

The cellular hydrophobicity threshold for the inversion of Sec-dependent Nin-Cout (type II) transmembrane domains is dictated by whether their membrane integration occurs cotranslationally or posttranslationally.
Abstract
Membrane insertion by the Sec61 translocon in the endoplasmic reticulum (ER) is highly dependent on hydrophobicity. This places stringent hydrophobicity requirements on transmembrane domains (TMDs) from single-spanning membrane proteins. On examining the single-spanning influenza A membrane proteins, we found that the strict hydrophobicity requirement applies to the Nout-Cin HA and M2 TMDs but not the Nin-Cout TMDs from the type II membrane protein neuraminidase (NA). To investigate this discrepancy, we analyzed NA TMDs of varying hydrophobicity, followed by increasing polypeptide lengths, in mammalian cells and ER microsomes. Our results show that the marginally hydrophobic NA TMDs (ΔGapp > 0 kcal/mol) require the cotranslational insertion process for facilitating their inversion during translocation and a positively charged N-terminal flanking residue and that NA inversion enhances its plasma membrane localization. Overall the cotranslational inversion of marginally hydrophobic NA TMDs initiates once ∼70 amino acids past the TMD are synthesized, and the efficiency reaches 50% by ∼100 amino acids, consistent with the positioning of this TMD class in type II human membrane proteins. Inversion of the M2 TMD, achieved by elongating its C-terminus, underscores the contribution of cotranslational synthesis to TMD inversion.
INTRODUCTION
Eukaryotic membrane protein maturation occurs cotranslationally within the endoplasmic reticulum (ER; Alder and Johnson, 2004; Braakman and Bulleid, 2011). The maturation process initiates when an N-terminal signal sequence or a hydrophobic segment of a nascent chain is recognized by the signal recognition particle (SRP; Walter and Blobel, 1981). SRP targets the ribosome-nascent chain complex to the SRP receptor located next to the Sec61 protein-conducting channel (the translocon) in the ER membrane (Gilmore et al., 1982; Deshaies and Schekman, 1987; Gorlich et al., 1992). Synthesis then resumes, and the translocon enables the passage of the nascent chain into the ER lumen and the partitioning of transmembrane domain (TMD) segments into the membrane (Bowie, 2005).
Membrane partitioning by the translocon occurs via the lateral gate and requires that TMD segments have the appropriate length and hydrophobicity to integrate into the lipid bilayer (Van den Berg et al., 2004; Hessa et al., 2007). However, it is quite common to find marginally hydrophobic TMD segments (ΔGapp for membrane insertion > 0 kcal/mol), which by themselves are expected to surpass the integration step and enter the ER lumen (Lu et al., 2000; Hessa et al., 2007; Zhang et al., 2007). The majority of these marginally hydrophobic TMDs exist in multispanning membrane proteins, where their integration has been associated with the orientation and hydrophobicity of neighboring TMDs (Ota et al., 1998b; Ojemalm et al., 2012), TMD–TMD interactions (Kanki et al., 2002; Zhang et al., 2007), flanking residue composition (Ota et al., 1998a), and the polypeptide length preceding a stop-transfer TMD (Hessa et al., 2003). Thus, in multispanning membrane proteins, the predominant compensatory mechanism for marginally hydrophobic TMD insertion appears to be interactions and associations with other TMDs.
For the majority of single-spanning membrane proteins, membrane partitioning occurs during the entry and passage of their lone TMD through the ER translocon. The exceptions are the few proteins whose TMDs are located ≤25 amino acids from the C-terminus and insert posttranslationally via the tail-anchored pathway (Kalbfleisch et al., 2007; Stefanovic and Hegde, 2007). Within Sec-dependent membrane proteins, the TMD positioning varies, and the position can determine whether their membrane integration occurs while attached to the ribosome (cotranslationally) or after release (posttranslationally).
The TMDs from single-spanning membrane proteins are generally more hydrophobic than those in multispanning membrane proteins (Arkin and Brunger, 1998; Hessa et al., 2007), a characteristic that is often linked to their dependence on one TMD for membrane integration. Nevertheless, marginally hydrophobic TMDs exist in single-spanning membrane proteins, and a recent study linked them to the quality control of heteromeric complex assembly, as their integration required TMD interactions with other subunits (Feige and Hendershot, 2013).
The processes that contribute to the insertion of Sec-dependent single-spanning membrane proteins have yet to be fully identified, especially with respect to changes in their TMD properties. In contrast, specific characteristics within this class of proteins are known to contribute to whether their TMDs acquire an Nout-Cin, or Nin-Cout orientation. Nout-Cin TMDs predominantly exist in single-spanning membrane proteins with a cleavable N-terminal signal sequence (type I membrane proteins; Daniels et al., 2003; Wang et al., 2005). The signal sequence of these proteins directs their ER targeting and translocation across the ER membrane, where the translocon inserts their TMD in an Nout-Cin orientation. Nin-Cout TMDs are generally located at the N-terminus of single-spanning membrane proteins and function as a signal anchor sequence (type II membrane proteins). These TMDs first target the protein to the ER, where they insert into the translocon head-on, and then achieve their Nin-Cout orientation by inverting end over end through a ribosomally mediated process (Goder and Spiess, 2003; Devaraneni et al., 2011). A small number of TMDs that function as signal anchor sequences do not undergo this inversion, which results in these TMDs acting as stop-transfer sequences and inserting in an Nout-Cin orientation (Hull et al., 1988).
Here we combined a computational and experimental analysis of the TMDs from the available single-spanning influenza A membrane protein sequences to identify relationships between TMD hydrophobicity, positioning, and orientation. Our results demonstrate that the contribution of the cotranslational membrane insertion process to the inversion of Nin-Cout type II membrane proteins is influenced by their TMD hydrophobicity. This process is essential for neuraminidase (NA) inversion with a marginally hydrophobic TMD (ΔGapp > 0 kcal/mol), which enhances their proper plasma membrane localization. In contrast, NA with a hydrophobic TMD inverts relatively well after ribosomal release. During translocation, an inversion efficiency of 50% required synthesis of ∼100 amino acids past the marginally hydrophobic TMD, consistent with the topology and hydrophobicity analysis of the human type II membrane proteome. These results, and the inversion of the Nout-Cin M2 TMD by elongating its C-terminus, indicate that ribosomal association likely enables less ideal TMDs to orientate and access the membrane, whereas synthesis contributes to the inversion process.
RESULTS
TMD hydrophobicity in influenza A virus membrane proteins depends on positioning and orientation
The analysis of protein sequence homology, property, and structural conservation has proved extremely useful in identifying protein topology and localization, functional domains, and the existence of certain cellular machinery (von Heijne, 2006; Daniels et al., 2010). However, the limited number of homologous sequences for human single-spanning membrane protein has hindered these approaches. Therefore we used the extensive human influenza A virus (IAV) sequence database to perform a comparative analysis of the TMD characteristics from natural single-spanning membrane proteins with Nout-Cin (HA and M2) and Nin-Cout (NA) orientations (Figure 1A).
FIGURE 1:
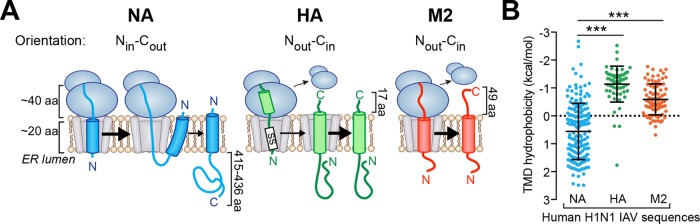
Nin-Cout (type II) NA TMDs from human H1N1 IAVs have a broad hydrophobicity range. (A) Topologies for the IAV membrane proteins NA, HA, and M2 and how their TMD positioning influences ribosomal involvement during the membrane integration step. (B) Dot plot showing predicted hydrophobicity (ΔGapp) variation in the unique NA, HA, and M2 TMDs from human H1N1 IAV sequences. Note that TMD hydrophobicity decreases with increasing positive ΔGapp.
