.01 compared with values for +/+ mice.
Figure 1. MYPT1 subunit mRNA and protein expression are attenuated in MYPT1-depleted bladder tissues.

MLCP subunit mRNA and protein extract from MYPT1SM+/+ and MYPT1SM−/− bladder tissues were analysed in parallel by deep sequencing (A) and immunoblot (B). The values from MYPT1SM−/− are expressed relative to those obtained in tissues from MYPT1SM+/+. Data used to calculate relative mRNA expression are from Table 1. *P < 0.05, **P < 0.01, ***P < 0.001 compared with values for MYPT1SM+/+ mice as indicated by Student's unpaired two-tailed t test; n = 3 for deep sequencing and 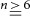 for immunoblot analysis.
for immunoblot analysis.
MLCP activity and subunit distribution in smooth muscle homogenates
Figure 2A and B show representative blots demonstrating the residual MYPT1 and reduced PP1cδ proteins in bladders from MYPT1SM−/− mice compared to bladders from MYPT1SM+/+ mice. MLCP activity was measured in detrusor homogenates from MYPT1SM+/+ and MYPT1SM−/− bladder smooth muscle with purified smooth muscle heavy meromyosin to determine if the decreased PP1cδ expression decreased MLCP activity similarly. In a separate set of tissues, measurements document a significant decrease in MLCP activity in MYPT1-deficient tissues (Fig. 2C), resulting from 19 ± 3% PP1cδ protein relative to tissues from wild-type mice (data not shown). The calculated specific activity for PP1cδ in bladder homogenates from wild-type and MYPT1-deficient mice was the same (Fig. 2D). Similar results (data not shown) were obtained with purified RLC as a substrate with 1 nm okadaic acid included to inhibit PP2A activity (Khromov et al. 2009). We attempted to measure MLCP activity in homogenates with constitutive phosphorylation of MYPT1, but both Thr696 and Thr853 were rapidly dephosphorylated even in the presence of 100 mm sodium pyrophosphate (data not shown). Although calyculin A inhibited dephosphorylation of MYPT1 in homogenates, it also inhibited phosphatase activity towards phosphorylated smooth muscle heavy meromyosin.
Figure 2. MYPT1 knockout affects MLCP activity without affecting subunit distribution in bladder smooth muscle from MYPT1SM+/+ (+/+) and MYPT1SM−/− (−/−) mice.
MYPT1 (A) and PP1cδ (B) expression was measured by immunoblotting the extracts prepared from MYPT1SM+/+ and MYPT1SM−/− bladders. Representative blots are shown above the quantified results with data presented as means ± SEM from 12 animals in each group. ***P < 0.001 compared with values for +/+ mice as indicated by Student's unpaired two-tailed t test. C, phosphatase activity (pmol 32P hydrolysed min–1 (μg protein)–1) in bladder homogenates from MYPT1SM+/+ and MYPT1SM−/− animals assayed with smooth muscle heavy meromyosin as described under Methods; immunoblots were also used to measure PP1cδ content in these samples (n ≥ 4, ***P < 0.001, t test, two-tailed). D, phosphatase activity normalized to PP1cδ protein expression. P = 0.23, t test, two-tailed. E, relative distribution of proteins in supernatant fractions after ultracentrifugation. PP1cδ and total PP1c showed significant solubility with no significant difference between tissues from MYPT1SM+/+ (open bars, n = 5) and MYPT1SM−/− (filled bars, n = 6) animals. All values were significantly less than soluble GAPDH (P < 0.01). **P < 0.01 compared with RLC, MYPT1 and MBS85 respectively. F, comparison of phosphatase activity of total (n = 5) and supernatant (n = 4) fractions from wild-type bladders after ultracentrifugation. **P < 0.01, t test, two-tailed.
MYPT1 binds and targets PP1cδ to myosin in smooth muscle myofilaments (Pato & Kerc, 1985; Alessi et al. 1992; Somlyo & Somlyo, 2003; Hartshorne et al. 2004). Therefore, tissue fractionation was performed to examine whether the MYPT1 knockout affects PP1cδ distribution between myofilaments and cytosolic fractions (Fig. 2E). As expected, GAPDH was completely soluble whereas RLC, MYPT1 and MBS85 were insoluble in tissues from MYPTSM+/+ mice. Similar results were obtained with tissues from MYPTSM−/− mice. Thus, the residual amount of MYPT1 in MYPTSM−/− mice showed a similar distribution compared to MYPT1 in tissues from MYPTSM+/+ mice. Interestingly, total PP1c and PP1cδ showed significant solubility in tissues from both groups of mice compared to PP1cα and PP1cγ, which were similarly insoluble compared to RLC, MYPT1 and MBS85. Thus, not all of PP1cδ was bound to MYPT1 in bladder tissues from MYPTSM+/+ mice. Furthermore, this cytosolic fraction containing no MYPT1 had MLCP activity, consistent with the amount of PP1cδ (Fig. 2F). The reduced amount of PP1cδ in tissues from MYPTSM−/− mice also showed a lesser amount in the supernatant fraction compared to the insoluble fraction.
Contractile responses of isolated tissues from MYPTSM+/+ and MYPTSM−/− mice
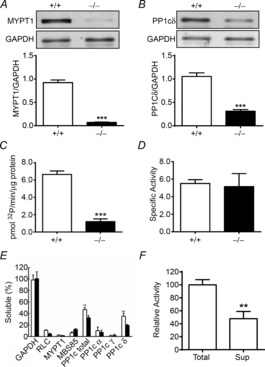
Contractile responses of bladder smooth muscle strips to KCl or the muscarinic receptor agonist carbachol were measured to identify any compensatory changes in force development in gene-ablated tissues. The maximal force development with KCl stimulation was comparable between MYPT1SM+/+ and MYPT1SM−/− tissues (Fig. 3A and C). However, the bladder strips from MYPT1SM−/− mice maintained higher force at the sustained phase (5 min) after KCl treatment (Fig. 3A and C). In contrast, the MYPT1-deficient bladder strips exhibited significant reduction in maximal force development in response to 10 μm carbachol stimulation (Fig. 3B and D), whereas the lower sustained responses at 5 min were similar to those obtained in MYPT1SM+/+ bladder (Fig. 3B and D). Although the maximal forces developed in response to KCl were similar, the time-to-peak force development was slower in tissues from MYPT1SM−/− mice (41 ± 2 s) than in those from MYPT1SM+/+ mice (27 ± 1 s). A similar result was also observed in carbachol-treated tissues from MYPT1SM+/+ mice (31 ± 1 s) and MYPT1SM−/− mice (50 ± 3 s). Similar results were obtained with cumulative carbachol dose–response measurements. The carbachol concentration-dependent contractions in isolated bladder strips showed a modest decrease in maximal responses in the MYPT1SM−/− group compared to the MYPT1SM+/+ group (0.64 ± 0.08 and 0.85 ± 0.06 g (mg wet weight)−1, respectively; P < 0.05, n = 10). No significant differences for the pEC50 values were found between the MYPT1SM+/+ and MYPT1SM−/− groups (5.94 ± 0.09 and 5.85 ± 0.10, respectively). To further assess the effect of MYPT1 knockout on endogenous MLCP activity, we next compared the relaxation rate after the washout of a stimulus. The relaxation rate following the removal of KCl in MYPT1SM−/− mouse bladder strip (t½ = 13.9 ± 0.6 s) was significantly slower than that observed in tissue from MYPT1SM+/+ (t½ = 9.3 ± 0.2 s, Fig. 3E). However, the relaxation rates following the removal of carbachol were similar in both MYPT1SM+/+ and MYPT1SM−/− groups (32.6 ± 1.9 s and 36.7 ± 2.3 s, respectively, Fig. 3F). These results show that bladder tissues from MYPT1SM−/− mice contract rapidly in response to carbachol and KCl treatments, and relax rapidly with their removal. Changes in contractile and relaxation force responses were not consistently observed with both kinds of treatments.
Figure 3. Bladder smooth muscle strips from MYPT1SM−/− mice contract with some changes in force development properties in response to KCl and carbachol treatments.
