

SNAREs and the Dsl1 complex mediate an alternative, Sey1p-independent homotypic endoplasmic reticulum (ER) fusion pathway in yeast. When both pathways and the reticulons are simultaneously disrupted, cells are inviable. This demonstrates that homotypic ER fusion is an essential process in yeast and that the Dsl1 complex has vesicle trafficking-independent functions.
Abstract
The peripheral endoplasmic reticulum (ER) network is dynamically maintained by homotypic (ER–ER) fusion. In Saccharomyces cerevisiae, the dynamin-like GTPase Sey1p can mediate ER–ER fusion, but sey1Δ cells have no growth defect and only slightly perturbed ER structure. Recent work suggested that ER-localized soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs) mediate a Sey1p-independent ER–ER fusion pathway. However, an alternative explanation—that the observed phenotypes arose from perturbed vesicle trafficking—could not be ruled out. In this study, we used candidate and synthetic genetic array (SGA) approaches to more fully characterize SNARE-mediated ER–ER fusion. We found that Dsl1 complex mutations in sey1Δ cells cause strong synthetic growth and ER structure defects and delayed ER–ER fusion in vivo, additionally implicating the Dsl1 complex in SNARE-mediated ER–ER fusion. In contrast, cytosolic coat protein I (COPI) vesicle coat mutations in sey1Δ cells caused no synthetic defects, excluding perturbed retrograde trafficking as a cause for the previously observed synthetic defects. Finally, deleting the reticulons that help maintain ER architecture in cells disrupted for both ER–ER fusion pathways caused almost complete inviability. We conclude that the ER SNAREs and the Dsl1 complex directly mediate Sey1p-independent ER–ER fusion and that, in the absence of both pathways, cell viability depends upon membrane curvature–promoting reticulons.
INTRODUCTION
In all eukaryotes, the endoplasmic reticulum (ER) consists of a branched network of tubules and ribosome-dense sheets (cisternae) that is continuous with the outer nuclear envelope. In Saccharomyces cerevisiae, the majority of the ER network resides at the cell cortex along the plasma membrane (peripheral ER), with several tubules connecting to the outer nuclear envelope (nuclear ER). Functionally, the ER mediates multiple processes, including protein synthesis and secretion, lipid synthesis, and calcium regulation (reviewed in Schuldiner and Weissman, 2013). Recent research has identified contact sites between the ER and most other organelles, implicating the ER in additional processes, including lipid droplet biogenesis, lipid transfer, and mitochondrial division (reviewed in English and Voeltz, 2013).
ER tubules are shaped and maintained by the reticulons (Rtn1p and Rtn2p in yeast) and the reticulon-like protein Yop1p (Voeltz et al., 2006; reviewed in Chen et al., 2013). Rtn1p or Yop1p is sufficient to form tubules in proteoliposomes in vitro (Hu et al., 2008) and likely functions by forming a curvature-inducing wedge in the lipid bilayer (Voeltz et al., 2006; Shibata et al., 2008; Zurek et al., 2011). Simultaneous deletion of rtn1, rtn2, and yop1 results in an almost entirely cisternal, nontubulated peripheral ER (West et al., 2011). Surprisingly, however, this triple mutant displays only a minor growth defect (Voeltz et al., 2006; Chen et al., 2012).
Over time, the ER network must alter its structure dynamically, while maintaining lumenal continuity, by forming new branch points or by merging existing branches at three-way tubule junctions via homotypic (ER–ER) fusion (Lee and Chen, 1988). Homotypic ER fusion is mediated in mammals by the dynamin-like GTPase atlastin and in yeast by its functional orthologue Sey1p (Hu et al., 2009; Orso et al., 2009). Sey1p is enriched at three-way junctions in the peripheral ER and binds to the reticulons Rtn1p and Yop1p (Hu et al., 2009). Surprisingly, sey1Δ mutants have no growth defect and relatively normal ER structure, similar to rtn1Δ and yop1Δ single mutants. sey1Δ rtn1Δ and sey1Δ yop1Δ double mutants, however, display abnormal, nontubulated peripheral ER with enlarged cisternae (Hu et al., 2009). Despite the lack of a strong growth or structural defect, sey1Δ mutants exhibit a decreased (but not abolished) rate of homotypic ER fusion in vivo, whereas rtn1Δ rtn2Δ yop1Δ mutants, which have extremely perturbed ER structure, exhibit a wild-type homotypic ER fusion rate (Anwar et al., 2012). Consistent with a direct role in ER–ER fusion, Sey1p is sufficient to mediate proteoliposome fusion in vitro (Anwar et al., 2012). Another protein, Lnp1p, also resides at three-way tubule junctions and appears to antagonize Sey1p fusogenic activity, possibly by promoting ring closure (Chen et al., 2012).
Together these findings demonstrate that Sey1p mediates homotypic ER fusion, but the relatively weak sey1Δ phenotypes suggest there is a second, partially redundant, Sey1p-independent homotypic ER fusion pathway. Patel et al. (1998) demonstrated that ER membranes isolated from ufe1-1 cells were defective in an in vitro ER–ER fusion assay. More recently, strong synthetic growth and ER structure defects were demonstrated for double mutants of sey1Δ and the three ER-bound retrograde (Golgi-to-ER) SNAREs (soluble N-ethylmaleimide–sensitive factor attachment protein receptors) sec20, ufe1, and use1 (Anwar et al., 2012; Rogers et al., 2013). Moreover, sey1Δ ufe1-1 double mutants had an even slower rate of homotypic ER fusion than sey1Δ or ufe1-1 single mutants (Anwar et al., 2012). These data suggest that SNARE proteins mediate the hypothesized Sey1p-independent homotypic ER fusion pathway.
SNAREs mediate vesicle fusion during cargo trafficking between organelles (reviewed in Delic et al., 2013). To fuse vesicles, SNARE proteins on the incoming vesicle bind to SNAREs on the target membrane and form a four-helix coiled-coil SNARE complex that forces the lipid membranes into close apposition (Sutton et al., 1998). In vivo, unique sets of SNARE proteins (usually four SNAREs) mediate the different vesicle-trafficking steps, although some SNAREs are used in multiple pathways (Burri and Lithgow, 2004).