On completion of this analysis, a trend was found with respect to the TMD hydrophobicity, orientation, and positioning. The Nout-Cin TMDs from HA that are located closest to the C-terminus (within 17 residues) possessed the highest average hydrophobicity (ΔGapp = −1.1 kcal/mol; Figure 1B). The Nout-Cin TMDs from M2, located 49 residues from the C-terminus, had a slightly lower average hydrophobicity (ΔGapp = −0.6 kcal/mol), and the Nin-Cout TMDs from NA, located 415–436 residues from the C-terminus, had the lowest average hydrophobicity (ΔGapp = 0.6 kcal/mol). In addition to having a broader hydrophobicity distribution, ∼70% of the unique Nin-Cout NA TMDs are marginally hydrophobic (ΔGapp > 0 kcal/mol), which should not support efficient membrane insertion (Hessa et al., 2007). Based on these results in IAV single-spanning membrane proteins, it appears their TMD hydrophobicity requirements become less strict when they are positioned further from the C-terminus or in an Nin-Cout orientation.
IAV TMD hydrophobicity values potentially relate to how the TMDs integrate into the membrane
In eukaryotic cells, ∼40 amino acids (aa) are required to span the ribosomal exit tunnel (Malkin and Rich, 1967; Blobel and Sabatini, 1970). Thus secretory protein TMDs located within 40 aa of the C-terminus integrate into the membrane after ribosomal release (posttranslationally), whereas TMDs located more than 40 aa away have the potential to integrate during synthesis (cotranslationally). Based on these ribosomal length restrictions, the TMD hydrophobicity differences in the IAV proteins appear to relate to their membrane integration process. The most hydrophobic TMDs are from HA, which are followed by a 17-aa C-terminus or C-tail and presumably integrate into the membrane posttranslationally. Next are the M2 TMDs with a 49-aa C-tail that can integrate cotranslationally and/or posttranslationally, and the least hydrophobic TMDs are in NA, which likely integrate and invert cotranslationally because of their 415- to 436-aa C-tail (Figure 1, A and B).
Marginally hydrophobic NA TMDs followed by a short C-terminus mislocalize in cells
To investigate potential relationships between TMD hydrophobicity and their C-terminal positioning in mammalian cells, we sought to exploit the normal plasma membrane (PM) localization of NA and M2. Whereas PM localization does not differentiate alterations in membrane integration, trafficking, PM recycling, or degradation, all of these are indicative of abnormal NA and M2 TMD behavior. To assess the PM localization, we compared the mean fluorescence signal of a substrate at the PM to the intracellular (IC) region of each cell to determine a PM/IC ratio (Supplemental Figure S1A). Based on PM and IC controls, substrates with a ratio >1 can be classified as PM localized and those with a ratio <1 as IC localized (Supplemental Figure S1, B and C).
This approach was first applied to evaluate the localization of full-length NA with natural hydrophobic (ΔGapp = −0.7 kcal/mol) and marginally hydrophobic (ΔGapp = +1.3) Nin-Cout TMDs. Including the epitope tag, both constructs possess a long, 440-aa C-tail that follows the TMD, and from here on are referred to by the nomenclature TM∆G XNAZaa, where X is the predicted TMD hydrophobicity (ΔGapp value in kcal/mol) and Z is the C-tail length in amino acids (aa). As expected, both full-length NA constructs (TM∆G −0.7NA440aa and TM∆G +1.3NA440aa) localized to the PM with an average PM/IC ratio >2 (Figure 2A). Similarly, full-length M2 with a hydrophobic TMD (ΔGapp = −1.1 kcal/mol) and a C-tail of 70 aa (including the epitope tag) also localized to the PM.
FIGURE 2:

Marginally hydrophobic NA TMDs with a short C-tail mislocalize in cells. (A) Cell PM/IC ratios for the full-length Nin-Cout membrane protein NA440aa with a hydrophobic (ΔGapp = −0.7 kcal/mol) and a marginally hydrophobic (ΔGapp = +1.3 kcal/mol) TMD, as well as the Nout-Cin membrane protein M270aa with a hydrophobic (ΔGapp = −1.1 kcal/mol) TMD and the intracellular (IC) marker. Right, representative cell section images. (B) PM/IC ratios from cells expressing these membrane proteins with a C-tail truncated to 36 aa. Right, representative images. (C) Higher-magnification image showing localization of TM∆G +1.3NA36aa (white) in “ring-like” vesicle structures near the nucleus (blue). (D) Vesicles from cells transfected with the indicated constructs were separated from the cytoplasm (Cyto) and subjected to an alkaline extraction to separate integral (P) from peripheral (SN) membrane proteins. Representative immunoblots with GFP as the soluble cytoplasm control. (E) Vesicle and cytoplasmic fractions from transfected cells were analyzed by flotation on sucrose cushions of different density. The dot blot shows the intensity of TM∆G +1.3NA36aa and TM∆G +1.3NA440aa that floated (SN) or pelleted (P) at each density, and the enzymatic activity distribution for TM∆G +1.3NA440aa provides an added control. (F) Similar analysis as in E, except that TM∆G +1.3NA36aa was radiolabeled for 30 min and isolated after the separation using Ni-Sepharose before resolution by SDS–PAGE.
We then asked, how does truncating the C-tail of NA or M2 to 36 aa affect its localization? The short C-tail length was chosen such that if these TMDs can integrate into cellular membranes, they would likely do so after ribosomal release. This would minimize the contribution of synthesis and ribosomal attachment, and also avoid targeting via the tail-anchored pathway. The short, 36-aa C-tail did not affect PM localization of NA with a hydrophobic TMD (TM∆G −0.7NA36aa), as the PM/IC ratios were all >1, but the same construct with a marginally hydrophobic TMD (TM∆G +1.3NA36aa) completely mislocalized to intracellular punctate, “ring-like” structures creating PM/IC ratios <1 (Figure 2, B and C). Surprisingly, M2 with a hydrophobic TMD and a short C-tail (TM∆G −1.1M236aa) showed a bimodal localization, with most of the cells possessing a PM/IC ratio >1, with some also <1. Thus shortening the C-tail length to 36 aa can affect TMDs from single-spanning membrane proteins, with the most dramatic mislocalization occurring when NA encoded a marginally hydrophobic TMD.
The mislocalized TM∆G +1.3NA36aa resides within the membrane of intracellular vesicles
A variety of biochemical assays were used to determine whether the intracellular “ring-like” structures containing TM∆G +1.3NA36aa (shown in Figure 2C) were cytoplasmic aggregates of mistargeted polypeptides or intracellular vesicles. Compared to the PM-localized full-length construct (TM∆G +1.3NA440aa), the C-terminal epitope on the mislocalized TM∆G +1.3NA36aa was completely protected from extracellular trypsin, confirming its intracellular localization (Supplemental Figure S2A). Isolation of vesicles from cells followed by alkaline extraction showed that TM∆G +1.3NA36aa is integrated into the membranes of the intracellular vesicles and not aggregated within the cytoplasm (Figure 2D).
Finally, we investigated the vesicle density where TM∆G +1.3NA36aa localized by flotation on sucrose cushions of low (1.03 g/ml), medium (1.15 g/ml), and high (1.25 g/ml) densities. At low density, all intracellular vesicles should sediment, and the cytoplasm should remain in the supernatant; at medium density, vesicles derived from the ER, PM, and Golgi should float, and the mitochondria, lysosomes, peroxisomes, and rough ER vesicles should sediment; and at high density, the majority of vesicles should float (Beaufay et al., 1974; Smith and Peters, 1980). Based on protein and enzyme activity distributions at steady-state, the majority of vesicles with the full-length protein (TM∆G +1.3NA440aa) floated at medium (1.15 g/ml) density, whereas the majority of vesicles with TM∆G +1.3NA36aa floated at high (1.25 g/ml) density (Figure 2E). In contrast, pulse labeling revealed that a portion of newly synthesized TM∆G +1.3NA36aa localized to vesicles with ER density (Figure 2F, flotation at 1.15 g/ml). Together the vesicle density analysis and their morphology suggest that TM∆G +1.3NA36aa is initially targeted to the ER and likely trafficked to the lysosome instead of the PM.