A and B, representative force tracings of MYPT1SM+/+ (grey trace) and MYPT1SM−/− (black trace) mouse bladder strips in response to 65 mm KCl and 10 μm carbachol (CCh) stimulation, respectively. C and D, quantitative summary of force development at peak and 5 min following 65 mm KCl or 10 μm CCh treatment, respectively. E and F, the time course of relaxation is shown following the removal of KCl or carbachol. Force measurements were normalized to the force at the beginning of the relaxation period. The data were fitted to a one-phase exponential decay equation (GraphPad Prism 6) and the time to reach 50% relaxation (t½) was calculated and shown in the insets. Data are means ± SEM from 10–13 measurements each from different bladders. Open bars, tissues from MYPT1SM+/+; filled bars, tissues from MYPT1SM−/−. ***P < 0.001 compared with MYPT1SM+/+.
Phosphorylation of signalling proteins relevant to contractile responses in MYPT1-deficient smooth muscle
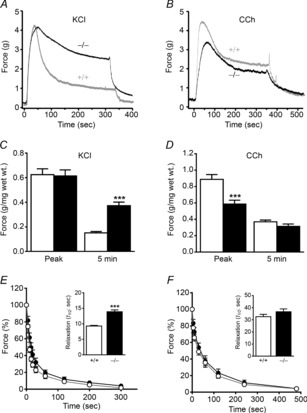
To pursue the mechanisms underlying the different contractile responses observed, the protein phosphorylation events converging on RLC phosphorylation were measured. As shown in Fig. 4A and B, RLC phosphorylation in MYPT1SM+/+ bladder strips increased rapidly and significantly to a maximal value of 0.46 ± 0.02 mol phosphate mol–1 RLC at 15 s after 10 μm carbachol treatment. RLC phosphorylation remained elevated at 1 min and then declined to 0.36 ± 0.03 mol phosphate mol–1 RLC at 5 min. Similar results were obtained in MYPT1SM−/− bladder tissues. Thus, there were no apparent differences in RLC phosphorylation in response to carbachol.
Figure 4. Carbachol-induced phosphorylation of proteins in the RLC signalling module shows modest differences between tissues from MYPT1SM−/− and MYPT1SM+/+ mice.
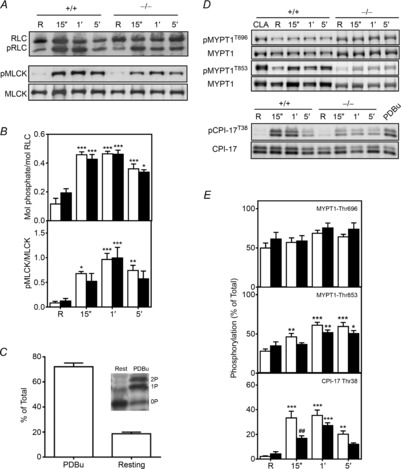
Representative immunoblots (A and D) and quantitative phosphorylation results (B and E) are shown for RLC, CPI-17, MYPT1 and MLCK for bladder tissues from MYPT1SM+/+ (open bars) and MYPT1SM−/− (filled bars) mice. Resting (R) smooth muscle strips were treated with 10 μm CCh and then rapidly frozen at 15 s, 1 min and 5 min. The frozen tissues were processed for protein phosphorylation analysis by immunoblotting as described under Methods. For more accurately measuring MYPT1 phosphorylation in MYPT1SM−/− tissues, the protein loaded was 5-fold greater (10 μg) than the amount loaded from MYPT1SM+/+ samples. CPI-17, and MYPT1 were quantified relative to results obtained with phorbol 12,13-dibutyrate (PDBu) or calyculin A (CLA) as described under Methods. C, Phos-tag PAGE was used to quantify CPI-17 phosphorylation in PDBu-treated tissues (n = 4) which separates CPI-17 according to the extent of phosphorylation) with no (0P), mono- (1P) or diphosphorylated (2P) species. Data are presented as means ± SEM for 6 to10 animals in each group. *P < 0.05, **P < 0.01, ***P < 0.001 vs.. resting value; ##P < 0.01 compared with MYPT1SM+/+ at 15 s as indicated by two-way ANOVA with Tukey's post hoc test for multiple comparisons.
We also analysed CPI-17 phosphorylation. Wild-type tissues were first treated with PDBu to activate PKC to obtain tissue samples with a high extent of CPI-17 phosphorylation. Phos-tag gel analysis of PDBu-treated tissues to separate different phosphorylated forms showed diphosphorylation, with the monophosphorylated species greater in amount than the diphosphorylated form (Fig. 4C). These results are consistent with an ordered hierarchical phosphorylation of Thr38 followed by partial phosphorylation of Ser12 (MacDonald et al. 2001). PDBu treatment increased the extent of CPI-17 Thr38 phosphorylation to 72 ± 3%, similar to previous studies. (Johnson et al. 2009; El-Yazbi et al. 2010; Moreno-Dominguez et al. 2013). Subsequent analyses of CPI-17 phosphorylation were performed by SDS–PAGE with phosphospecific antibody to CPI-17 Thr38, with PDBu-treated tissues used as standard for quantification. Analysis of CPI-17 phosphorylation in tissues from MYPT1SM+/+ mice showed prompt increases in CPI-17 phosphorylation with carbachol treatment, reaching a maximal value at 15 s (33 ± 5% vs. 2 ± 1% at rest, P < 0.001), and then declining by 5 min to an amount still significantly greater than resting phosphorylation (20 ± 2%, P < 0.01, Fig. 4D and E). In MYPT1SM−/− bladder strips, carbachol evoked smaller increases in CPI-17 phosphorylation. At the rising phase of contraction (15 s), the extent of CPI-17 phosphorylation was attenuated compared to responses obtained with tissues from MYPT1SM+/+ mice (Fig. 4D and E). Thus, CPI-17 was phosphorylated in MYPT1-deficient tissues, but showed some attenuation.
We developed procedures to quantify the extent of phosphorylation of MYPT1 Thr696 and Thr853. As shown in Fig. 5A, an expressed GST fragment of MYPT1 (residues 654–880) containing both Thr696 and Thr853 was phosphorylated by purified ROCK, resulting in diphosphorylation with prolonged incubation. This fragment was then used as a standard to quantify the extent of MYPT1 Thr696 and Thr853 phosphorylation of bladder smooth muscle tissues treated for 30 min with calyculin A. The total MYPT1 antibody raised to the MYPT1 sequence 723–840 allowed measurements of amount of the tissue MYPT1 relative to the GST-MYPT1 fragment for calculating the extent of MYPT1 phosphorylation with calyculin A treatment. Results show complete phosphorylation of both sites with calyculin A treatment of bladder tissues (Fig. 5B and C).
Figure 5. Quantitation of MYPT1 phosphorylation in calyculin A treated bladder strips.
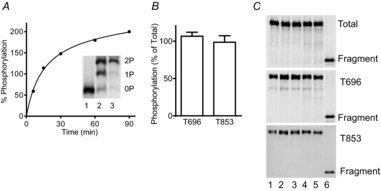
A, purified GST-MYPT1 (654–880) was maximally phosphorylated by ROCK1 in an in vitro kinase assay. Representative time course of phosphorylation is shown with immunoblot for total MYPT1 in the inset. Mono- (P) and diphosphorylated (PP) GST-MYPT1 (654–880) were separated from the non-phosphorylated (N) protein by Phos-tag gel in samples taken before (1), during (2), and end (3) of kinase assay. Samples which were confirmed to be fully diphosphorylated by Phostag gel analysis were used as controls for quantitation of endogenous MYPT1 Thr696 and Thr853 phosphorylation. B, amount of tissue MYPT1 phosphorylated at Thr696 per total Thr696 and tissue MYPT1 phosphorylated at 853 per total 853 are shown as ratios. C, representative blots for calyculin A-treated bladder strips from different wild-type mice (lanes 1–5) and diphosphorylated GST-MYPT1 fragment (lane 6) are shown for total proteins (top panel), phosphorylated Thr696 (middle panel), and phosphorylated Thr853 (bottom panel).