Although SNAREs are sufficient to fuse vesicles in vitro (Weber et al., 1998), vesicle trafficking in vivo additionally requires vesicle coats, Rab GTPases, and SM (Sec1/Munc18) proteins, and tethering complexes (reviewed in Hong and Lev, 2014). In retrograde vesicle trafficking, the v-SNARE Sec22p on cis-Golgi–derived cytosolic coat protein I (COPI)-coated vesicles binds the t-SNAREs Sec20p, Ufe1p, and Use1p on the ER to catalyze vesicle–ER fusion (reviewed in Spang, 2013). Retrograde trafficking additionally requires the Rab GTPase Ypt1p, the SM protein Sly1p, and the Dsl1 tethering complex (Ossig et al., 1991; Jedd et al., 1995; Kraynack et al., 2005; Li et al., 2005; Kamena et al., 2008). The Dsl1 tethering complex is a CATCHR (complex associated with tethering containing helical rods) family member comprising the subunits Tip20p, Dsl1p, and Sec39p/Dsl3. This complex is thought to mediate the initial connection between the ER membrane and an incoming vesicle. Specifically, the Dsl1 complex is anchored to the ER membrane through interactions of two of its subunits (Tip20p and Sec39p) with two ER t-SNAREs (Sec20p and Use1p; Figure 1A; Andag et al., 2001; Andag and Schmitt, 2003; Ren et al., 2009; Tripathi et al., 2009; Zink et al., 2009). The third Dsl1 complex subunit, Dsl1p, connects Tip20p to Sec39p and contains a flexible 80-residue segment called the “lasso.” The lasso, located at the tip of the 20-nm-tall Dsl1 complex, binds two subunits of COPI coats, α-COP (Cop1p) and δ-COP (Ret2p). By binding multiple ER SNAREs and tethering the incoming vesicle near the ER membrane, the Dsl1 complex appears to promote the assembly of productive trans-SNARE complexes; it may also facilitate vesicle uncoating (Ren et al., 2009; Zink et al., 2009; Diefenbacher et al., 2011).
FIGURE 1:

SNARE-mediated homotypic ER fusion requires DSL1. (A) The Dsl1 complex (Dsl1p-Sec39p-Tip20p) binds to the N-terminal regulatory domains of the SNAREs Use1p and Sec20p through interactions with Sec39p and Tip20p, respectively. The flexible lasso domain of Dsl1p binds to the COPI coatomer (not shown; adapted from Ren et al., 2009). (B) Growth assays (see Materials and Methods) of wild type (MY14008), dsl1ΔL (MY14012), sey1Δ (MY14016), sey1Δ dsl1ΔL (MY14020), dsl1ΔE (MY14024), and sey1Δ dsl1ΔE (MY14028), grown on YEPD for 2 d at 30°C or 6 d at 18°C. Each spot from left to right represents a 10-fold dilution.
Although it appeared from previous work (Anwar et al., 2012; Rogers et al., 2013) that the retrograde SNAREs Sec20p, Ufe1p, and Use1p mediate a redundant, Sey1p-independent homotypic ER fusion pathway, several fundamental questions remained. First, it is possible that the synthetic growth and ER structure defects are due to downstream effects of perturbed retrograde vesicle trafficking. Second, it is unclear whether the SNAREs act in isolation or in combination with their normal vesicle-trafficking accessory proteins, including the Dsl1 tethering complex and the COPI coat. Third, it remained possible that the Sey1p-mediated and putative SNARE-mediated homotypic ER fusion pathways are not truly redundant but might mediate distinct subsets of homotypic ER fusion events. In this study, we have further characterized SNARE-mediated homotypic ER fusion and determined that it requires the Dsl1 complex but not the COPI coat, is not a result of perturbed retrograde vesicle trafficking, and is only partially redundant with Sey1p-mediated homotypic ER fusion.
RESULTS
SNARE-mediated homotypic ER fusion requires DSL1
Cells containing both a sey1 deletion and a mutant retrograde SNARE allele (sec20-1, ufe1-1, use1-10AA, or use1-0layer) have strong synthetic growth and ER structural defects not seen in either single mutant (Anwar et al., 2012; Rogers et al., 2013). We hypothesized that homotypic ER fusion is mediated by two parallel pathways, one Sey1p mediated and the other SNARE mediated, and that only disruption of both pathways results in severe ER–ER fusion and growth defects.
To assess whether the SNAREs act with their normal accessory proteins or alone, we first created sey1Δ dsl1 double mutants and assayed possible synthetic growth defects. Dsl1p, like the other subunits of the Dsl1 complex, Tip20p and Sec39p/Dsl3p, is encoded by an essential gene. We assayed two dsl1 mutants in this study: dsl1ΔE and dsl1ΔL (Figure 1A). The C-terminal E domain is the most highly conserved region of Dsl1p (Zink et al., 2009), while the flexible L (lasso) region mediates α-COP (Cop1p)- and δ-COP (Ret2p) binding (Andag et al., 2001; Andag and Schmitt, 2003; Zink et al., 2009); remarkably, neither the E domain nor the lasso is needed in vivo for normal growth (Zink et al., 2009; R. W. Baker and F. M. H., unpublished data). Interestingly, sey1Δ dsl1ΔE double mutants exhibited a strong synthetic growth defect at 30°C and an even more severe defect at 18°C, whereas sey1Δ dsl1ΔL mutants exhibited no growth defect at 30°C and a minor growth defect at 18°C (Figure 1B). These data suggest that SNARE-mediated ER–ER fusion requires Dsl1p but not the COPI-binding lasso.
SNARE-mediated homotypic ER fusion requires the entire Dsl1 complex but not the COPI coat
Because dsl1ΔE but not dsl1ΔL mutants exhibited synthetic growth defects with sey1Δ, we screened for additional genetic interactions to gain insight into the function of the Dsl1p E domain. Additionally, we reasoned that this screen might identify novel components of the Sey1p-mediated ER–ER fusion pathway, as any pathway-specific components should, like sey1Δ, display negative genetic interactions with dsl1ΔE but not with dsl1ΔL. To perform the screen, we used synthetic genetic array (SGA) technology. We crossed each strain to a genome-wide set of nonessential deletion mutants (∼4300 gene deletion strains) and a large-scale temperature-sensitive mutant collection (∼1200 temperature-sensitive strains). We assessed the growth of double-mutant haploids isolated from these crosses using previously published methodology (Baryshnikova et al., 2010; Costanzo et al., 2010). Briefly, we assigned each double mutant an SGA score based on colony size measurements and a multiplicative fitness model. A negative SGA score indicates that the double mutant grew more poorly than expected (synthetic growth defect), whereas a positive score indicates more robust growth than expected.