PM localization of NA36aa is linked to its inversion, which requires a hydrophobic TMD
The NA construct with a short C-tail mislocalized with a marginally hydrophobic TMD (TM∆G +1.3NA36aa), but not a hydrophobic TMD (TM∆G −0.7NA36aa). To determine whether this is a general effect, we increased the hydrophobicity of TM∆G +1.3NA36aa in four incremental steps by substituting Ala residues into the TMD and determined the PM/IC ratios (Figure 3 and Table 1). More cells had a PM/IC ratio >1 with the increasing TMD hydrophobicity in these constructs, and PM localization was favored in the most hydrophobic ones (TM∆G −1.4NA36aa and TM∆G −2.5NA36aa). These results confirmed that PM localization of NA with a short, 36-aa C-tail is dependent on TMD hydrophobicity and that it is also related to the sequence context, as the natural TMD (TM∆G−0.7NA36aa) had more PM localization (Figure 2B) than the engineered, more hydrophobic mutant (TM∆G −1.4NA36aa).
FIGURE 3:
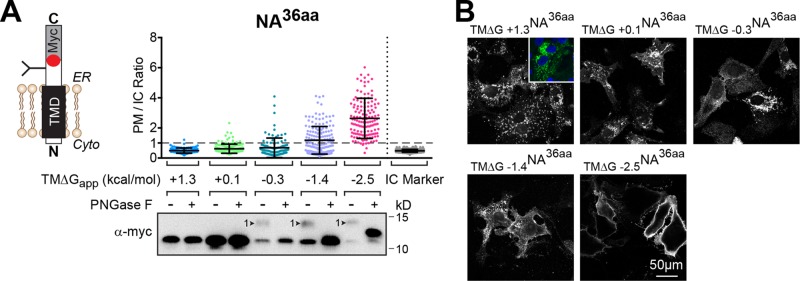
NA TMDs with a short C-tail have a stricter hydrophobicity requirement. (A) PM/IC ratios of individual cells expressing NA with a short, 36-aa C-tail and TMDs that range from being marginally hydrophobic (+1.3 kcal/mol) to hydrophobic (−2.5 kcal/mol). The immunoblot shows the constructs that received the expected N-linked glycan (arrowhead), which was confirmed by digestion with PNGase F. (B) Representative cell section images of the constructs analyzed in A. Inset, the mislocalized TM∆G +1.3NA36aa (green) with respect to the nucleus (blue).
TABLE 1:
NA and M2 TMD sequences used in this study and their respective hydrophobicity values.
| Construct name | ∆Gapp (kcal/mol) | TMD sequence |
|---|---|---|
| TM ΔG −2.5 NA | −2.5 | IITIGSVCMTIGMAALILAIGAIISIWI |
| TM ΔG −1.4 NA | −1.4 | IITIGSVCMTIGMAALILQIGAIISIWI |
| TM ΔG −0.7 NA | −0.7 | IITIGSICMVVGIISLILQIGNIISIWI |
| TM ΔG −0.3 NA | −0.3 | IITIGSVCMTIGMAALILQIGNIISIWI |
| TM ΔG +0.1 NA | +0.1 | IITIGSVCMTIGMANLILAIGNIISIWI |
| TM ΔG +1.3 NA | +1.3 | IITIGSVCMTIGMANLILQIGNIISIWI |
| TM ΔG −1.1 M2 | −1.1 | LVIAANIIGILHLILWILDRLFF |
The inversion of Nin-Cout TMDs was previously shown to become less efficient with increasing hydrophobicity (Goder and Spiess, 2003). Therefore we examined the orientation of the NA TMDs with increasing hydrophobicity and the short, 36-aa C-tail to determine whether their topology changed in relation to the PM localization. Inversion to the proper Nin-Cout orientation was assayed by the ∼2.5 kDa shift caused by the N-linked glycan addition to the C-tail glycosylation site. According to the molecular weight shift and digestion of the N-linked glycan by peptide-n-glycosidase F (PNGase F), only the three most hydrophobic constructs were glycosylated (Figure 3A, immunoblot). Thus the inversion and ER membrane integration of NA TMDs with a short, 36-aa C-tail increase with hydrophobicity and do not occur properly with marginally hydrophobic TMDs, which suggests that the inversion is necessary for their PM trafficking and accumulation.
Long C-tails help marginally hydrophobic NA TMDs to invert and localize to the PM
Our results thus far indicate that full-length NA with a marginally hydrophobic TMD localizes to the PM, likely because the longer C-tail facilitates inversion and potentially ER targeting. By making smaller increases in the C-tail length after the marginally hydrophobic NA TMD, we observed a stepwise increase in the average PM/IC ratios (Figure 4A). The increase slightly began with a C-tail length of 41 aa, which approximates the length of the ribosomal tunnel (Malkin and Rich, 1967; Blobel and Sabatini, 1970), and increased until a length of 76 aa, which showed a PM/IC profile distribution similar to the full-length construct (TM∆G +1.3NA440aa) in this steady-state assay. These results confirmed that the C-tail length contributes to PM localization of the marginally hydrophobic NA TMD.
FIGURE 4:

Inversion and PM localization of marginally hydrophobic NA TMDs is C-tail-length dependent. (A) PM/IC ratios of cells expressing the marginally hydrophobic NA construct (TM∆G +1.3NA) with increasing C-tail lengths and representative cell section images. (B) Orientation of the marginally hydrophobic NA TMD construct with increasing C-tail lengths was determined by the expected N-linked glycosylation pattern (depicted by the construct diagrams) at steady state (top, immunoblots) and after metabolic labeling (bottom, [35S]Met/Cys autoradiographs). Cell lysates were harvested 48 h posttransfection or after a 15-min pulse, and, where indicated, N-linked glycans were digested with PNGase F. The N-linked glycan number (arrowheads), partial PNGase F digestion (asterisks), and SDS-resistant TMD tetramer (Tet) are indicated. (C) The percent glycosylation of the marginally hydrophobic NA TMDs with respect to C-tail length from steady-state and 35S-labeling experiments is shown. Glycosylation indicates that the constructs have the proper Nin-Cout orientation. (D) Analysis of the indicated 35S-labeled constructs by nonreducing and reducing SDS–PAGE reveals that only the glycosylated species form the expected intermolecular disulfide. Note the glycosylated-band molecular weight shifts ± DTT and the partial SDS-resistant tetrameric species.
The inversion of Nin-Cout TMDs has been shown to occur through a ribosomally mediated process (Goder and Spiess, 2003; Devaraneni et al., 2011), which raised the question of whether the C-tail aids in the inversion of the marginally hydrophobic NA TMD. Therefore we examined the orientation of the marginally hydrophobic NA TMD with elongated C-tails by their N-linked glycosylation patterns. At steady-state levels, an extremely small percentage of glycosylated chains were first observed with a C-tail of 46 aa, and the percentage slowly increased with the C-tail length until a significant jump (15–90%) occurred between 66 and 71 aa (Figure 4, B, top, immunoblots, and C). To ensure that the low positioning of the first glycosylation site (∼10 aa from the TMD) did not affect these results, we shifted this native site up 6 aa on two short C-tail constructs, which also yielded similar low glycosylation levels (Supplemental Figure S2C).
The strong inflection point for TMD inversion at steady-state could be due to the differential stability of glycosylated versus unglycosylated membrane proteins (Buck and Skach, 2005; Mochizuki et al., 2007). Thus the orientation analysis was also performed on newly synthesized chains that were pulse labeled with [35S]Met and Cys (Figure 4, B, bottom, audioradiographs, and C). After labeling for 15 min, minor glycosylation of the marginally hydrophobic NA TMD first occurred with a C-tail of 61 aa. In contrast to the steady-state analysis, the percentage of nascent glycosylated chains increased in a linear manner as the C-tail was elongated from 61 aa, and the 50% inversion point required a C-tail of ∼100 aa.
As a second test for orientation, we exploited the C-tail cysteine that creates intermolecular disulfide-bonded NA TMD dimers in a glycan-independent manner (da Silva et al., 2013). Under nonreducing conditions (without dithiothreitol [DTT]), the majority of the pulse-labeled, marginally hydrophobic NA TMDs with N-linked glycans existed in disulfide-bonded dimers and SDS-resistant tetramers (Figure 4D). Upon addition of the reducing agent DTT, the oligomeric species fully (dimer) and partially (tetramer) collapsed to monomeric glycosylated species. In contrast, the band intensities of the unglycosylated species did not change, which showed that their C-tails were located in the cytoplasm. Together these results demonstrate that the marginally hydrophobic NA TMDs require the synthesis of ∼70 aa after the TMD to initiate their inversion during translocation, and the efficiency reaches 50% after the synthesis of ∼100 aa. Therefore ribosomal attachment and/or protein synthesis is essential for the cellular inversion of marginally hydrophobic Nin-Cout (type II) NA TMDs.