Using this quantitative procedure, we found that MYPT1 Thr696 and Thr853 sites were both substantially phosphorylated under resting conditions. MYPT1 Thr696 phosphorylation was greater than MYPT1 Thr853 phosphorylation in tissues from MYPT1SM+/+ mice (Fig. 4C and D). The amount of constitutive phosphorylation was similar in the residual amount of MYPT1 remaining in tissues from MYPT1SM−/− mice. Treatment of tissues with 3 μm GF 109203X or 1 μm H-1152 under resting conditions did not change MYPT1 Thr696 phosphorylation. However, MYPT1 Thr853 was decreased from 22 ± 3% to 7 ± 1% with H-1152, but not with GF 109203X treatment (data not shown), consistent with constitutive ROCK activity acting on MYPT1 Thr853. Agonist treatment with 10 μm carbachol did not increase MYPT1 Thr696 phosphorylation significantly in tissues from either MYPT1SM+/+ or MYPT1SM−/− mice (Fig. 4C and D). In contrast, MYPT1 Thr853 was significantly increased with carbachol treatment at 15 s, reaching a maximal response at 1 min which was maintained for 5 min in tissues from MYPT1SM+/+ mice (Fig. 4C and D). Similar phosphorylation results were obtained at 1 and 5 min after carbachol stimulation for the smaller amount of MYPT1 in tissues from MYPT1SM−/− mice (Fig. 4C and D). Thus, MYPT1 showed significant constitutive phosphorylation at Thr696 and Thr853, which increased at Thr853 with carbachol treatment. Although MYPT1 content was greatly diminished in tissues from MYPT1SM−/− mice, the temporal and relative phosphorylation of the residual MYPT1 were similar to those found in tissues from MYPT1SM+/+ mice.
The MYPT1 Thr696 phosphorylation site conserved in MBS85 (Thr560), one of the MYPT family members (Grassie et al. 2011), is also phosphorylated by ROCK, resulting in the inhibition of phosphatase activity of PP1cδ (Tan et al. 2001). Like MYPT1, MBS85 possesses the capacity for binding to phosphorylated myosin II and targeting PP1cδ catalytic activity to dephosphorylate RLC, although it is present in much lower amounts compared to MYPT1. We evaluated the possibility that MBS85 may be phosphorylated to compensate partially for MYPT1 deficiency during agonist-induced smooth muscle contraction. Although we did not confirm that calyculin A treatment resulted in 100% phosphorylation of MBS85 Thr560, we noted that calyculin A treatment increased MBS85 phosphorylation substantially and therefore we assumed that calculated phosphorylation was similar to results presented for MYPT1 phosphorylation. Like MYPT1 Thr696, MBS85 was constitutively phosphorylated (28 ± 3%) and the extent of phosphorylation did not increase with carbachol treatment (data not shown). Application of carbachol did not affect MBS85 phosphorylation in bladder smooth muscles from either MYPT1SM+/+ or MYPT1SM−/− mice.
Ca2+-dependent phosphorylation of Ser1760 in the C-terminus of the short MLCK calmodulin binding sequence desensitizes the kinase to activation by Ca2+–calmodulin (Kamm & Stull, 2001; Ding et al. 2009; Tsai et al. 2012). Therefore, we determined if there were any compensatory changes in MYPT1-deficient tissues. MLCK Ser1760 phosphorylation was low under resting conditions in both MYPT1SM+/+ and MYPT1SM−/− bladder smooth muscles (Fig. 4A and B). Exposure of bladder strips to carbachol induced an immediate phosphorylation of MLCK at 15 s, reaching a maximal value at 1 min and then decreasing in both MYPT1SM+/+ and MYPT1SM−/− mice by 5 min. Thus, MLCK phosphorylation was not modified in MYPT1-deficient tissues.
Signalling mechanisms underlying KCl-induced contraction were examined in the presence of 1 μm atropine to block muscarinic effects of acetylcholine which may be released from parasympathetic nerves upon depolarization. In parallel with force development, phosphorylation of RLC increased rapidly from a resting value of 0.09 ± 0.02 mol phosphate per mol RLC to 0.29 ± 0.02 mol phosphate per mol RLC then declined slightly by 5 min after 65 mm KCl treatment (Fig. 6A). Despite the greater sustained contractile force in MYPT1-deficient smooth muscles compared to those from MYPT1SM+/+ mice, the extent of RLC phosphorylation was not greatly enhanced (0.26 ± 0.01 mol phosphate per mol RLC and 0.21 ± 0.02 mol phosphate per mol RLC in tissues from MYPT1SM−/− and MYPT1SM+/+ mice, respectively). Emerging evidence indicates that KCl-induced contraction may also activate Ca2+ sensitization responses (Sakurada et al. 2003; Urban et al. 2003) so phosphorylation of CPI-17 and MYPT1 was examined. As illustrated in Fig. 6B, treatment of MYPT1SM−/− and MYPT1SM+/+ bladder smooth muscles with 65 mm KCl both showed small, transient increases in CPI-17 phosphorylation which were comparable between two groups. However, phosphorylation of MYPT1 at Thr853 and Thr696 were unaffected by KCl treatment in both MYPT1SM+/+ and MYPT1SM−/− bladder tissues (data not shown). Membrane depolarization by KCl also promptly increased MLCK phosphorylation which lasted for 5 min (Fig. 6C). The responses were comparable between MYPT1SM+/+ and MYPT1SM−/− bladder smooth muscles.
Figure 6. KCl-induced phosphorylation of proteins in the RLC signalling module shows no significant differences in tissues from MYPT1SM−/− mice.
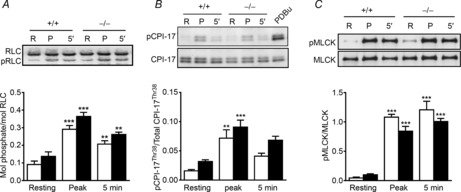
Representative immunoblots and quantitative phosphorylation results shown for strips rapidly frozen at peak and 5 min following 65 mm KCl treatment. The frozen tissues were processed for RLC (A), CPI-17 (B) and MLCK (C) phosphorylation measurements as described under Methods for bladder tissues from MYPT1SM+/+ (open bars) and MYPT1SM−/− (filled bars) mice. Data are presented as means ± SEM for 6 to 10 animals in each group. **P < 0.01, ***P < 0.001 vs. resting value as indicated by two-way ANOVA with Tukey's post hoc test for multiple comparisons.
Protein phosphorylation affecting actin dynamics in response to carbachol
In addition to Ca2+-dependent MLCK activation and Ca2+ sensitization by MLCP inhibition, a growing body of studies provides evidence for dynamic changes in the actin cytoskeleton that play a fundamental role in the regulation of airway and vascular smooth muscle contractile response (Gerthoffer & Gunst, 2001; Gunst & Zhang, 2008; Walsh & Cole, 2013). Tyrosine phosphorylation of FAK and paxillin at membrane-associated dense plaques occurs during force development with agonists or induction of a myogenic response. These phosphorylations are thought to play a role in the polymerization of submembranous cytoskeletal actin filaments that increases the rigidity of adhesion complexes in the membrane, allowing force transmission from phosphorylated, cycling myosin cross-bridges. Therefore, the phosphorylations of FAK and paxillin in response to contractile stimuli were measured. FAK-Tyr397 phosphorylation increased slowly in a time-dependent manner after bladder strips were treated with 10 μm carbachol or 65 mm KCl (Fig. 7A and C). In carbachol-treated strips, maximal FAK-Tyr397 phosphorylation was obtained by 1 min and maintained for 5 min. However, there were no significant differences in the phosphorylation responses between tissues from MYPT1SM+/+ and MYPT1SM−/− mice. Although a similar pattern was observed in KCl-treated strips, the phosphorylation of FAK-Tyr397 was significantly enhanced in MYPT1SM−/− bladders when compared to MYPT1SM+/+ tissues at 5 min. Deep sequencing analysis of mRNA and immunoblot for total FAK revealed no differences in the FAK expression levels between MYPT1SM+/+ and MYPT1SM−/− bladder smooth muscles.
Figure 7. Phosphorylation of proteins in the adhesion complex in response to carbachol and KCl treatments differs in tissues from MYPT1SM−/− and MYPT1SM+/+ mice.