To identify the most informative genetic interactions, we grouped the interactions into three classes: interactions shared between dsl1ΔE and dsl1ΔL, dsl1ΔL-specific interactions, and dsl1ΔE-specific interactions (Figure 2A; full data set available in Supplemental Table S1; classes determined via a cutoff score and a difference threshold; see Materials and Methods). As expected, the strongest interactions shared between dsl1ΔE and dsl1ΔL consisted of known components of vesicle trafficking. In the dsl1ΔL-specific class, we identified five negative interactions: pse1-41, hir1Δ, mps3-1, chk1Δ, and cse2Δ. Four of these genes have nuclear roles: Pse1p interacts with nuclear pore complexes, Mps3p resides at the half-bridge and mediates spindle pole body formation, Hir1p is a subunit of the histone regulation complex, and Cse2p is a subunit of the RNA polymerase II mediator complex. Interestingly, while pse1-41 is by far the strongest dsl1ΔL-specific negative genetic interaction, pse1-34 is among the strongest dsl1ΔL-specific positive interactions. The final interaction, chk1Δ, uniquely interacts both negatively with dsl1ΔL and positively with dsl1ΔE. Chk1p is a checkpoint kinase that mediates cell cycle arrest.
FIGURE 2:

DSL1 and SEY1 SGAs. (A) Scatterplot of the SGA scores between dsl1ΔL, dsl1ΔE, and the indicated alleles. Genes without an allele designation are deletions. Identical scores would fall on the diagonal line. Alleles listed in purple have similar scores for both Dsl1 alleles (see Materials and Methods for classification metric), alleles listed in blue are classified as dsl1ΔL-specific, and alleles listed in red are classified as dsl1ΔE-specific. A few genes were tested multiple times independently during the SGA experiment (e.g., bre5Δ) and therefore appear multiple times in the scatterplot. (B) As in A, except dsl1ΔE is compared with independently derived sey1Δ SGA data (Costanzo et al., 2010; unpublished data [version 13-04-22], Boone lab). Only alleles with data present for both dsl1ΔE and sey1Δ are plotted. Alleles of genes encoding subunits of the COPI coat complex are shown in red. The strongest sey1Δ-interacting alleles are shown in black.
Among the 22 dsl1ΔE-specific negative interactions (Table 1), eight were related to trafficking (although not exclusively retrograde trafficking), and seven were related to transcriptional regulation. The final seven interactions consist of sey1Δ, whose dsl1ΔE-specific negative interaction (Figure 1B) motivated the genome-wide screen; ice2Δ, a gene involved in inheritance of cortical ER (Estrada de Martin et al., 2005) that also has a negative genetic interaction with sey1Δ (Figure 2B); act1-2, an actin allele; pyc2Δ, a pyruvate carboxylase isoform; atg15Δ, a lipase involved in autophagy; and two dubious open reading frames (ORFs), ygr139wΔ and ypr050cΔ. Of note, although act1-2 displayed the strongest negative genetic interaction, 19 other act1 alleles tested displayed no genetic interaction. Allele-specific genetic interactions often result from physical interactions (Sandrock et al., 1997), raising the possibility that Dsl1p and Act1p interact physically, although it also possible that act1-2 specifically disrupts Sey1p-mediated ER–ER fusion. It is possible that any of the aforementioned DSL1-interacting genes could be new members of the Sey1p-mediated ER–ER fusion pathway; however, we have not yet studied these genes further.
TABLE 1:
SGA scores of dsl1ΔE-specific negative genetic interactions, grouped by known functions.
| Allele | dsl1ΔE | dsl1ΔL | Description |
|---|---|---|---|
| Vesicular traffic | |||
| bre5Δ | −0.83 | −0.09 | Ubiquitin protease cofactor; regulates anterograde/retrograde transport |
| ufe1-1 | −0.81 | −0.09 | ER-localized SNARE required for retrograde vesicular traffic |
| use1-TS | −0.80 | 0.01 | ER-localized SNARE required for retrograde vesicular traffic |
| ret3-1 | −0.69 | −0.38 | Zeta subunit of the COPI vesicle coatomer complex |
| sec22-1 | −0.59 | −0.22 | SNARE required for retrograde and anterograde vesicular traffic |
| ubp3Δ | −0.53 | −0.20 | Ubiquitin-specific protease involved in transport and osmotic response |
| cop1-1 | −0.45 | −0.04 | Alpha subunit of COPI vesicle coatomer complex |
| sec26-F856A,W860A | −0.36 | 0.00 | Beta subunit of COPI vesicle coatomer complex |
| Transcription | |||
| ydr290WΔ | −0.73 | −0.33 | ORF overlaps RTT103 |
| rsc9-PH | −0.70 | −0.06 | Component of the RSC chromatin remodeling complex |
| swc5Δ | −0.65 | −0.15 | Component of the SWR1 chromatin remodeling complex |
| rtt103Δ | −0.63 | −0.19 | Protein involved in transcription termination by RNA polymerase II |
| brf1-W107R | −0.56 | 0.12 | RNA polymerase III transcription initiation factor TFIIIB B–related factor |
| sen1-1 | −0.55 | −0.19 | Presumed helicase and subunit of the Nrd1 complex |
| ask10Δ | −0.54 | 0.12 | Component of RNA polymerase II holoenzyme |
| Other | |||
| act1-2 | −1.01 | 0.02 | Actin |
| pyc2Δ | −0.93 | −0.50 | Pyruvate carboxylase isoform |
| atg15Δ | −0.88 | −0.24 | Lipase required for intravacuolar lysis of autophagic and Cvt bodies |
| sey1Δ | −0.67 | −0.01 | Dynamin-like GTPase that mediates homotypic ER fusion |
| ypr050CΔ | −0.49 | −0.17 | Dubious ORF |
| ygr139WΔ | −0.40 | 0.00 | Dubious ORF |
| ice2Δ | −0.29 | 0.01 | Integral ER membrane protein with type-III transmembrane domains |
Class specificity as in Figure 2A, determined via cutoff score (−0.25) and difference threshold (0.25); see Materials and Methods for full classification metric.
Following similar logic, we reasoned that any additional components in the SNARE/Dsl1-mediated ER–ER fusion pathway should, like dsl1ΔE, display negative genetic interactions with sey1Δ. Therefore we examined independently generated sey1Δ SGA data (Costanzo et al., 2010; unpublished data [version 13-04-22], C. Boone, University of Toronto). As expected, the strongest negative synthetic genetic interactions with sey1Δ include use1-TS (temperature sensitive), a retrograde SNARE implicated previously in SNARE-mediated ER–ER fusion; sec39-1, a Dsl1 complex subunit; and sly1-TS, an SM protein required for normal retrograde trafficking (Table 2, interactions with SGA score ≤ −0.25). These results suggest that SNARE-mediated homotypic ER fusion may require the SM protein Sly1p and possibly the entire Dsl1 complex. We discuss the potential role of the remaining five sey1Δ interactions in the Discussion.