Inversion of the hydrophobic NA TMD does not require ribosomal attachment
Because the hydrophobic NA TMDs (ΔGapp < 0 kcal/mol) with short, 36-aa C-tails properly localized to the PM (Figures 2 and 3), we also examined the hydrophobic NA TMD orientation with increasing C-tail lengths. At steady-state, these constructs were already ∼65% glycosylated, with a C-tail of 36 aa, and their glycosylation percentage increased further once the C-tail length was >61 aa (Figure 5A, top, immunoblots, and B). The analysis of the nascent chains after a 15-min pulse showed that the percentage of glycosylated species increased with the C-tail length beginning from 36 aa (Figure 5A, bottom, audioradiographs, and B). These results demonstrate that hydrophobic NA TMDs benefit from, but do not require, the cotranslational membrane insertion process (ribosomal attachment and/or synthesis) for their inversion, which explains their PM localization with short C-tails.
FIGURE 5:
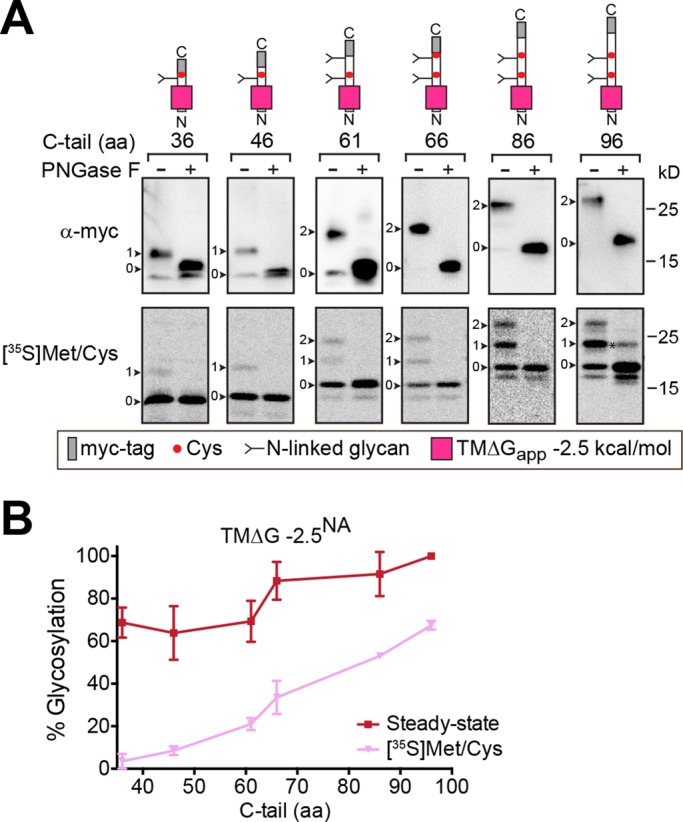
Hydrophobic NA TMDs are less dependent on C-tail length for inversion. (A) The orientation of the hydrophobic NA TMD construct with increasing C-tail lengths was analyzed at steady-state (top, immunoblots) and after metabolic labeling for 15 min (bottom, [35S]Met/Cys autoradiographs) as described in Figure 4B. The N-linked glycan number (arrowheads) and partial PNGase F digested species (asterisk) are shown. (B) The percent glycosylation observed for the hydrophobic NA TMDs with respect to C-tail length from steady-state and 35S-labeling experiments is shown.
Marginally hydrophobic NA TMDs can facilitate cotranslational ER targeting
In addition to affecting membrane integration and orientation, decreased hydrophobicity in Nin-Cout (type II) TMDs and shorter C-tail length could also potentially affect their SRP recognition and hence their ER targeting capability (Bird et al., 1990; Hatsuzawa et al., 1997). Therefore we created a series of ribosomally arrested chains with elongated C-tails using the hydrophobic and marginally hydrophobic NA TMDs and assayed their ability to direct targeting to rough ER microsomes. As expected, no targeting to the ER microsomes was observed when the marginally hydrophobic TMD was largely sequestered in the ribosome with a 22-aa C-tail (Figure 6A). With a 36-aa C-tail, a higher proportion of the marginally hydrophobic NA TMD ribosomally arrested chains sedimented with the ER microsomes, and the proportion substantially increased with a 46-aa C-tail, which is similar to the 49-aa C-tail on the SRP-dependent M2 TMD (Hull et al., 1988). A comparable but more efficient ER targeting profile was observed with the hydrophobic NA TMD ribosomally arrested chains (Figure 6B).
FIGURE 6:

Marginally hydrophobic NA TMDs with a short C-tail can target to the ER cotranslationally. Ribosomally arrested chains with the indicated C-tail lengths and the marginally hydrophobic (A) and hydrophobic (B) NA TMD were in vitro translated in the absence and presence of rough ER microsomes (MS) before sedimentation through a sucrose cushion to isolate the microsomally associated chains (left). Representative experiments are shown for each construct, with the corresponding total amount of synthesized protein (Lysate) for the shortest constructs. To test for inversion and arrest, the glycosylation pattern of the 46-aa and 96-aa C-tail constructs were released with 1 mM puromycin (Puro) before sedimentation (right). The N-linked glycan number, unglycosylated chains (circles), and a potential SDS-resistant NA TMD dimer (asterisk) are indicated.
To confirm the chains were arrested, we examined the glycosylation pattern of the 46-aa and 96-aa C-tail constructs after release by puromycin. With the marginally hydrophobic NA TMD, the single glycan on the 46-aa C-tail construct did not appear after release, indicating that its inversion was defective (Figure 6A, right). In contrast, puromycin release of the 96-aa C-tail construct resulted in the expected enhanced recognition of its second glycosylation site, demonstrating the chains were inverted and arrested. Puromycin release of both constructs with the hydrophobic NA TMD enhanced their glycosylation, confirming their ability to properly invert (Figure 6B, right). These in vitro experiments demonstrate that both types of NA TMDs can facilitate ER targeting, albeit with potentially different efficiencies, and that the ER-targeted marginally hydrophobic NA TMDs do not effectively invert with short C-tails, which supports our cellular observations.
N-terminal length and charge contribute to cotranslational type II NA TMD inversion
The orientation of Nin-Cout (type II) TMDs can be influenced by the positioning of positively charged flanking residues and the length of the N-terminal flanking region (von Heijne, 1989; Kocik et al., 2012). In line with these findings, the 6-aa N-terminus is a highly conserved region in NA and includes a positively charged residue (K or, less commonly, R) adjacent to the TMD (Figure 7A). To investigate whether these N-terminal features aid the marginally hydrophobic NA TMD inversion, we examined the glycosylation pattern of the TM∆G +1.3NA76aa construct with several N-terminal deletions and mutations (Figure 7B).
FIGURE 7:

N-terminal flanking residues help marginally hydrophobic NA TMDs to invert. (A) Logo plot displaying the conservation of the cytoplasmic-localized, N-terminal NA TMD flanking residues in the human H1N1 IAV sequences. (B) Diagram of the N-terminal mutations and deletions that were analyzed in the TMΔG+1.3NA76aa construct. (C) Immunoblots showing the orientation of the constructs depicted in B by the absence or presence of the expected two N-linked glycans (arrowheads). Cell lysates were harvested 48 h posttransfection, and a portion was treated with PNGase F before resolution by reducing Tris-tricine SDS–PAGE. (D) PM/IC ratios from cells expressing the constructs shown in B with representative cell section images. Insets, cellular localization of the constructs (green) with respect to the nucleus (blue).