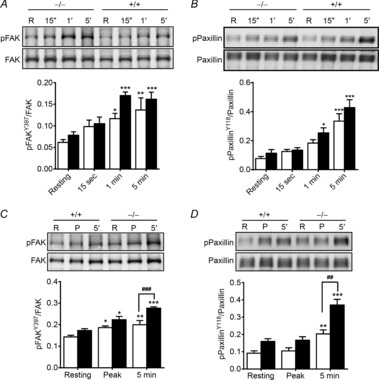
Representative immunoblots and quantitation shown for FAK (A and C) and paxillin (B and D) phosphorylation for bladder tissues from MYPT1SM+/+ (open bars) and MYPT1SM−/− (filled bars) mice. Smooth muscle strips were treated with 10 μm CCh (A and B) or 65 mm KCl (C and D) and then snap-frozen at indicated times. The frozen tissues were processed for protein phosphorylation analysis by immunoblotting as described under Methods. Data are presented as means ± SEM from at least 6 animals in each group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. resting value; ##P < 0.01, ###P < 0.001 vs. MYPT1SM+/+ strips at 5 min as indicated by two-way ANOVA with Tukey's post hoc test for multiple comparisons.
Paxillin phosphorylation at Tyr118 also occurs during agonist-stimulated vascular and airway smooth muscle contraction (Tang et al. 2003; Min et al. 2012). Moreover, FAK binds to paxillin to induce the phosphorylation of paxillin at Tyr118 (Bellis et al. 1995; Schaller & Parsons, 1995). In parallel with FAK Tyr397 phosphorylation, paxillin Tyr118 phosphorylation increased slowly to a maximal extent by 5 min following carbachol treatment (Fig. 7B). There were no significant differences between tissue responses from MYPT1SM+/+ and MYPT1SM−/− mice. Similarly, membrane depolarization with 65 mm KCl also evoked a slow increase in paxillin Tyr118 phosphorylation, and this phosphorylation was significantly enhanced in MYPT1SM−/− bladders when compared to MYPT1SM+/+ tissues at 5 min (Fig. 7D). The mRNA and protein expression levels of paxillin were also comparable between MYPT1SM+/+ and MYPT1SM−/− mice (data not shown).
Pharmacological characterization of ROCK and PKC signalling during carbachol-induced contraction in MYPT1-deficient smooth muscle
Proposed agonist-induced Ca2+-sensitization mechanisms generally involve phosphorylation of MYPT1 Thr696 and Thr853 as well as CPI-17 by ROCK and PKC (Somlyo & Somlyo, 2000; Hartshorne et al. 2004; Grassie et al. 2011). To gain further insights into signalling mechanisms during carbachol-induced bladder smooth muscle contraction, a pharmacological approach was used to characterize the respective contributions of PKC and ROCK in tissues from MYPT1SM+/+ and MYPT1SM−/− mice. Figure 8 illustrates representative force traces showing the contractile responses to 10 μm carbachol in the presence of 1 μm ROCK inhibitor H-1152 (Fig. 8A and C) or 3 μm PKC inhibitor GF 109203X (Fig. 8B and D). The efficacy of both inhibitors was altered in bladder strips from MYPT1SM−/− compared to MYPT1SM+/+ tissues. GF 109203X markedly inhibited the initial peak force, with less inhibition of the sustained force phase of carbachol-induced contraction from wild-type animals (Fig. 8E and F). H-1152 partially suppressed peak contractile response and sustained force development (Fig. 8E and F). The extent of inhibition of the sustained phase from H-1152 treatment was similar in magnitude to that obtained with GF 109203X treatment. Collectively, these results suggest PKC makes a larger contribution to the initial rising phase while both PKC and ROCK equally contribute to the sustained phase of carbachol-activated bladder smooth muscle contraction. The inhibitory effects of the two protein kinase inhibitors on carbachol-induced contraction were additive in tissues from wild-type animals (Fig. 8E and F). The inhibitory effect of GF 109203X was attenuated in bladder smooth muscles from MYPT1SM−/− mice. As predicted, the inhibitory effect of H-1152 was almost abolished in MYPT1-deficient tissues.
Figure 8. ROCK and PKC inhibitors have greater effects on carbachol-induced force development in bladder strips from MYPT1SM+/+ mice than from MYPT1SM−/− mice.
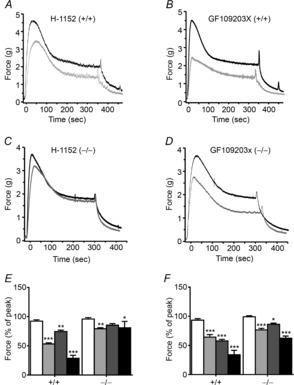
Representative force traces (grey) show the effects of ROCK inhibitor H-1152 (A and C) or PKC inhibitor GF109203X (B and D) on contractile responses to 10 μm carbachol treatment in tissue strips from MYPT1SM+/+ (A and B) and MYPT1SM−/− (C and D) mice. Force traces in the absence of inhibitors are shown in black. Quantitative summary of the effects of ROCK and PKC inhibitor on the force development at peak (E) and 5 min (F) following 10 μm CCh stimulation are shown. Force values are expressed relative to the peak and plateau forces recorded in the absence of inhibitors. Open bars, vehicle; grey bars, GF109203X; dark grey bars, H-1152; black bars, GF109203X+H-1152. Data are means ± SEM for 4–7 measurements from different animals. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle value.
As ROCK and PKC have been reported to be involved in FAK and paxillin tyrosine phosphorylation (Heidkamp et al. 2003; Li et al. 2003), the effects of PKC and ROCK inhibitors on carbachol-induced FAK and paxillin tyrosine phosphorylation were also examined. However, neither GF 109203X nor H-1152 exerted any effect on carbachol-evoked FAK and paxillin phosphorylation (data not shown).
Discussion
MYPT1 holds a central position in the regulation of MLCP activity (Hartshorne, 1987; Somlyo & Somlyo, 2000, 2004; Matsumura & Hartshorne, 2008; Grassie et al. 2011). Biochemically, MYPT1 binds PP1cδ and myosin, resulting in an increase in phosphatase activity towards RLC in myosin (Alessi et al. 1992). MYPT1 binding is restricted to the PP1cδ isoform where the central region of PP1cδ confers the isoform specific binding (Scotto-Lavino et al. 2010). This structural reshaping of PP1cδ is also necessary for the selective inhibitory effect of phosphorylated CPI-17 for MLCP (Eto, 2009). Thus, MYPT1 functions as a scaffolding protein that directs PP1cδ activity towards phosphorylated smooth muscle myosin, and provides selectivity for inhibition of the activity of PP1cδ bound to MYPT1 by phosphorylated CPI-17. Although MYPT1 binds PP1cδ stoichiometrically, not all of the PP1cδ may be bound to MYPT1 because other PP1c targeting proteins contain the RVXF motif (Hendrickx et al. 2009).
We found MLCP activity was proportional to the amount of PP1cδ with or without MYPT1, using phosphorylated smooth muscle heavy meromyosin as a MLCP specific substrate (Pato & Adelstein, 1983; Hartshorne et al. 1998; Somlyo & Somlyo, 2000). Similar results were obtained with phosphorylated RLC if okadaic acid was used to inhibit PP2A activity. PP2A and PP1c both dephosphorylate isolated RLC, but not RLC bound to myosin heavy chain as in smooth muscle heavy meromyosin (Pato et al. 1983). It is noteworthy that there was significant MLCP activity with smooth muscle heavy meromyosin without MYPT1 in a soluble cytosolic fraction containing PP1cδ from wild-type tissues, in addition to the significant phosphatase activity in MYPT1-deficient tissues. We were surprised that the specific activity of PP1c was the same with or without MYPT1 in bladder homogenates considering the biochemical stimulatory effects when purified MYPT1 is added to PP1c. One possibility is that PP1c is bound to another protein that enhances its activity. This and other possibilities need to be pursued. The decrease in the amount of PP1cδ in tissues from MYPT1SM−/− mice is probably due to the lack of the formation of a stable protein complex of PP1cδ with MYPT1 bound to myosin, because the cellular stability of PP1cδ depends on MYPT1 (Scotto-Lavino et al. 2010). Thus, MYPT1 was not biochemically necessary for MLCP activity in bladder tissues.