TABLE 2:
SGA scores of sey1Δ-negative genetic interactions.
| Allele | sey1Δ | dsl1ΔE | Description |
|---|---|---|---|
| scs3Δ | −0.84 | −0.05 | Protein required for inositol prototrophy |
| use1-TS | −0.65 | −0.80 | ER-localized SNARE required for retrograde vesicular traffic |
| sec39-1 | −0.58 | −0.91 | Component of the Dsl1 tethering complex |
| sly1-TS | −0.54 | −0.50 | SM-family protein involved in ER/Golgi traffic |
| pom33Δ | −0.37 | 0.00 | Transmembrane nucleoporin |
| sec23-1 | −0.36 | −0.05 | COPII coat subunit, also stimulates the GTPase activity of Sar1p |
| yop1Δ | −0.32 | −0.05 | Membrane protein that interacts with Sey1p to maintain ER morphology |
| ice2Δ | −0.31 | −0.29 | Integral ER membrane protein with type-III transmembrane domains |
Sorted list of sey1Δ-negative SGA scores, cutoff at −0.25.
Notably, we also observed that sey1Δ exhibited no genetic interactions with the members of the COPI coat (cop1-1, ret2-1, ret3-1, sec21-1, sec26F856A,W860A, sec26-2, sec27-1, and sec28Δ), whereas both dsl1ΔL and dsl1ΔE exhibited strong interactions with most members (Figure 2B). Additionally, with the exception of sec23-1, other nonretrograde vesicle-trafficking pathway components showed no synthetic interactions with sey1Δ (see Table S1 for a listing of SGA data available for vesicle-trafficking components). Therefore it is unlikely that the observed genetic interactions between sey1Δ and the SNAREs, dsl1ΔE, and sec39-1 are simply a downstream result of perturbed vesicle trafficking. Instead, our data suggest that the SNAREs and the Dsl1 complex together directly mediate Sey1p-independent ER–ER fusion, whereas the COPI coat functions specifically in retrograde vesicle trafficking.
We manually verified the most important SGA results and checked for synthetic growth defects between sey1Δ and candidates that were missing from the SGA arrays. We confirmed the synthetic growth defect in sey1Δ sec39-1 mutants (Figure 3A) and the lack of a COPI interaction (no growth defects in sey1Δ sec27-1, sey1Δ ret2-1, and sey1Δ cop1-1 mutants; Figure 3B and Supplemental Figure S1A). We also tested the third and final subunit of the Dsl1 complex, Tip20p, which was missing from the SGA analysis; sey1Δ tip20-5 mutants exhibited strong synthetic growth defects, implicating the entire Dsl1 complex in ER–ER fusion (Figure 3C).
FIGURE 3:

SNARE-mediated homotypic ER fusion requires the entire Dsl1 complex but not the COPI coat. (A) Growth assays of wild type (MY14289), sey1Δ (MY14291), sec39-1 (MY14293), and sey1Δ sec39-1 (MY14296) grown for 2 d at 30°C. (B) Top, wild type (MY14653), sey1Δ (MY14655), sec27-1 (MY14657), and sey1Δ sec27-1 (MY14659) grown for 3 d at 23°C and 2 d at 30°C. Bottom, wild type (MY14653), sey1Δ (MY14655), ret2-1 (MY14661), and sey1Δ ret2-1 (MY14663) grown at the indicated temperatures for 2 d. (C) Indicated genotypes derived from parent MY15008 grown for 2 d at 27°C. (D) Indicated genotypes with plasmid pRY270 (dsl1-A533D) or pRY261 (dsl1-L55E/L58D). Top, wild type (MY14365), sey1Δ (MY14369), dsl1Δ (MY14373), and sey1Δ dsl1Δ (MY14377). Bottom, wild type (MY14384), sey1Δ (MY14388), dsl1Δ (MY14392), and sey1Δ dsl1Δ (MY14396).
To test whether Dsl1p, Sec39p, and Tip20p function as a complex during ER–ER fusion, as they do during vesicle trafficking, we tested two additional sey1Δ dsl1 mutants: sey1Δ dsl1-A533D and sey1Δ dsl1-L55E/L58D. In vitro, Dsl1p-A533D has a weakened interaction with Sec39p and Dsl1p-L55E/L58D has a weakened interaction with Tip20p (Ren et al., 2009; Tripathi et al., 2009). The sey1Δ dsl1-A533D double mutants exhibited a synthetic growth defect (Figure 3D), suggesting that the Dsl1-Sec39 interaction is required for SNARE-mediated ER–ER fusion. On the other hand, sey1Δ dsl1-L55E/L58D did not exhibit a synthetic growth defect (Figure 3D). This result is consistent with the ability of the same allele to support retrograde vesicle trafficking, as judged by its wild-type growth rate, and suggests that the dsl1-L55E/L58D mutation is not effective in disrupting the Dsl1 complex in vivo. Overall we conclude that the Dsl1 complex needs to be at least partially intact in order to participate in SNARE-mediated ER–ER fusion.
The Dsl1 complex is directly required for ER–ER fusion
It remained possible that the synthetic growth defects observed in sey1Δ dsl1ΔE double mutants were due to disruption of some other process required for normal growth, other than ER–ER fusion or vesicle trafficking. We therefore examined these mutants for ER structure defects that should arise from disrupted homotypic ER fusion. In wild-type cells, Sec63-GFP marks the entire ER network, which includes the nucleus, tubules extending from the nucleus to the cell cortex, and the tubules and sheets at the cell periphery. Sec63–green fluorescent protein (GFP) distribution appeared similar to wild type in sey1Δ and dsl1ΔE single mutants, but sey1Δ dsl1ΔE double mutants exhibited severely disrupted peripheral ER, lacking an organized tubular network and mostly appearing as patches or aggregates of peripheral ER (Figure 4A). The pool of ER near the plasma membrane is consistent with an inability for growing ER tubules to fuse at other parts of the ER network. Additionally, whereas in wild-type cells the ER forms an almost continuous network close to the plasma membrane, in sey1Δ dsl1ΔE mutant cells, we frequently observed large sections of the cortex with no ER and regions of ER internal to the cell.