When the initiation Met was followed by five Ala (MAA), subtle glycosylation was observed compared with wild type (WT), but if the positively charged Lys remained (MAK), the glycosylation levels matched those of WT (Figure 7C). No glycosylation was observed when the initiation Met was placed next to the TMD (M), which indicated that the subtle glycosylation of MAA, due to its inversion, was facilitated by the length of the N-terminus. However, the construct with a Met and Lys next to the TMD (MK) showed substantial glycosylation, demonstrating that the positively charged residue was the most dominant feature. From a PM localization perspective, the N-terminal, positively charged Lys residue (MK) made the largest contribution, and this contribution was even greater in the context of the proper N-terminal length (MAK; Figure 7D). Thus the N-terminal flanking residues increase the inversion efficiency of marginally hydrophobic NA TMDs, with positively charged residues making the most predominant contribution.
Cotranslational protein synthesis contributes to the TMD inversion process
To examine whether the cotranslational integration process contributes to TMD inversion more directly, we tested whether elongating the C-tail following the Nout-Cin M2 TMD could facilitate its inversion. As shown by the oxidation of M2 into homodimers and tetramers via its N-terminal Cys residues, full-length M2 with a 70-aa C-tail (including the epitope tag) possessed an Nout-Cin orientation at steady-state (Figure 8A). Similarly, when the 76-aa NA C-tail was fused to the M2 N-terminus and TMD, the Nout-Cin orientation remained unchanged, as no glycosylation of the NA C-tail was observed (Figure 8B, lanes 3 and 4). However, when the full-length 440-aa NA C-tail was fused to the M2 N-terminus and TMD, a reasonable amount of inversion to an Nin-Cout orientation occurred, based on glycosylation (Figure 8B, lanes 7 and 8) and the production of enzymatically active NA (Figure 8C). The ability to invert the hydrophobic M2 TMD in the absence of its single C-terminal positive charge upon substantially lengthening the 76-aa NA C-tail to 440 aa indicates that cotranslational synthesis and/or integration contributes to the inversion process. However, the production of mixed orientated species also implies that TMDs likely possess their own topology determinants in addition to their cytoplasmic-localized flanking residues.
FIGURE 8:

An elongated C-tail can invert the Nout-Cin M2 TMD and is associated with marginally hydrophobic human Nin-Cout (type II) TMDs. (A) Immunoblot of reduced and nonreduced cell lysates showing the Nout-Cin orientation of M2 based on the intermolecular disulfide bonds formed by its N-terminal Cys residues in the ER lumen. (B) Immunoblots of untreated and PNGase F–treated lysates showing the glycosylation patterns for TMΔG+1.3NA76aa, M2 with the 76-aa NA C-tail, full-length NA (TMΔG+1.3NA440aa), and M2 with the full-length 440-aa NA C-tail with the enzymatic domain. The number of N-linked glycans for each species is indicated. (C) Enzymatic activity was used to confirm M2 TMD inversion, as NA only folds within the ER lumen. The activity rate of M2 with the full-length 440-aa NA C-tail was calculated in comparison to lysates from cells expressing full-length NA as described in da Silva et al. (2013). (D) TMD hydrophobicity for the annotated type II human membrane proteins with respect to the length of the C-terminus after their TMD (C-tail). Dashed line at 100 aa corresponds to the ∼50% inversion point for newly synthesized NA with a marginally hydrophobic TMD. Regions covering the marginally hydrophobic TMDs and potential tail-anchored pathway substrates are highlighted. Protein sequences were obtained from Uniprot and analyzed using MPEx. The raw data and references are given in Supplemental Table S1.
Marginally hydrophobic TMDs in human type II membrane proteins insert cotranslationally
Overall our findings indicate that the cotranslational insertion via the ER translocon can lower the hydrophobicity requirement for the proper inversion and membrane integration of the Nin-Cout (type II) NA TMDs. Specifically, the inversion initiated when ∼70 aa past the marginally hydrophobic NA TMD had been synthesized, and it required ∼100 aa to reach an efficiency of 50% for newly synthesized polypeptides. Using the 50% inversion efficiency point (a C-tail of 100 aa), we found these results to be consistent with the hydrophobicity and positioning of the ∼400 predicted human type II membrane proteins (Figure 8D and Supplemental Table 1). A number of marginally hydrophobic TMDs (ΔGapp > 0 kcal/mol) are found in human type II membrane proteins, and all of them possess a C-tail >100 aa. Together the data on marginally hydrophobic TMDs in NA, and their positioning in type II human membrane proteins, imply that the cotranslational insertion process facilitates the integration and inversion of marginally hydrophobic Nin-Cout (type II) TMDs.
DISCUSSION
The insertion of TMDs from single-spanning membrane proteins differs from those in multispanning membrane proteins on the basis that their positioning largely dictates whether their membrane integration occurs while attached to the ribosome or upon release. Previous studies on these TMDs showed that orientation can add additional complexity to their membrane integration process, as Nin-Cout (type II) TMDs use a two-step insertion and inversion mechanism (Goder and Spiess, 2003; Devaraneni et al., 2011). More recently, marginally hydrophobic Nout-Cin (type I) TMDs, which primarily integrate after ribosomal release, were found to require TMD–TMD interactions with their heteromeric complex subunits to integrate into the membrane (Feige and Hendershot, 2013).
The results presented here build on these previous findings by showing that the Nin-Cout (type II) NA TMDs exploited the cotranslational membrane integration process to overcome the strict hydrophobicity threshold for single-spanning membrane protein TMDs (Arkin and Brunger, 1998; Hessa et al., 2007). The consistency of our results with the hydrophobicity and positional analysis of human type II membrane protein TMDs suggests that lowering the hydrophobicity membrane integration threshold may be a general property of cotranslational insertion. If true, this property could rationalize why the NA TMDs in IAVs, but not the HA and M2 TMDs, have been able to evolve against the hydrophobicity threshold (Nordholm et al., 2013). In contrast, the short C-tails after the marginally hydrophobic Nout-Cin TMDs in the T-cell receptor subunits preclude this option, possibly explaining why they use TMD–TMD interactions for their integration, which is common in multispanning membrane proteins (Kanki et al., 2002; Zhang et al., 2007; Feige and Hendershot, 2013). Although the latter mechanism is hard to reconcile for the cotranslational maturation of a homo-oligomer such as NA (Wang et al., 2008), it reveals that a number of mechanisms potentially exist for the integration of this large class of TMDs.
The correlation between the glycosylation inversion assay and the qualitative PM localization measurements for the marginally hydrophobic NA TMD was not always ideal. For instance, the constructs with C-tails from 46 to 61 aa showed PM/IC ratio increases and only incremental increases in the low glycosylation levels at steady-state. This implied that either these NA TMDs invert after synthesis and translocation or noninverted TMDs also traffic to the PM but less efficiently. Trypsin treatment of cells expressing the glycosylated construct with a C-tail of 76 aa digested a significant portion of the C-terminal myc epitope, indicating that the NA TMD was inverted, whereas the majority of the epitope was protected on the surface-localized construct with a C-tail of 61 aa (Supplemental Figure S2B). This indicates that noninverted NA TMDs can also localize to the PM, and the observed increases in the PM/IC ratio are more indicative of changes in the efficiency of PM localization that accompanies being in the proper orientation.
Our results showed that efficient cotranslational insertion and inversion required a C-tail of 70–100 aa, which indicates that 30–60 aa outside of the ribosome are needed as “slack” for this process, or that synthesis actively aids in the inversion, as the M2 TMD experiments indicated. If one considers recent findings (Devaraneni et al., 2011) and the ∼20 aa needed to span the translocon (Whitley et al., 1996), these results suggest that the additional 10–40 aa provide time (∼2–8 s at a cellular synthesis rate of 5 aa/s; Hershey, 1991) and a potential driving force for less ideal type II TMDs to invert. However, this does not explain why the hydrophobic NA TMDs inverted relatively well upon ribosomal release. An alternative explanation for these TMDs is that the added hydrophobicity slows the translocation kinetics due to association with the translocon (Goder and Spiess, 2003). Similar to ribosomal association, translocon binding could provide the necessary time for the TMD to orientate and integrate but in a less efficient manner, resulting in the observed mixed orientations.