In our previous report showing that MYPT1 knockout in ileal tissues results in no decrease in PP1cδ content (He et al. 2013), a different knockout animal model was used. Mice containing floxed MYPT1 alleles were crossed with SMA-Cre transgenic mice, which express non-inducible Cre recombinase under the control of the α-SMA promoter during gestation (Wu et al. 2007). In the studies reported herein mice containing floxed Mypt1 alleles were crossed with a SMMHC-CreERT2 transgenic mouse line expressing a fusion protein of the Cre recombinase with the modified oestrogen receptor binding domain (CreERT2) under the control of the smooth muscle myosin heavy chain (SMMHC) promoter (Wirth et al. 2008; Gao et al. 2013). Thus, MYPT1 expression was extinguished in these adult mice with tamoxifen treatment, resulting in the decrease in PP1cδ content. PP1cδ was also decreased in ileal and aortic tissues in these mice (data not shown). We have confirmed the lack of a significant decrease in PP1cδ in ileal tissues from the SMA-Cre transgenic mice. The knockout of MYPT1 during gestation in the SMA-Cre transgenic mice may result in developmental compensation in the amount of PP1cδ expressed whereas the more acute knockout in adult animals with tamoxifen injection provides a response similar to the acute knockdown of MYPT1 in cells in culture (Scotto-Lavino et al. 2010).
Phosphorylation of MYPT1 Thr696 and Thr853 inhibits PP1cδ activity (Hartshorne, 1987; Somlyo & Somlyo, 2000; Matsumura & Hartshorne, 2008; Grassie et al. 2011). Stimulation of the RhoA pathway by various agonists activates ROCK to phosphorylate MYPT1 Thr853 (Somlyo & Somlyo, 2003; Dimopoulos et al. 2007). Constitutive Thr853 phosphorylation observed under resting conditions in smooth muscles is reduced with a ROCK inhibitor, but a more robust phosphorylation is generally observed with agonists (Somlyo & Somlyo, 2004; Dimopoulos et al. 2007; Wang et al. 2009; Mori et al. 2011). We have confirmed that the constitutive phosphorylation of MYPT1 Thr853 is reduced with a ROCK inhibitor and agonist-induced Thr853 is robust in bladder smooth muscle.
Phosphorylation of Thr696 in response to agonists is variable in terms of whether there is additional phosphorylation over a high extent of basal phosphorylation (Kitazawa et al. 2003; Niiro et al. 2003; Seko et al. 2003; Mizuno et al. 2008; Neppl et al. 2009). It is not clear which kinase(s) phosphorylate this site. We found the significant constitutive phosphorylation of MYPT1 Thr696 was not significantly reduced with PKC or ROCK inhibitors. Although zipper-interacting protein kinase (ZIPK) biochemically phosphorylates MYPT1 Thr696 and Thr853 (Grassie et al. 2011), recent evidence shows MYPT1 is not a target substrate for this kinase in permeable fibres (Moffat et al. 2011). Biochemically, integrin-linked kinase (ILK) also phosphorylates MYPT1 (Muranyi et al. 2002). However, there are no selective inhibitors of this kinase so whether it phosphorylates MYPT1 Thr696 in intact smooth muscle tissues is unknown. Thus, the protein kinase phosphorylating Thr696 remains unidentified in bladder smooth muscle.
In terms of MYPT1 regulating RLC dephosphorylation and relaxation of different smooth muscles serving diverse physiological functions, a genetic approach of knocking out MYPT1 in adult mouse smooth muscle tissues surprisingly did not result in a severe phenotype (He et al. 2013) like the MLCK knockout which produces smooth muscle contractile failure in intestinal, airway, mesenteric, aortic and urinary bladder smooth muscles, leading to death (He et al. 2008, 2011; Zhang et al. 2010; Gao et al. 2013). The smooth muscle-specific knockout of MYPT1 resulted in viable mice with normal body sizes, no apparent changes in relevant contractile or regulatory proteins, normal intestinal motility in vivo, modest changes in isolated intestinal muscle contractions, and mild hypertension (He et al. 2013). Isolated bladder tissues from MYPT1SM−/− mice contracted, and then relaxed rapidly. Similar results were observed with intestinal tissues (He et al. 2013). There were differences in some contractile properties but they were not consistent in relation to KCl versus carbachol treatments. For example, the t½ for relaxation following the washout of carbachol was unaffected by MYPT1 deletion, and the t½ for relaxation in KCl-precontracted tissues was increased only 1.5-fold in MYPT1SM−/− mice compared to that in wild-type tissues. As the KCl-induced sustained force was greater in MYPT1SM−/− mice, the t½ of relaxation increased. Phosphorylation of different proteins in the myosin RLC signalling module, including phosphorylation of RLC, CPI-17 and MLCK, was also not profoundly impacted. In addition, the phosphorylation of MYPT1 Thr696 or Thr853 was not disrupted for the residual MYPT1 remaining in smooth muscle tissue after tamoxifen treatment. The residual MYPT1 (less than 5%) may have resulted from incomplete DNA recombination or MYPT1 present in non-smooth muscle cells in the bladder smooth muscle tissue strips without urothelium. The normal phosphorylation of the residual MYPT1 demonstrates that signalling pathways are not greatly perturbed, although the inhibitory effects of these phosphorylations would be minimal on phosphatase activity.
One interesting observation consistent with MYPT1 regulating RLC phosphorylation is related to the effects of the PKC and ROCK inhibitors. The inhibitory actions of GF 109203X and H-1152 were attenuated in smooth muscle tissues from MYPT1SM−/− mice, consistent with phosphorylated CPI-17 inhibiting PP1cδ bound to MYPT1, and phosphorylated MYPT1 Thr853 also inhibiting PP1cδ bound to MYPT1 in tissues from MYPT1SM+/+ mice. Although we do not know why CPI-17 phosphorylation appears to be slower in carbachol-treated tissues, its potential importance appears to be minimal in terms of inhibiting PP1cδ activity in the absence of MYPT1. An obvious reason for the mild phenotype and modest modifications in contractile properties could be related to redundancy or compensation by other members of the MYPT1 family. Based on deep mRNA sequencing and Western blot results, we found no evidence for abundant expression of MYPT2, MYPT3 and MBS85 in bladder smooth muscle tissue. MBS85 was expressed at 16% of the amount of MYPT1 while MYPT2 and MYPT3 appeared to be much lower. Importantly, there was also no increased expression of any of these family members in MYPT1-deficient tissues.
We also considered the possibility that changes in actin dynamics may have compensated secondarily. Dynamic polymerization of the cortical actin in airway and vascular smooth muscle cells mediates assembly of extracellular matrix–cytoskeletal membrane junctions (adhesomes) at the sarcolemma (Gunst & Zhang, 2008; Walsh & Cole, 2013). Activated FAK induces tyrosine phosphorylation of paxillin (Opazo Saez et al. 2004) which is required for actin polymerization and contraction in airway smooth muscle (Wang et al. 1996; Tang et al. 2003; Opazo Saez et al. 2004). Although it is not yet clear if actin polymerization plays a dynamic role in bladder smooth muscle contraction, we evaluated this possibility by measuring phosphorylation of FAK Tyr397 and paxillin Tyr118. Consistent with our previous report (Ding et al. 2009), carbachol-induced paxillin phosphorylation increased slower than the contractile responses without significant differences between tissues from MTPT1SM+/+ and MTPT1SM−/− mice. The slow increase in paxillin phosphorylation is similar to the agonist-induced, slow phosphorylation of paxillin previously reported in carotid artery (Rembold et al. 2007). Both FAK and paxillin phosphorylation were enhanced in strips from MTPT1SM−/− mice compared to MTPT1SM+/+ mice treated with KCl, but only after 5 min. The potential importance of this difference with prolonged KCl treatment needs further investigation.