FIGURE 4:

sey1Δ dsl1ΔE double mutants exhibit severe ER structure defects. (A) Cells expressing integrated Sec63p-GFP were grown to mid– to late log phase at 30°C in YEPD and imaged live (see Materials and Methods for image acquisition and analysis). Arrowheads depict large stretches of the cell periphery lacking ER. Haploid strains of the indicated genotype were derived from diploid parent MY14907. Enlarged panel images were sharpened in ImageJ (unsharp mask, 2.0 pixels, weight = 0.60). Scale bar: 2 μm. (B) Images as in A, but cells are expressing integrated Rtn1-GFP. Strains were derived from diploid parent MY15008. Scale bar: 2 μm. Top right graph: average (arbitrary units) and SD (error bars) of the maximum pixel intensity of each cell for the indicated genotype. Mean value and number of cells measured: WT, 2763 (22 cells); sey1Δ, 4586 (32 cells); dsl1ΔE, 3000 (37 cells); and sey1Δ dsl1ΔE, 26,439 (50 cells). p values vs. WT (two-sided Kolmogorov-Smirnov test): sey1Δ, 5 × 10−6; dsl1ΔE, 0.28, sey1Δ dsl1ΔE, 4 × 10−13. Bottom right graph: as above, but the sum of all pixel intensities was measured (including all z-slices). Mean value and p value vs. WT: WT, 0.75 × 106; sey1Δ, 0.57 × 106, p = 0.03; dsl1ΔE, 0.74 × 106, p = 0.62; and sey1Δ dsl1ΔE, 1.7 × 106, p = 3 × 10−6. All quantifications were performed in background-subtracted, raw images (not deconvolved). (C) Representative electron micrograph of ultrathin-sectioned sey1Δ dsl1ΔE cells (parent diploid MY14907), imaged at 10,000×. Scale bar: 500 nm. Right, 22,500× image of the region outlined in the black box in the left panel. Scale bar: 200 nm.
We also assessed ER structure using Rtn1-GFP, which in wild-type cells localizes specifically to the tubular ER and the edges of sheets at the cell periphery and is excluded from the nucleus. Again, sey1Δ and dsl1ΔE single mutants appeared similar to wild type (Figure 4B). sey1Δ dsl1ΔE double mutants, however, exhibited a striking defect in which Rtn1-GFP accumulated in a few large, bright puncta, often at or near the bud neck (Figure 4B). As a measure of accumulation, the maximum fluorescence intensity of Rtn1-GFP in sey1Δ dsl1ΔE cells was ∼10-fold higher than wild type (Figure 4B, top right), despite having only twofold higher total GFP (Figure 4B, bottom right). We occasionally observed a few long, unbranched ER tubules at the cell periphery (Figure 4B, sey1Δ dsl1ΔE peripheral enlargement), but the bright Rtn1-GFP aggregates near the bud neck usually obscured fluorescence signal at the periphery in most cells. Importantly, similar ER network defects were observed in sey1Δ tip20-5 mutants but not sey1Δ cop1-1 mutants (Figure S1B). Finally, we examined sey1Δ dsl1ΔE cells by electron microscopy and, consistent with our findings by fluorescence microscopy, observed abnormal ER in 53% of cells (random single sections through cells, n = 38; Figure S2A) and dense aggregates of ER-like membranes in 21% of cells (Figures 4C and S2B). Together these data demonstrate a perfect correlation between synthetic growth defects and synthetic ER structure defects, consistent with a causal relationship.
To address whether the Dsl1 complex directly mediates ER–ER fusion, rather than another step that modifies ER structure, we used a direct in vivo ER–ER fusion assay adapted from Anwar et al. (2012). In this assay, we mated cells containing cytosolic GFP and cells containing ER–lumenal mCherry (directed by a signal sequence and C-terminal ER-retention sequence, HDEL). For each mating pair, we collected images at 1-min intervals and determined the duration of the delay between cell fusion (marked by cytosolic GFP transfer) and ER–ER fusion (marked by mCherry transfer; Figure 5A). As previously demonstrated (Anwar et al., 2012), ER–ER fusion required significantly more time in sey1Δ cells than in wild type (mean delay 8.7 vs. 4.9 min, p = 0.01, two-sample t test; Figure 5B). dsl1ΔE cells were not significantly different from wild type (5.3 vs. 4.9 min, p = 0.81). In contrast, ER–ER fusion in sey1Δ dsl1ΔE mating pairs occurred ∼24 min after wild type, an approximately fivefold increase (p < 0.001; Figure 5B). Because of the low-throughput nature of time-lapse microscopy, we performed a second assay in which we fixed the mating mixtures in paraformaldehyde and imaged the ER–lumenal mCherry distribution in many unbudded zygotes (n > 50 for each genotype for each experiment). While this assay is less sensitive to minor delays in ER–ER fusion, it allows a higher number of zygotes to be scored. We found that wild type, sey1Δ, and dsl1ΔE all appeared similar (<2% zygotes with unfused ER), whereas, in agreement with the results of the live-cell assay, sey1Δ dsl1ΔE zygotes displayed a dramatic deficiency in ER–ER fusion (62 ± 4% contained unfused ER; Figure 5C). Of note, ER–ER fusion was largely restored when sey1Δ dsl1ΔE cells were mated against wild type (Figure 5C). This initially seemed surprising, as ER–ER fusion presumably requires fusogens in both membranes; however, Dsl1p is soluble in the cytoplasm and likely equilibrates quickly between ER membranes. Together the ER structural defects and decreased ER–ER fusion rate demonstrate that the Dsl1 complex, in conjunction with the SNAREs and in parallel to Sey1p, plays a direct role in ER–ER fusion.