The potential benefit of the kinetic component for membrane insertion was previously alluded to by posttranslational insertion assays with Escherichia coli membranes, for which lowering ATP levels decreased the translocation rate, enabling less hydrophobic TMDs to integrate (Duong and Wickner, 1998). For mammalian cells, the slow synthesis rate (5 aa/s) likely provides an analogous mechanism during cotranslational translocation. This could provide marginally hydrophobic TMDs with the additional time and a possible driving force to orientate, potentially via a Sec62-dependent (Reithinger et al., 2013) or TRAP-dependent manner (Sommer et al., 2013), so they accommodate the hydrophobicity filter of the lateral gate (Junne et al., 2010; Trueman et al., 2012).
Positively charged N-terminal flanking residues, and to a lesser extent the length of this region, also contributed to Nin-Cout (type II) TMD inversion. With regard to IAVs, the inefficient inversion of NA in the absence of the conserved N-terminal Lys potentially explains the significantly deformed IAV morphology that has been observed upon deleting the entire NA N-terminal region (Garcia-Sastre and Palese, 1995; Mitnaul et al., 1996; Jin et al., 1997). Although this study offers new insight into the relationship between hydrophobicity and the mode of membrane integration in cells, further studies are required to investigate what cellular machinery and characteristics drive TMD inversion and how insertion pathways contribute to cotranslational membrane protein folding.
MATERIALS AND METHODS
Plasmids and constructs
Mammalian expression vectors were created by PCR overlap cloning (Mellroth et al., 2012) using pCDNA3.1A (Life Technologies, Foster City, CA) containing the full-length NA (TM∆G −0.7NA440aa) and M2 (TM∆G −1.1M270aa) genes from influenza A/WSN/33 (H1N1) fused to a 21-aa C-terminal myc-his tag. Site-directed mutagenesis was used to replace the WSN NA TMD in TM∆G −0.7NA440aa with the indicated TMD sequences (Table 1) and to make the N-terminal NA flanking-region substitutions and deletions. For the C-terminal tail (C-tail) truncations NAZaa and M2Zaa, the indicated length Z minus 21 aa following the N-terminal TMD were fused to the C-terminal myc epitope by PCR using the full-length constructs as templates. The TM∆G −1.1M2NA76aa and TM∆G −1.1M2NA440aa were created by fusing M2 residues 1–48 with 76-aa and 440-aa NA C-tails. All constructs were verified by sequencing (Eurofins MWG Operon, Ebersberg, Germany).
Cell culture and transfections
Vero and HeLa cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and streptomycin (all from Life Technologies) and maintained at 37°C in a 5% CO2 humidified incubator. For each transfection, 1.5 μg of plasmid DNA was incubated with 5 μl of LT-1 (Mirus, Madison, WI) in 500 μl of Opti-MEM (Life Technologies) for 20 min. During the incubation period, trypsinized cells were sedimented (500 × g, 5 min) and resuspended at a density of ∼5 × 105 cells/ml in Opti-MEM 10% FBS, and 1 ml of cells was added to each transfection mixture and seeded on 3.5 cm dishes.
Single-cell immunostaining analysis for PM localization
Vero cells (∼1 × 105) were transfected with 1 μg of plasmid DNA of the indicated construct and 0.5 μg of the intracellular marker (red fluorescent protein) plasmid DNA and seeded onto coverslips in 3.5 cm dishes. At 48 h posttransfection, coverslips were washed with cold phosphate-buffered saline (PBS), pH 7.4, fixed for 20 min with 4% paraformaldehyde (PFA) in PBS, permeabilized with 0.05% saponin in 4% PFA for 5 min, and fixed again with 4% PFA for 10 min. Cells were incubated with a monoclonal myc antibody (Cell Signaling Technology, Beverly, MA) at 1:100 in 1% bovine serum albumin (BSA) PBST-Az (PBS, pH 7.4, 0.1% Tween-20, 0.2 mg/l sodium azide) for 2 h at 37°C, washed three times with PBST-Az, and labeled with Alexa Fluor 488 secondary (Life Technologies) for 1 h at 25°C in the dark. DNA was labeled with 1 μg/ml Hoechst 33258 (Life Technologies) in 1% BSA PBST-Az at 25°C for 10 min, and the cells were washed and mounted with Vectashield. For PM controls, 48 h posttransfection with green fluorescent protein (GFP), Vero cells were washed three times with 37°C PBS and incubated with 5 μl/ml wheat germ agglutinin (WGA) Alexa Fluor 594 (Life Technologies) in PBS for 2 min at 37°C. Unbound WGA was removed by washing, and the cells were placed in fresh Opti-MEM for live image acquisition. Fixed and live-cell section images were acquired with a Zeiss LSM700 inverted microscope using a 40× object with 0.6-μm pinhole, and image acquisition was performed with Zen 2011. Individual cell PM/IC ratios were calculated with Cell Profiler 2.0 (Kamentsky et al., 2011) as outlined in Supplemental Figure S1.
Metabolic labeling, cell harvesting, and protein isolation by Ni-Sepharose
HeLa cells (∼1 × 105 cells) seeded in 24-well plates were infected with recombinant vaccinia virus expressing T7 RNA polymerase (Fuerst et al., 1986). At 40 min postinfection, each well was transfected with 2 μg of plasmid DNA mixed with 6 μl of LT-1 in 0.5 ml of Opti-MEM. At 4 h posttransfection, medium was replaced with starvation medium (0.5 ml DMEM lacking Met and Cys) for 20 min before labeling for 15 min with 35 μCi of [35S]Met/Cys (Perkin Elmer, Waltham, MA). Cells were harvested by scraping in 50 μl of lysis buffer (TBS [50 mM Tris, pH 7.4, 200 mM NaCl] with 1% Triton X-100, 10 mM N-ethylmaleimide, and 1× protease inhibitor cocktail [Sigma-Aldrich, Steinheim, Germany]), the whole-cell lysates were sonicated (15 s) on ice and sedimented (20,000 × g; 1 min), and the postnuclear supernatants were retained. For protein isolation by Ni-Sepharose, postnuclear supernatants were incubated with 500 μl of 5% Ni-Sepharose beads (GE Healthcare, Uppsala, Sweden) in TBS with 0.1% Triton X-100 and 40 mM imidazole for 1 h at 4ºC and washed three times with TBS, 0.1% Triton X-100, and 50 mM imidazole. Beads were resuspended in 500 μl of PNGase F denaturing buffer (TBS, 0.1% Triton X-100, and 0.5% SDS), heated at 37ºC for 10 min, and divided in two, and one sample received 50 U of PNGase F (NEB, Frankfurt am M., Germany). Both samples were incubated for 1 h at 37ºC, the beads were sedimented (20,000 × g; 3 min), and the protein was released with 50 μl of Tris-tricine sample buffer (Bis-Tris 360 mM, bicine 160 mM, 1% SDS, 15% glycerol, 100 mM DTT) containing 10 mM EDTA before Tris-tricine SDS–PAGE.
Cell surface trypsinization and postnuclear supernatant PNGase F digestions
The medium was removed from transfected cells in a 3.5 cm dish, and the cells were incubated with 200 μl of TBS containing 0.5 mg/ml trypsin (Sigma-Aldrich) at 37ºC for 15 min, after which the cells were washed three times in cold PBS, pH 7.4, and the postnuclear supernatants were obtained for immunoblotting. As a positive control, the trypsin digestion was carried out in the presence of 1% Triton X-100, and the postnuclear supernatants were extracted directly from the trypsin-treated lysate mixture. For PNGase F treatment, 15 μl of transfected-cell postnuclear supernatant was adjusted to 0.5% SDS and heated to 37ºC for 10 min before incubation for 1 h at 37ºC with 50 U of PNGase F (Jejcic et al., 2009).
Intracellular vesicle isolation, alkaline extraction, and flotation
Cells from a 6 cm dish were washed 48 h posttransfection or after metabolic labeling with PBS, harvested by scraping in 300 μl of ice-cold buffer A (50 mM triethanolamine, pH 7.5, 50 mM KOAc, 6 mM Mg(OAc)2, 1× protease inhibitor cocktail) with 0.25 M sucrose, passed through a 27-gauge needle 30 times, sonicated for 30 s on ice, and centrifuged at (2000 × g; 10 min) to remove the nuclei. The resulting intracellular vesicle and cytoplasmic fractions were layered on 800 μl of buffer A with 0.25 M sucrose and sedimented (200,000 × g; 45 min). The supernatant (cytoplasm) was precipitated with 20% trichloroacetic acid, washed with 95% acetone, and resuspended in 100 μl of sample buffer. Alkaline extraction of the membrane vesicle pellet in 800 μl of ice-cold 0.1 M Na2CO3 pH 11.5, has been previously described (Daniels et al., 2006). For density flotation analysis, the intracellular vesicle and cytoplasmic fractions were equally divided on 800 μl cushions of buffer A with 0.25, 1.2, and 2 M sucrose and sedimented (200,000 × g; 45 min). The supernatants were retained, the pellets were resuspended in equivalent volumes of their cushion buffer, and equal portions were analyzed by dot blotting or incubated with Ni-Sepharose beads before SDS–PAGE and autoradiography.