It became apparent in this investigation that we needed more quantitative approaches to understand the physiological role of MYPT1 in relation to RLC dephosphorylation, and the reason why the MYPT1 knockout did not result in marked phenotypic changes. We used established procedures to quantify RLC and CPI-17 phosphorylation by separating the non-phosphorylated form of each protein from phosphorylated RLC Ser19 and CPI-17 Thr38 using urea–glycerol or Phos-tag PAGE (Persechini et al. 1986; Mizuno et al. 2008; Takeya et al. 2008; Johnson et al. 2009). Treatment of bladder strips with PDBu induced a high extent of CPI-17 phosphorylation, consistent with previous results with vascular smooth muscle (Johnson et al. 2009; El-Yazbi et al. 2010; Moreno-Dominguez et al. 2013). Previous phosphorylation analyses of MYPT1 Thr696 and Thr853 have relied on measurements with site specific phospho-antibodies relative to some other measurement such as total MYPT1 or actin (Mizuno et al. 2008; Takeya et al. 2008; Johnson et al. 2009). This general approach is sufficient for demonstrating relative changes with pharmacological treatments, but no information is provided about the extents of phosphorylation of each site. Therefore, we developed a new procedure involving a fragment of MYPT1 containing both phosphorylation sites that could be completely phosphorylated by purified ROCK, which we then used as a standard for tissue measurements. This procedure also relied on the separation of the non-phosphorylated, monophosphorylated and diphosphorylated fragments to verify complete phosphorylation of both MYPT1 Thr696 and Thr853. The MYPT1 fragment with both sites fully phosphorylated was used to quantify phosphorylation of endogenous MYPT1 Thr696 and Thr853 in bladder tissues treated with calyculin A, and 100% phosphorylation of both sites was found. Tissue controls could thus be prepared routinely with calyculin A to standardize MYPT1 Thr696 and Thr853 phosphorylation measurements under resting conditions and with treatments.
Under resting conditions, the constitutive phosphorylation of CPI-17 Thr38 and MYPT1 Thr696 and Thr853 were different in bladder tissue from wild-type mice. CPI-17 was less than 5% phosphorylated whereas MYPT1 Thr696 was 45% phosphorylated. The same Thr696 site present in MBS85 was also significantly phosphorylated. Although the extent of MYPT1 Thr853 phosphorylation was less than Thr696, the amount was significant (28%). A recent study proposed a model involving intramolecular inhibition of PP1cδ catalytic activity bound to MYPT1 with phosphorylation at either Thr696 or Thr853 (Khromov et al. 2009). The direct docking of a single phosphorylation site with the active site of PP1cδ inhibited phosphatase activity. The interaction between the active site of PP1cδ bound to MYPT1 with either inhibitory site phosphorylated would be sufficient for inhibition of myosin light chain phosphatase activity. Thus, the significant constitutive phosphorylation of either site in bladder tissues is predicted to inhibit the activity of PP1cδ bound to phosphorylated MYPT1. Additionally, phosphorylated CPI-17 has a high affinity for MLCP and any constitutively phosphorylated CPI-17 is expected to contribute to inhibition of MLCP (Eto, 2009). These considerations lead to a physiological model for constitutive and agonist induced Ca2+ sensitization (Fig. 9). MLCP is active without binding phosphorylated MYPT1 or CPI-17 or it is inhibited by binding of one of the three phosphorylated sites, including MYPT1 Thr696 or Thr853 or CPI-17 Thr38. Thus, the constitutive phosphorylation would attenuate MLCP activity. Agonist-induced Ca2+ sensitization increases phosphorylation of MYPT1 Thr853 and CPI-17 Thr38, and thus, would result in additional inhibition of MLCP activity (Fig. 9). It is noteworthy that the amount of constitutive phosphorylation of these three phosphorylation sites is about half of the total amount measured with agonist stimulation. The amount of proposed inhibition should be considered as an upper limit from these calculations because it assumes no diphosphorylated MYPT1, and does not consider inhibition of the significant amount of PP1cδ that is not bound to MYPT1. It seems unlikely there would be no diphosphorylated MYPT1 and other approaches are needed to measure this.
Figure 9. Physiological model for constitutive and agonist-induced inhibition of myosin light chain phosphatase.
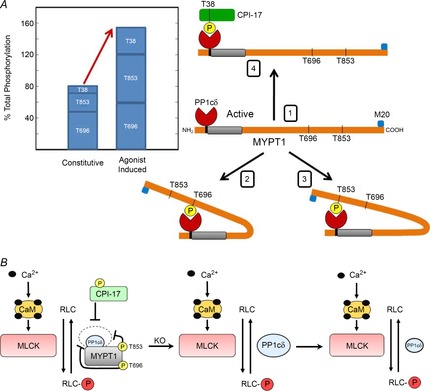
A, MLCP is predicted to exist physiologically in four states; (1) active MLCP, or inhibited by (2) MYPT1 Thr696 phosphorylation, (3) MYPT1 Thr853 phosphorylation, or (4) CPI-17 Thr38 phosphorylation. This model is based on intramolecular autoinhibition of MLCP activity by MYPT1 Thr696 phosphorylation or MYPT1 Thr853 phosphorylation (Khromov et al. 2009). The active site of PP1cδ will thus be inhibited by the binding of a phosphate from any one of the three phosphorylation sites on MYPT1 and CPI-17 where complete phosphorylation of all three sites is 300%. Quantitative measurements herein show that substantial constitutive phosphorylation is similar to the amount of agonist-induced phosphorylation of MYPT1 and CPI-17. B, signalling schema for RLC phosphorylation by Ca2+–calmodulin-dependent myosin light chain kinase and dephosphorylation by myosin light chain phosphatase containing MYPT1 and PP1cδ (left panel). Phosphorylated MYPT1 and CPI-17 inhibits PP1cδ activity (inner circle above MYPT1) compared to activity in the absence of phosphorylation (outer dashed circle). Knockout of MYPT1 in smooth muscle tissues is predicted to disinhibit PP1cδ activity (centre panel), but the reduction in PP1cδ protein decreases phosphatase activity (right panel) so that its activity is similar to MLCP activity inhibited by constitutively phosphorylated MYPT1.
The significant constitutive phosphorylation of MYPT1 may participate in regulation of RLC phosphorylation in relation to MLCK activation. Free Ca2+–calmodulin is limiting for MLCK activation in many smooth muscles, despite the abundance of total calmodulin (Tansey et al. 1994; Zimmermann et al. 1995; Isotani et al. 2004; Gao et al. 2013). In bladder smooth muscle quantitative measurements showed only 30% MLCK activation upon treatment with a high concentration of carbachol (Isotani et al. 2004; Mizuno et al. 2008). Although only a fraction of MLCK appears to be activated to initiate maximal contractile responses dependent on RLC phosphorylation, the attenuation of MLCP activity by constitutive phosphorylation of MYPT1 would enhance the initial RLC phosphorylation and force development. Considering neurostimulation of the murine urinary bladder (Tsai et al. 2012) and gastric fundus (Bhetwal et al. 2013) did not enhance MYPT1 phosphorylation, the constitutive phosphorylation of MYPT1 may provide a primary physiological signalling mechanism for Ca2+ sensitization in these tissues. It is not yet clear if constitutive phosphorylation of MYPT1 may change to affect smooth muscle responses under different physiological or pathophysiological conditions. The new quantitative approach described herein provides a tool to assess these possibilities.
With inactivation of MYPT1 alleles, the elimination of MYPT1 by cell degradation processes was associated with the secondary loss of PP1cδ. If the amount of PP1cδ protein was not decreased, its phosphatase activity would not be inhibited constitutively by phosphorylated MYPT1. However, there was a marked decrease in PP1cδ protein and activity. If the decreased PP1cδ activity resulting from MYPT1 deficiency was similar to the PP1cδ activity with constitutive phosphorylation of MYPT1 in wild-type tissues, RLC phosphorylation and smooth muscle contractions would not be physiologically impaired in vivo. Any potential differences in contractile performance may be functionally minimized, considering the adaptability of physiological systems that modulate smooth muscle tone with the autonomic nervous system and other signalling systems.
In summary, smooth muscle MYPT1 was not necessary for adult mouse viability. Isolated MYPT1-deficient intestinal (He et al. 2013) and bladder (herein) smooth muscle tissues contracted rapidly with KCl and agonist treatments, and relaxed rapidly with their washout. Novel quantitative measurements show significant constitutive MYPT1 phosphorylation predicted to attenuate MLCP activity in normal tissues. Depletion of MYPT1 in smooth muscle tissues reduced the amount of PP1cδ so that its activity may be similar to the reduced MLCP activity resulting from constitutively phosphorylated MYPT1. Thus, the loss of MYPT1 does not cause significant derangement of smooth muscle contractility and function in vivo. Potential physiological and pathophysiological mechanisms for regulation of constitutive MYPT1 phosphorylation need investigation.
Acknowledgments
We thank Tara Billman for assistance with maintaining mice and Xiaoyan Liu for purification of smooth muscle heavy meromyosin. We also appreciate the gifts of SMMHC-CreERT2 transgenic mice from Stefan Offermanns, and biochemical reagents from Masumi Eto.