FIGURE 5:
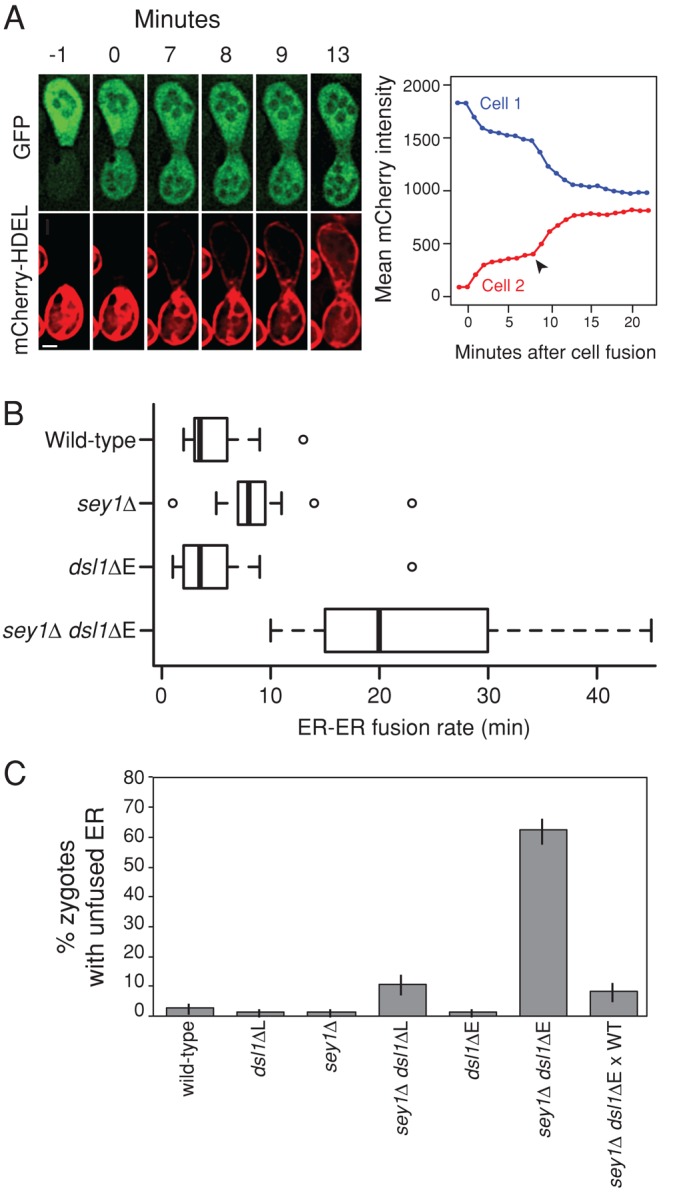
ER–ER fusion is severely delayed in sey1Δ dsl1ΔE mutants. (A) Representative example of the ER–ER fusion assay. Identical genotypes were mated to each other at room temperature; MATa strains expressed mCherry-HDEL (pMR6474), MATα strains expressed cytosolic GFP (pMR3619). sey1Δ cells are shown. The first peak in mCherry transfer at 0–2 min corresponds to the transfer of cytosolic mCherry-HDEL after cell fusion. Arrowhead depicts the point of ER–ER fusion (8 min). Scale bar: 2 μm. (B) Box plot of the times required for ER–ER fusion after cell fusion. Data are pooled from at least two independent experiments for each genotype. Mean values: wild type, 4.9 min (MY14509 × MY14513, n = 16); sey1Δ, 8.8 min (MY14510 × MY14514, n = 16); dsl1ΔE, 5.3 min (MY14511 × MY14515, n = 14); and sey1Δ dsl1ΔE, 23.4 min (MY14512 × MY14516; n = 10). Each box shows the interquartile range (25–75% of the data), black bars represent the median, and outliers are shown as open circles beyond the 1.5 * interquartile range. (C) Percentage of zygotes with unfused ER (binary scoring). The data are pooled from two independent experiments; error bars show ± SE for a binomial distribution. Strains used: wild type (MY14059 × MY14009), dsl1ΔL (MY14061 × MY14013), sey1Δ (MY14063 × MY14017), sey1Δ dsl1ΔL (MY14065 × MY14021), dsl1ΔE (MY14067 × MY14025), sey1Δ dsl1ΔE (MY14071 × MY14031), and sey1Δ dsl1ΔE (MY14071) × wild type (MY14009). All MATa strains expressed GFP-HDEL (pMR6473).
The reticulons are required for viability in a sey1Δ dsl1ΔE background
The reticulons Rtn1p, Rtn2p, and Yop1p are required for normal peripheral ER structure in vivo and can mediate ER tubule formation in vitro (Voeltz et al., 2006; Hu et al., 2008). Previous studies focusing only on Sey1p-mediated ER–ER fusion were unable to explain why sey1Δ rtn1Δ rtn2Δ yop1Δ mutants have only minor growth defects, despite having extremely perturbed ER structure (Voeltz et al., 2006; West et al., 2011). It was posited that either ER–ER fusion is not essential for viability under normal growth conditions in yeast or that there exists an alternative ER–ER fusion pathway. Despite the identification of an alternative SNARE/Dsl1-mediated ER–ER fusion pathway, sey1Δ dsl1ΔE double mutants, which have lost two ER–ER fusion pathways, remain viable (albeit slow growing). It therefore remained possible that ER–ER fusion is a nonessential process in yeast.
To address this possibility and to explore the relationship, if any, between the reticulons and the SNARE/Dsl1-mediated ER–ER fusion pathway, we dissected ∼100 tetrads from a diploid heterozygous for sey1Δ, rtn1Δ, rtn2Δ, yop1Δ, lnp1Δ, and dsl1ΔE, and quantified colony growth over time of the different genetic combinations, averaged by genotype. We observed a minimal effect of rtn2Δ on the growth rate relative to any comparable genetic combination (Figure S3, A and B; see Table S2 for full growth data), consistent with Rtn2p's low level of expression under normal growth conditions (De Craene et al., 2006; Voeltz et al., 2006). We therefore averaged colony sizes independently of RTN2. Additionally, our results recapitulated published growth defects of lnp1Δ rtn1Δ and suppression of lnp1Δ rtn1Δ by sey1Δ (Figure S3C; Chen et al., 2012). lnp1Δ was included in our analysis, as Lnp1p was previously shown to antagonize Sey1p activity via an unknown mechanism (Chen et al., 2012). However, there were no synthetic genetic interactions between lnp1Δ and dsl1ΔE, and we therefore do not discuss LNP1 further (Figure S3C).
We next compared growth rates related only to sey1Δ, rtn1Δ, yop1Δ, and dsl1ΔE genotypic combinations (Figure 6A). As expected, all single mutants grew about as well as wild type (compare strain 1 with strains 2–5 in Figure 6A). Among double mutants (strains 6–11), sey1Δ dsl1ΔE exhibited a strong growth defect, as expected from our initial work, and yop1Δ rtn1Δ exhibited a minor growth defect, as previously reported. Surprisingly, rtn1Δ dsl1ΔE exhibited an intermediate growth defect, implying that Rtn1p and Dsl1p share functional redundancy. All triple mutants (strains 12–15) showed intermediate to severe growth defects (see Figure 6B for colony sizes after 6 d of growth). The quadruple-mutant sey1Δ yop1Δ rtn1Δ dsl1ΔE (strain 16) was almost completely inviable (four out of eight spores did not form colonies after 6 d of growth, and the remaining spores formed extremely tiny colonies; Figure 6B), suggesting that there is an unexpected partial redundancy between the function of the reticulons and the two ER–ER fusion pathways, which together are essential for cell viability.