In vitro analysis of ribosomally arrested chains
Ribosomally arrested constructs were generated as previously described (Gilmore et al., 1991), with the exception that PCR was used to create the DNA templates for transcription. Briefly, each NA construct was amplified from their respective vector using a 3′ primer that changed the last three codons to Met (to enable detection) and excluded the stop codon. The products were purified, and 200 ng of each construct was translated in 7 μl of TNT T7 PCR reticulocyte lysate (Promega, Madison, WI) with 3 μCi of [35S]Met/Cys in the absence and presence of canine pancreas microsomes at 30ºC for 30 min. Equal volume (5 μl) of each translation reaction was layered on 800 μl of buffer A with 0.25 M sucrose and sedimented (157,000 × g; 10 min), the supernatant was removed, and the pellet containing the microsomally associated arrested chains was resuspended in Tris-tricine sample buffer. Where indicated, the arrested chains were released with 1 mM puromycin for 10 min at 30ºC before sedimentation.
SDS–PAGE, autoradiography, immunoblotting, and dot blotting
Constructs >40 kDa were resolved using standard reducing 9% Tris-glycine SDS–PAGE, whereas the smaller constructs were mixed with reducing Tris-tricine sample buffer and separated using 14% Tris-tricine SDS–PAGE. Autoradiography gels were developed using a phosphoimager (Fuji FLA-9000), and the 35S-labeled protein was quantified with ImageJ software. Where indicated, DTT was omitted from the sample buffer to analyze disulfide bond formation by molecular weight shifts on nonreducing SDS–PAGE (Francis et al., 2002). Gels for immunoblotting were transferred to a 0.2 μm pore polyvinylidene fluoride membrane (GE Healthcare) for 30 min at 15 V. For dot blots, twofold serial dilutions of each sample were prepared and loaded onto a nitrocellulose membrane using a Bio-dot apparatus (Bio-Rad, Munich, Germany). Both membranes were processed and developed as previously described (da Silva et al., 2013). Dot-blot samples with the same dilution in the linear signal range were used for comparative purposes.
Sequence analysis
The NA, M2, and HA protein sequences from human H1N1 IAVs were retrieved from the National Center for Biotechnology Information Influenza Virus Resource Database, and the hydrophobicity and location of the unique TMDs from NA (residues 7–34), M2 (residues 26–48), and HA (residues 529–556) were determined using the Membrane Protein Explorer (MPEx; Hessa et al., 2007; Snider et al., 2009). The protein sequences of the human type II single-spanning membrane proteins were obtained from UniProtKB/Swiss-Prot and processed with a length window (19–33 amino acids) that corresponds to the predicted TMDs from eukaryotes (Sharpe et al., 2010). The C-tail distance was computed by the position of the most probable TMD with regard to the protein length. The data for the human type II TMDs are given in Supplemental Table S1 with the referenced literature used to identify the proteins that localize to the mitochondria, likely have an Nout-Cin orientation, and have been shown to be soluble.
Supplementary Material
Acknowledgments
We are thankful to IngMarie Nilsson for help with the in vitro microsome experiments and to the members of the Center for Biomembrane Research in the Department of Biochemistry and Biophysics at Stockholm University for their suggestions. This work was supported by grants from the Swedish Research Council, Swedish Foundation for Strategic Research, Carl Trygger Foundation, and Harald Jeanssons Stiftelse to R.D., Helge Ax:Son Johnsons and Lindhés Advokatbyrå AB grants to D.S. and J.N., and the Sven and Lilly Lawskis Fund to J.N.
Abbreviations used:
- DTT
dithiothreitol
- IAV
influenza A virus
- IC
intracellular
- NA
neuraminidase
- PM
plasma membrane
- PNGase F
peptide-n-glycosidase F
- TMD
transmembrane domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www .molbiolcell.org/cgi/doi/10.1091/mbc.E14-04-0874) on August 27, 2014.
REFERENCES
- Alder NN, Johnson AE. Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J Biol Chem. 2004;279:22787–22790. doi: 10.1074/jbc.R400002200. [DOI] [PubMed] [Google Scholar]
- Arkin IT, Brunger AT. Statistical analysis of predicted transmembrane alpha-helices. Biochim Biophys Acta. 1998;1429:113–128. doi: 10.1016/s0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- Beaufay H, Amar-Costesec A, Thines-Sempoux D, Wibo M, Robbi M, Berthet J. Analytical study of microsomes and isolated subcellular membranes from rat liver. 3. Subfractionation of the microsomal fraction by isopycnic and differential centrifugation in density gradients. J Cell Biol. 1974;61:213–231. doi: 10.1083/jcb.61.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird P, Gething MJ, Sambrook J. The functional efficiency of a mammalian signal peptide is directly related to its hydrophobicity. J Biol Chem. 1990;265:8420–8425. [PubMed] [Google Scholar]
- Blobel G, Sabatini DD. Controlled proteolysis of nascent polypeptides in rat liver cell fractions. I. Location of the polypeptides within ribosomes. J Cell Biol. 1970;45:130–145. doi: 10.1083/jcb.45.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Buck TM, Skach WR. Differential stability of biogenesis intermediates reveals a common pathway for aquaporin-1 topological maturation. J Biol Chem. 2005;280:261–269. doi: 10.1074/jbc.M409920200. [DOI] [PubMed] [Google Scholar]
- Daniels R, Kurowski B, Johnson AE, Hebert DN. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell. 2003;11:79–90. doi: 10.1016/s1097-2765(02)00821-3. [DOI] [PubMed] [Google Scholar]
- Daniels R, Mellroth P, Bernsel A, Neiers F, Normark S, von Heijne G, Henriques-Normark B. Disulfide bond formation and cysteine exclusion in gram-positive bacteria. J Biol Chem. 2010;285:3300–3309. doi: 10.1074/jbc.M109.081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels R, Rusan NM, Wadsworth P, Hebert DN. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol Cell. 2006;24:955–966. doi: 10.1016/j.molcel.2006.11.001. [DOI] [PubMed] [Google Scholar]
- da Silva DV, Nordholm J, Madjo U, Pfeiffer A, Daniels R. Assembly of subtype 1 influenza neuraminidase is driven by both the transmembrane and head domains. J Biol Chem. 2013;288:644–653. doi: 10.1074/jbc.M112.424150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Schekman R. A yeast mutant defective at an early stage in import of secretory protein precursors into the endoplasmic reticulum. J Cell Biol. 1987;105:633–645. doi: 10.1083/jcb.105.2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraneni PK, Conti B, Matsumura Y, Yang Z, Johnson AE, Skach WR. Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell. 2011;146:134–147. doi: 10.1016/j.cell.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong F, Wickner W. Sec-dependent membrane protein biogenesis: SecYEG, preprotein hydrophobicity and translocation kinetics control the stop-transfer function. EMBO J. 1998;17:696–705. doi: 10.1093/emboj/17.3.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige MJ, Hendershot LM. Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol Cell. 2013;51:297–309. doi: 10.1016/j.molcel.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis E, Daniels R, Hebert DN. Analysis of protein folding and oxidation in the endoplasmic reticulum. Curr Protoc Cell Biol. 2002 doi: 10.1002/0471143030.cb1506s14. Chapter 15, Unit 15.16. [DOI] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Palese P. The cytoplasmic tail of the neuraminidase protein of influenza A virus does not play an important role in the packaging of this protein into viral envelopes. Virus Res. 1995;37:37–47. doi: 10.1016/0168-1702(95)00017-k. [DOI] [PubMed] [Google Scholar]
- Gilmore R, Collins P, Johnson J, Kellaris K, Rapiejko P. Transcription of full-length and truncated mRNA transcripts to study protein translocation across the endoplasmic reticulum. Methods Cell Biol. 1991;34:223–239. doi: 10.1016/s0091-679x(08)61683-0. [DOI] [PubMed] [Google Scholar]