Glossary
- CPI-17
protein kinase C (PKC)-potentiated protein phosphatase 1 inhibitor protein of 17 kDa
- FAK
focal adhesion kinase
- MBS85
myosin binding subunit 85
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- MYPT1
myosin phosphatase targeting subunit-1
- PDBu
phorbol 12,13-dibutyrate
- RLC
myosin regulatory light chain
- ROCK
RhoA-associated protein kinase
Additional information
Competing interests
The authors have no conflicts of interest to declare.
Author contributions
M.Z., W.H., H.L.S., K.E.K. and J.T.S. conceived the experimental approaches; M.-H.T., A.N.C., J.H., K.E.K and J.T.S. designed experiments; M.-H.T, A.N.C., J.H. and W.H. performed the experiments; M.-H.T, A.N.C., J.H., M.Z., H.L.S., K.E.K. and J.T.S. analysed data and wrote the manuscript. All authors approved the final version.
Funding
This work was supported by National Institutes of Health Grants R01 HL112778 and P01 HL110869, Moss Heart Fund, and the Fouad A. and Val Imm Bashour Distinguished Chair in Physiology, National Basic Research Program of China Grant 2009CB942602, and National Natural Science Funding of China Grant 31272311.
References
- Alessi D, MacDougall LK, Sola MM, Ikebe M, Cohen P. The control of protein phosphatase-1 by targeting subunits. Eur J Biochem. 1992;210:1023–1035. doi: 10.1111/j.1432-1033.1992.tb17508.x. [DOI] [PubMed] [Google Scholar]
- Bellis SL, Miller JT, Turner CE. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- Bhetwal BP, Sanders KM, An C, Trappanese DM, Moreland RS, Perrino BA. Ca2+ sensitization pathways accessed by cholinergic neurotransmission in the murine gastric fundus. J Physiol. 2013;591:2971–2986. doi: 10.1113/jphysiol.2013.255745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal DK, Stull JT. Effects of pH, ionic strength, and temperature on activation by calmodulin an catalytic activity of myosin light chain kinase. Biochemistry. 1982;21:2386–2391. doi: 10.1021/bi00539a017. [DOI] [PubMed] [Google Scholar]
- Chang AN, Chen G, Gerard RD, Kamm KE, Stull JT. Cardiac myosin is a substrate for zipper-interacting protein kinase (ZIPK) J Biol Chem. 2010;285:5122–5126. doi: 10.1074/jbc.C109.076489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn JC, Michnoff CH, Hsu LC, Slaughter CA, Kamm KE, Stull JT. Sites phosphorylated in myosin light chain in contracting smooth muscle. J Biol Chem. 1988;263:19166–19173. [PubMed] [Google Scholar]
- Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121:11–18. doi: 10.1242/jcs.007112. [DOI] [PubMed] [Google Scholar]
- Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res. 2007;100:121–129. doi: 10.1161/01.RES.0000253902.90489.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding HL, Ryder JW, Stull JT, Kamm KE. Signaling processes for initiating smooth muscle contraction upon neural stimulation. J Biol Chem. 2009;284:15541–15548. doi: 10.1074/jbc.M900888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Yazbi AF, Johnson RP, Walsh EJ, Takeya K, Walsh MP, Cole WC. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol. 2010;588:1747–1762. doi: 10.1113/jphysiol.2010.187146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem. 2009;284:35273–35277. doi: 10.1074/jbc.R109.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter EF, Eto M, Wardle RL, Brautigan DL, Murphy RA. Activation of myosin light chain phosphatase in intact arterial smooth muscle during nitric oxide-induced relaxation. J Biol Chem. 2001;276:34681–34685. doi: 10.1074/jbc.M104737200. [DOI] [PubMed] [Google Scholar]
- Gao N, Huang J, He W, Zhu M, Kamm KE, Stull JT. Signaling through myosin light chain kinase in smooth muscles. J Biol Chem. 2013;288:7596–7605. doi: 10.1074/jbc.M112.427112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol. 2001;91:963–972. doi: 10.1152/jappl.2001.91.2.963. [DOI] [PubMed] [Google Scholar]
- Grassie ME, Moffat LD, Walsh MP, MacDonald JA. The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch Biochem Biophys. 2011;510:147–159. doi: 10.1016/j.abb.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol. 2008;295:C576–C587. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne DA. Physiology of the Gastrointestinal Tract. 2nd edn. New York: Raven Press; 1987. Biochemistry of the contractile process in smooth muscle; pp. 423–482. [Google Scholar]
- Hartshorne DJ, Ito M, Erdodi F. Myosin light chain phosphatase: subunit composition, interactions and regulation. J Muscle Res Cell Motil. 1998;19:325–341. doi: 10.1023/a:1005385302064. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ, Ito M, Erdodi F. Role of protein phosphatase Type 1 in contractile functions: Myosin phosphatase. J Biol Chem. 2004;279:37211–37214. doi: 10.1074/jbc.R400018200. [DOI] [PubMed] [Google Scholar]
- He WQ, Peng YJ, Zhang WC, Lv N, Tang J, Chen C, Zhang CH, Gao S, et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology. 2008;135:610–620. doi: 10.1053/j.gastro.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WQ, Qiao YN, Peng YJ, Zha JM, Zhang CH, Chen C, Chen CP, Wang P, et al. Altered contractile phenotypes of intestinal smooth muscle in mice deficient in myosin phosphatase target subunit 1. Gastroenterology. 2013;144:1456–1465. doi: 10.1053/j.gastro.2013.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WQ, Qiao YN, Zhang CH, Peng YJ, Chen C, Wang P, Gao YQ, Chen X, et al. Role of myosin light chain kinase in regulation of basal blood pressure and maintenance of salt-induced hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H584–H591. doi: 10.1152/ajpheart.01212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidkamp MC, Bayer AL, Scully BT, Eble DM, Samarel AM. Activation of focal adhesion kinase by protein kinase C epsilon in neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2003;285:H1684–H1696. doi: 10.1152/ajpheart.00016.2003. [DOI] [PubMed] [Google Scholar]
- Hendrickx A, Beullens M, Ceulemans H, Den Abt T, Van Eynde A, Nicolaescu E, Lesage B, Bollen M. Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem Biol. 2009;16:365–371. doi: 10.1016/j.chembiol.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Hirano K, Derkach DN, Hirano M, Nishimura J, Kanaide H. Protein kinase network in the regulation of phosphorylation and dephosphorylation of smooth muscle myosin light chain. Mol Cell Biochem. 2003;248:105–114. doi: 10.1023/a:1024180101032. [DOI] [PubMed] [Google Scholar]
- Isotani E, Zhi G, Lau KS, Huang J, Mizuno Y, Persechin A, Geguchadze R, Kamm KE, Stull JT. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc Natl Acad Sci U S A. 2004;101:6279–6284. doi: 10.1073/pnas.0308742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol. 2009;587:2537–2553. doi: 10.1113/jphysiol.2008.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- Khromov A, Choudhury N, Stevenson AS, Somlyo AV, Eto M. Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J Biol Chem. 2009;284:21569–21579. doi: 10.1074/jbc.M109.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T. G protein-mediated Ca2+-sensitization of CPI-17 phosphorylation in arterial smooth muscle. Biochem Biophys Res Comm. 2010;401:75–78. doi: 10.1016/j.bbrc.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem. 2000;275:9897–9900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Khalequzzaman M. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J Physiol. 2003;546:879–889. doi: 10.1113/jphysiol.2002.029306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Kitazawa K. Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J Physiol. 2012;590:5401–5423. doi: 10.1113/jphysiol.2012.241315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang SQ, Kwartler CS, Byanova KL, Pham J, Gong L, Prakash SK, Huang J, Kamm KE, et al. Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res. 2012;110:1411–1422. doi: 10.1161/CIRCRESAHA.111.261743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wernig F, Leitges M, Hu Y, Xu Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J. 2003;17:2106–2108. doi: 10.1096/fj.03-0150fje. [DOI] [PubMed] [Google Scholar]