FIGURE 6:

The reticulons are required for viability in a sey1Δ dsl1ΔE background. (A) Colony sizes after tetrad dissection (parent MY14454) and 48 h of growth at 30°C on YEPD for the indicated genotypes. All genotypes are LNP1+ and averaged independently of RTN2. Number of colonies averaged for each genotype ranged from 6 to 18 (see Table S2 for full data). Errors bars show ± SE of the mean. (B) As in A, but after 6 d of growth at 30°C.
It is thought that the reticulons both generate and maintain ER tubules and thus may act genetically in the same pathway with Sey1p (Hu et al., 2009; Chen et al., 2012). The relationship between SNARE-mediated ER–ER fusion and the reticulons, however, is unclear. Because the sey1Δ yop1Δ rtn1Δ dsl1ΔE quadruple mutant (strain 16) is almost inviable, The Dsl1 pathway must be responsible for the relatively normal growth rate of the sey1Δ yop1Δ rtn1Δ triple mutant (strain 12). We conclude, therefore, that SNARE/Dsl1-mediated ER–ER fusion does not require the reticulons Rtn1p and Yop1p (Figure 6A). Conversely, Sey1p is less able than Dsl1p to support reticulon-independent growth, as cells that are only SEY1+ (yop1Δ rtn1Δ dsl1ΔE, strain 15) have a more severe growth defect than cells that are only DSL1+ (sey1Δ yop1Δ rtn1Δ, strain 12). Notably, however, SEY1 still confers a growth advantage on cells lacking reticulons and wild-type DSL1 (Figure 6B, compare strains 15 and 16). Finally, when dsl1ΔE is combined with sey1Δ, rtn1Δ, or both sey1Δ and rtn1Δ, the resulting growth defects are all similar (Figure 6A, compare strains 8, 11, and 14), suggesting that Sey1p and Rtn1p share a genetic pathway distinct from Dsl1p.
A surprising observation was that cells that are only YOP1+ (sey1Δ rtn1Δ dsl1ΔE, strain 14) have an intermediate growth defect, whereas cells that are only RTN1+ (sey1Δ yop1Δ dsl1ΔE, strain 13) are almost inviable (Figure 6B). Therefore Yop1p but not Rtn1p can contribute substantially to cell growth independently of the other reticulons and of the Sey1p- and SNARE/Dsl1-dependent ER–ER fusion pathways.
DISCUSSION
Characterization of SNARE-mediated homotypic ER fusion
On the basis of synthetic growth defects and ER–ER fusion experiments in this study, we conclude that SNARE-mediated homotypic ER fusion additionally requires the Dsl1 complex, consisting of Sec39p, Dsl1p, and Tip20p. Furthermore, genetic analysis indicates that the observed synthetic defects are not indirectly caused by perturbed vesicle trafficking; if they were, then other mutations that disrupt vesicle trafficking, such as mutations in vesicle coat components or other SNAREs, should have resulted in growth and ER structure defects in a sey1Δ background (see Table S1 for full list of trafficking components tested). These results imply that SNARE-mediated ER–ER fusion occurs independently of vesicle fusion and relies on direct ER–ER interactions, similar to the model for Sey1p-mediated ER–ER fusion (Figure 7).
FIGURE 7:

Model for homotypic ER fusion. The left-most panel depicts the ER network (blue) within a typical yeast cell (black outline). A few tubules connect the nuclear ER to the peripheral ER, which is discontinuous when viewed as a slice. The two middle panels depict alternative homotypic ER fusion pathways for an ER tubule. Either pathway leads to the same outcome, depicted in the right-most panel: the creation of a new three-way junction. The top middle panel depicts canonical Sey1p-mediated ER–ER fusion. The bottom middle panel depicts SNARE/Dsl1 complex–mediated homotypic ER fusion, as defined in this study. The two dotted-line SNAREs represent uncertainty in the exact composition and topology of the final SNARE complex.
In the Dsl1 complex mutants examined in this study, there was a perfect correlation between synthetic growth defects and synthetic ER structure and ER–ER fusion defects. It is conceivable that aberrant ER structures could inhibit the observed rate of ER–ER fusion in our assay, but we note that previous reports demonstrated that rtn1Δ rtn2Δ yop1Δ mutants, which have severe ER structure defects, have a normal rate of ER–ER fusion (Anwar et al., 2012). Therefore we conclude that the SNAREs most likely act with the Dsl1 complex as true fusogens for homotypic ER fusion.
The full composition of the ER–ER fusion SNARE complex is not yet clear. Only three SNARE genes—SEC20, USE1, and UFE1—have shown genetic interactions with SEY1, whereas a normal four-helix SNARE complex requires four SNARE proteins. In retrograde trafficking, Sec22p is the fourth SNARE, but Ykt6p can compensate in cells lacking Sec22p (Liu and Barlowe, 2002). Sec22p/Ykt6p could also redundantly mediate ER–ER fusion, but this is difficult to test, as known sec22 ykt6 double mutants are either inviable or severely slow growing (Liu and Barlowe, 2002; Rogers et al., 2013).
During retrograde trafficking, the Dsl1 complex tethers ER-bound SNAREs to the incoming COPI-coated vesicle and promotes trans-SNARE complex assembly. Our results imply that the Dsl1 complex uses a coat-independent function during ER–ER fusion. In agreement with this conclusion, dsl1-4 mutants accumulate ER and vesicles at the nonpermissive temperature, whereas dsl1-7 mutants accumulate only ER, suggesting that Dsl1p has two different functions (VanRheenen et al., 2001). Furthermore, overexpression of Sec21p (γ-COP), a COPI complex subunit, suppressed the lethality of dsl1-4 but not dsl1-7, supporting a COPI-independent role for Dsl1p (VanRheenen et al., 2001). Mechanistically, because the Dsl1 complex promotes SNARE complex formation and stabilization, this mechanism could apply to ER–ER fusion as well, independent of a vesicle-tethering function (Ren et al., 2009; Diefenbacher et al., 2011). An attractive hypothesis is that the Dsl1p complex might adopt an extended conformation that tethers SNAREs residing on the two apposing ER membranes before fusion.