- Gilmore R, Walter P, Blobel G. Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J Cell Biol. 1982;95:470–477. doi: 10.1083/jcb.95.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V, Spiess M. Molecular mechanism of signal sequence orientation in the endoplasmic reticulum. EMBO J. 2003;22:3645–3653. doi: 10.1093/emboj/cdg361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Hartmann E, Kalies KU, Rapoport TA. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 1992;71:489–503. doi: 10.1016/0092-8674(92)90517-g. [DOI] [PubMed] [Google Scholar]
- Hatsuzawa K, Tagaya M, Mizushima S. The hydrophobic region of signal peptides is a determinant for SRP recognition and protein translocation across the ER membrane. J Biochem. 1997;121:270–277. doi: 10.1093/oxfordjournals.jbchem.a021583. [DOI] [PubMed] [Google Scholar]
- Hershey JW. Translational control in mammalian cells. Annu Rev Biochem. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- Hessa T, Monne M, von Heijne G. Stop-transfer efficiency of marginally hydrophobic segments depends on the length of the carboxy-terminal tail. EMBO Rep. 2003;4:178–183. doi: 10.1038/sj.embor.embor728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull JD, Gilmore R, Lamb RA. Integration of a small integral membrane protein, M2, of influenza virus into the endoplasmic reticulum: analysis of the internal signal-anchor domain of a protein with an ectoplasmic NH2 terminus. J Cell Biol. 1988;106:1489–1498. doi: 10.1083/jcb.106.5.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jejcic A, Daniels R, Goobar-Larsson L, Hebert DN, Vahlne A. Small molecule targets Env for endoplasmic reticulum-associated protein degradation and inhibits human immunodeficiency virus type 1 propagation. J Virol. 2009;83:10075–10084. doi: 10.1128/JVI.01700-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Leser GP, Zhang J, Lamb RA. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16:1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junne T, Kocik L, Spiess M. The hydrophobic core of the Sec61 translocon defines the hydrophobicity threshold for membrane integration. Mol Biol Cell. 2010;21:1662–1670. doi: 10.1091/mbc.E10-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbfleisch T, Cambon A, Wattenberg BW. A bioinformatics approach to identifying tail-anchored proteins in the human genome. Traffic. 2007;8:1687–1694. doi: 10.1111/j.1600-0854.2007.00661.x. [DOI] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Sakaguchi M, Kitamura A, Sato T, Mihara K, Hamasaki N. The tenth membrane region of band 3 is initially exposed to the luminal side of the endoplasmic reticulum and then integrated into a partially folded band 3 intermediate. Biochemistry. 2002;41:13973–13981. doi: 10.1021/bi026619q. [DOI] [PubMed] [Google Scholar]
- Kocik L, Junne T, Spiess M. Orientation of internal signal-anchor sequences at the Sec61 translocon. J Mol Biol. 2012;424:368–378. doi: 10.1016/j.jmb.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Lu Y, Turnbull IR, Bragin A, Carveth K, Verkman AS, Skach WR. Reorientation of aquaporin-1 topology during maturation in the endoplasmic reticulum. Mol Biol Cell. 2000;11:2973–2985. doi: 10.1091/mbc.11.9.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin LI, Rich A. Partial resistance of nascent polypeptide chains to proteolytic digestion due to ribosomal shielding. J Mol Biol. 1967;26:329–346. doi: 10.1016/0022-2836(67)90301-4. [DOI] [PubMed] [Google Scholar]
- Mellroth P, Daniels R, Eberhardt A, Ronnlund D, Blom H, Widengren J, Normark S, Henriques-Normark B. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J Biol Chem. 2012;287:11018–11029. doi: 10.1074/jbc.M111.318584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitnaul LJ, Castrucci MR, Murti KG, Kawaoka Y. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J Virol. 1996;70:873–879. doi: 10.1128/jvi.70.2.873-879.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Kagawa T, Numari A, Harris MJ, Itoh J, Watanabe N, Mine T, Arias IM. Two N-linked glycans are required to maintain the transport activity of the bile salt export pump (ABCB11) in MDCK II cells. Am J Physiol. Gastrointest Liver Physiol. 2007;292:G818–G828. doi: 10.1152/ajpgi.00415.2006. [DOI] [PubMed] [Google Scholar]
- Nordholm J, da Silva DV, Damjanovic J, Dou D, Daniels R. Polar residues and their positional context dictate the transmembrane domain interactions of influenza a neuraminidases. J Biol Chem. 2013;288:10652–10660. doi: 10.1074/jbc.M112.440230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojemalm K, Halling KK, Nilsson I, von Heijne G. Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol Cell. 2012;45:529–540. doi: 10.1016/j.molcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota K, Sakaguchi M, Hamasaki N, Mihara K. Assessment of topogenic functions of anticipated transmembrane segments of human band 3. J Biol Chem. 1998a;273:28286–28291. doi: 10.1074/jbc.273.43.28286. [DOI] [PubMed] [Google Scholar]
- Ota K, Sakaguchi M, von Heijne G, Hamasaki N, Mihara K. Forced transmembrane orientation of hydrophilic polypeptide segments in multispanning membrane proteins. Mol Cell. 1998b;2:495–503. doi: 10.1016/s1097-2765(00)80149-5. [DOI] [PubMed] [Google Scholar]
- Reithinger JH, Kim JE, Kim H. Sec62 protein mediates membrane insertion and orientation of moderately hydrophobic signal anchor proteins in the endoplasmic reticulum (ER) J Biol Chem. 2013;288:18058–18067. doi: 10.1074/jbc.M113.473009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Stevens TJ, Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell. 2010;142:158–169. doi: 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Peters TJ. Analytical subcellular fractionation of rat liver with special reference to the localisation of putative plasma membrane marker enzymes. Eur J Biochem. 1980;104:305–311. doi: 10.1111/j.1432-1033.1980.tb04429.x. [DOI] [PubMed] [Google Scholar]
- Snider C, Jayasinghe S, Hristova K, White SH. MPEx: a tool for exploring membrane proteins. Protein Sci. 2009;18:2624–2628. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer N, Junne T, Kalies KU, Spiess M, Hartmann E. TRAP assists membrane protein topogenesis at the mammalian ER membrane. Biochim Biophys Acta. 2013;1833:3104–3111. doi: 10.1016/j.bbamcr.2013.08.018. [DOI] [PubMed] [Google Scholar]
- Stefanovic S, Hegde RS. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell. 2007;128:1147–1159. doi: 10.1016/j.cell.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Trueman SF, Mandon EC, Gilmore R. A gating motif in the translocation channel sets the hydrophobicity threshold for signal sequence function. J Cell Biol. 2012;199:907–918. doi: 10.1083/jcb.201207163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature. 1989;341:456–458. doi: 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol. 1981;91:557–561. doi: 10.1083/jcb.91.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Daniels R, Hebert DN. The cotranslational maturation of the type I membrane glycoprotein tyrosinase: the heat shock protein 70 system hands off to the lectin-based chaperone system. Mol Biol Cell. 2005;16:3740–3752. doi: 10.1091/mbc.E05-05-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Glidden EJ, Murphy SR, Pearse BR, Hebert DN. The cotranslational maturation program for the type II membrane glycoprotein influenza neuraminidase. J Biol Chem. 2008;283:33826–33837. doi: 10.1074/jbc.M806897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley P, Nilsson IM, von Heijne G. A nascent secretory protein may traverse the ribosome/endoplasmic reticulum translocase complex as an extended chain. J Biol Chem. 1996;271:6241–6244. doi: 10.1074/jbc.271.11.6241. [DOI] [PubMed] [Google Scholar]
- Zhang L, Sato Y, Hessa T, von Heijne G, Lee JK, Kodama I, Sakaguchi M, Uozumi N. Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain. Proc Natl Acad Sci USA. 2007;104:8263–8268. doi: 10.1073/pnas.0611007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.