- Lowey S, Trybus KM. Common structural motifs for the regulation of divergent class II myosins. J Biol Chem. 2010;285:16403–16407. doi: 10.1074/jbc.R109.025551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JA, Eto M, Borman MA, Brautigan DL, Haystead TA. Dual Ser and Thr phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by MYPT-associated kinase. FEBS Lett. 2001;493:91–94. doi: 10.1016/s0014-5793(01)02277-3. [DOI] [PubMed] [Google Scholar]
- Matsumura F, Hartshorne DJ. Myosin phosphatase target subunit: Many roles in cell function. Biochem Biophys Res Commun. 2008;369:149–156. doi: 10.1016/j.bbrc.2007.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Reznichenko M, Poythress RH, Gallant CM, Vetterkind S, Li Y, Morgan KG. Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol. 2012;227:3585–3592. doi: 10.1002/jcp.24062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Isotani E, Huang J, Ding H, Stull JT, Kamm KE. Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am J Physiol Cell Physiol. 2008;295:C358–C364. doi: 10.1152/ajpcell.90645.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat LD, Brown SB, Grassie ME, Ulke-Lemee A, Williamson LM, Walsh MP, MacDonald JA. Chemical genetics of zipper-interacting protein kinase reveal myosin light chain as a bona fide substrate in permeabilized arterial smooth muscle. J Biol Chem. 2011;286:36978–36991. doi: 10.1074/jbc.M111.257949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Dominguez A, Colinas O, El-Yazbi A, Walsh EJ, Hill MA, Walsh MP, Cole WC. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol. 2013;591:1235–1250. doi: 10.1113/jphysiol.2012.243576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori D, Hori M, Murata T, Ohama T, Kishi H, Kobayashi S, Ozaki H. Synchronous phosphorylation of CPI-17 and MYPT1 is essential for inducing Ca2+ sensitization in intestinal smooth muscle. Neurogastroenterol Motil. 2011;23:1111–1122. doi: 10.1111/j.1365-2982.2011.01799.x. [DOI] [PubMed] [Google Scholar]
- Muranyi A, MacDonald JA, Deng JT, Wilson DP, Haystead TA, Walsh MP, Erdodi F, Kiss E, Wu Y, Hartshorne DJ. Phosphorylation of the myosin phosphatase target subunit by integrin-linked kinase. Biochem J. 2002;366:211–216. doi: 10.1042/BJ20020401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Ann Rev Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- Neppl RL, Lubomirov LT, Momotani K, Pfitzer G, Eto M, Somlyo AV. Thromboxane α2-induced Bi-directional regulation of cerebral arterial tone. J Biol Chem. 2009;284:6348–6360. doi: 10.1074/jbc.M807040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiro N, Koga Y, Ikebe M. Agonist-induced changes in the phosphorylation of the myosin-binding subunit of myosin light chain phosphatase and CPI17, two regulatory factors of myosin light chain phosphatase, in smooth muscle. Biochem J. 2003;369:117–128. doi: 10.1042/BJ20021040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol. 2004;286:C433–C447. doi: 10.1152/ajpcell.00030.2003. [DOI] [PubMed] [Google Scholar]
- Pato MD, Adelstein RS. Purification and characterization of a multisubunit phosphatase from turkey gizzard smooth muscle. The effect of calmodulin binding to myosin light chain kinase on dephosphorylation. J Biol Chem. 1983;258:7047–7054. [PubMed] [Google Scholar]
- Pato MD, Adelstein RS, Crouch D, Safer B, Ingebritsen TS, Cohen P. The protein phosphatases involved in cellular regulation. 4. Classification of two homogeneous myosin light chain phosphatases from smooth muscle as protein phosphatase-2A1 and 2C, and a homogeneous protein phosphatase from reticulocytes active on protein synthesis initiation factor eIF-2 as protein phosphatase-2A2. Eur J Biochem. 1983;132:283–287. doi: 10.1111/j.1432-1033.1983.tb07360.x. [DOI] [PubMed] [Google Scholar]
- Pato MD, Kerc E. Purification and characterization of a smooth muscle myosin phosphatase from turkey gizzards. J Biol Chem. 1985;260:12359–12366. [PubMed] [Google Scholar]
- Persechini A, Kamm KE, Stull JT. Different phosphorylated forms of myosin in contracting tracheal smooth muscle. J Biol Chem. 1986;261:6293–6299. [PubMed] [Google Scholar]
- Rembold CM, Tejani AD, Ripley ML, Han S. Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol. 2007;293:C993–C1002. doi: 10.1152/ajpcell.00090.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto-Lavino E, Garcia-Diaz M, Du G, Frohman MA. Basis for the isoform-specific interaction of myosin phosphatase subunits protein phosphatase 1c beta and myosin phosphatase targeting subunit 1. J Biol Chem. 2010;285:6419–6424. doi: 10.1074/jbc.M109.074773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522:177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+-sensitivity of smooth and non-muscle myosin II: modulation by G Proteins, kinases and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction through the RhoA/Rho-kinase pathway in smooth muscle. J Muscle Res Cell Motil. 2004;25:613–615. doi: 10.1007/s10974-004-3146-1. [DOI] [PubMed] [Google Scholar]
- Takeya K, Loutzenhiser K, Shiraishi M, Loutzenhiser R, Walsh MP. A highly sensitive technique to measure myosin regulatory light chain phosphorylation: the first quantification in renal arterioles. Am J Physiol Renal Physiol. 2008;294:F1487–F1492. doi: 10.1152/ajprenal.00060.2008. [DOI] [PubMed] [Google Scholar]
- Tan I, Ng CH, Lim L, Leung T. Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. J Biol Chem. 2001;276:21209–21216. doi: 10.1074/jbc.M102615200. [DOI] [PubMed] [Google Scholar]
- Tang DD, Turner CE, Gunst SJ. Expression of non-phosphorylatable paxillin mutants in canine tracheal smooth muscle inhibits tension development. J Physiol. 2003;553:21–35. doi: 10.1113/jphysiol.2003.045047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Luby-Phelps K, Kamm KE, Stull JT. Ca2+-dependent phosphorylation of myosin light chain kinase decreases the Ca2+ sensitivity of light chain phosphorylation within smooth muscle cells. J Biol Chem. 1994;269:9912–9920. [PubMed] [Google Scholar]
- Tsai MH, Kamm KE, Stull JT. Signalling to contractile proteins by muscarinic and purinergic pathways in neurally stimulated bladder smooth muscle. J Physiol. 2012;590:5107–5121. doi: 10.1113/jphysiol.2012.235424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol. 2003;285:C1377–C1385. doi: 10.1152/ajpcell.00501.2002. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Cole WC. The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab. 2013;33:1–12. doi: 10.1038/jcbfm.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Kendig DM, Smolock EM, Moreland RS. Carbachol-induced rabbit bladder smooth muscle contraction: roles of protein kinase C and Rho kinase. Am J Physiol Renal Physiol. 2009;297:F1534–F1542. doi: 10.1152/ajprenal.00095.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Pavalko FM, Gunst SJ. Tyrosine phosphorylation of the dense plaque protein paxillin is regulated during smooth muscle contraction. Am J Physiol Cell Physiol. 1996;271:C1594–C1602. doi: 10.1152/ajpcell.1996.271.5.C1594. [DOI] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, et al. G12-G13-LARG-mediated signalling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X, Xu J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respir Res. 2007;8:1. doi: 10.1186/1465-9921-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki K, Ito M, Machida H, Moriki N, Okamoto R, Isaka N, Shimpo H, Kohda A, et al. Identification of human CPI-17, an inhibitory phosphoprotein for myosin phosphatase. Biochem Biophys Res Commun. 2001;285:1040–1045. doi: 10.1006/bbrc.2001.5290. [DOI] [PubMed] [Google Scholar]
- Zhang WC, Peng YJ, Zhang GS, He WQ, Qiao YN, Dong YY, Gao YQ, Chen C, et al. Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J Biol Chem. 2010;285:5522–5531. doi: 10.1074/jbc.M109.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann B, Somlyo AV, Ellis-Davies GC, Kaplan JH, Somlyo AP. Kinetics of prephosphorylation reactions and myosin light chain phosphorylation in smooth muscle. Flash photolysis studies with caged calcium and caged ATP. J Biol Chem. 1995;270:23966–23974. doi: 10.1074/jbc.270.41.23966. [DOI] [PubMed] [Google Scholar]