The SNARE/Dsl1-mediated homotypic ER fusion pathway characterized here may have additional components that are not shared with retrograde vesicle trafficking. Such components should exhibit negative genetic interactions with sey1Δ. Only five candidates were identified (Table 2): yop1Δ, ice2Δ, scs3Δ, pom33Δ, and sec23-1. Yop1p and Ice2p broadly affect ER structure and function and may not be specific to SNARE-mediated ER–ER fusion. Scs3p is required for normal ER membrane biosynthesis and exhibits genetic interactions with many cellular processes, suggesting that it too may not be specific for SNARE-mediated ER–ER fusion (Hosaka et al., 1994; Moir et al., 2012). Pom33p is a transmembrane nucleoporin (Chadrin et al., 2010). Interestingly, some reticulons have additional roles in nuclear pore complex biogenesis in addition to ER tubulation, probably via a common curvature-inducing mechanism (Dawson et al., 2009). Furthermore, Rtn1p and Yop1p physically interact with Ndc1p, a component of nuclear pore complexes (Casey et al., 2012). Therefore sey1Δ pom33Δ double mutants may have disrupted nuclear pore complex functions. However, as SNARE-mediated ER–ER fusion must occur at the peripheral ER, away from the nucleus, a role for Pom33p at the peripheral ER would be surprising. Finally, Sec23p is a component of the COPII coat that stimulates the GTPase activity of Sar1p and is required for ER-to-Golgi anterograde vesicle trafficking. However, all other anterograde trafficking components, including the rest of the COPII coat, do not genetically interact with sey1Δ (except sar1, for which we have no data; Table S1). It is therefore possible that Sec23p has a novel, vesicle-independent function in SNARE-mediated ER–ER fusion.
Finally, we note that all identified members of SNARE-mediated ER–ER fusion have conserved mammalian orthologues, raising the possibility that this pathway is conserved among eukaryotes (conservation reviewed in Schmitt, 2010).
Functional overlap between Sey1p-mediated and SNARE-mediated ER–ER fusion
An open question is whether Sey1p and SNAREs mediate distinct ER–ER fusion events. sey1Δ and dsl1ΔE single mutants exhibit no growth defects, and sey1Δ mutants have only minor defects in ER structure and ER–ER fusion rate compared with sey1Δ dsl1ΔE double mutants. This suggests that under normal growth conditions the two pathways are largely, but not entirely, redundant and independent. However, it is possible that one pathway is favored under certain environmental conditions, such as different osmolarities or temperatures, or that they mediate fusion at ER domains that have subtle differences.
The function of ER structure and fusion in yeast cells
An early mystery in our study was why sey1Δ dsl1ΔE double mutants are viable if both ER–ER fusogens have been disrupted. We reasoned that this could be because 1) the dsl1ΔE mutation only partially disrupts SNARE-mediated ER–ER fusion; 2) there is a third, redundant ER–ER fusion pathway; or 3) ER–ER fusion is not an essential process for yeast viability under normal growth conditions. The first possibility is difficult to test, as all of the genes in the SNARE/Dsl1 pathway are essential and cannot be deleted. The second possibility, that there is a third, albeit inefficient, fusogen, is supported by the observation that Yop1p is required for most of the remaining growth in the sey1Δ dsl1ΔE double mutant. Therefore Yop1p may be an essential member of a third ER–ER fusion pathway, or Yop1p itself may function as both a reticulon and a weak fusogen. The latter interpretation is supported by the finding that Yop1p is sufficient to form both tubules and three-way ER junctions in vitro, whereas Rtn1p only forms tubules and no junctions (Hu et al., 2008). The third possibility, that ER–ER fusion is not an essential process under our growth conditions, remains formally plausible, as even sey1Δ rtn1Δ yop1Δ dsl1ΔE mutants occasionally formed tiny colonies after ∼6 d of growth at 30°C (four out of eight spores). However, this conclusion assumes there is no fourth fusogen, that ER–ER fusion will not spontaneously occur without a fusogen, and that the dsl1ΔE mutation fully disrupts SNARE-mediated ER–ER fusion.
A remaining problem is identifying the most proximal cause of the sey1Δ dsl1ΔE growth defect. Disrupted ER–ER fusion could affect multiple downstream processes, including ER inheritance, lipid homeostasis between the nuclear and peripheral ER, and protein recycling. In such cases, one should be able to restore normal growth by rescuing the downstream processes without rescuing ER–ER fusion itself. More indirectly, accumulated ER could physically block exo/endocytosis at the plasma membrane, or organelle or nuclear segregation, as was observed for sey1Δ mutants during nuclear congression during mating (Rogers et al., 2013). Discerning these mechanisms should prove fruitful in future studies.
MATERIALS AND METHODS
Strains and general yeast methods
Strains and plasmids used are listed in Table S3. Standard methods including cell culture and transformations were performed as described by Amberg et al. (2005). In general, many of the slow-growing strains described in this study were prone to accumulating intermediate suppressors of growth rate if used for many generations. To avoid this confounding issue, we sometimes generated desired genotypes for an experiment from a heterozygous diploid parent as stated in the figure legends but did not freeze the haploids as separate strains.
Growth assays
Cultures were grown to saturation in yeast extract/peptone/dextrose (YEPD) at 23°C (except 30°C for the assay in Figure 1B) and then 0.2 OD600 unit of cells was pelleted and resuspended in 200 μl dH2O. Five 10-fold serial dilutions were made in a 96-well plate and then spotted on YEPD plates and grown at various temperatures as indicated.
SGA experiments and analysis
dsl1ΔE:NatMX (MY15059), dsl1ΔLasso:NatMX (MY15060), and DSL1+::NatMX (MY15058) strains were created using parent strain Y7092, and SGA screens were performed and analyzed as previously described (Tong and Boone, 2006; Baryshnikova et al., 2010). SGA screens with temperature-sensitive alleles were performed at 26°C. To separate the alleles into classes, we used a cutoff score (requiring at least one SGA score with an absolute value ≥ 0.25) and a difference threshold (requiring that SGA scores differed by at least an absolute value of 0.25 to be classified as specific). Additionally, for the alleles in the shared class, we required that both SGA scores had an absolute value of at least 0.25.
Image acquisition and analysis
All microscopy was performed on a DeltaVision deconvolution microscope (Applied Precision, Issaquah, WA), based on a Nikon TE200 (Melville, NY) with an inverted 100× NA 1.4 objective, a 50-W mercury lamp, and a Photometrics Cool Snap HQ CCD camera (Photometrics, Tucson, AZ). All images were deconvolved using the Applied Precision SoftWoRx imaging software.
ER structure microscopy
Cells were imaged live in growth medium at room temperature on a standard glass slide and #1.5 coverslip. Typically, imaging began at the bottom of a cell (nearest the objective and touching the coverslip) and 30–40 slices were imaged with 0.15-μm z-spacing. Exposure times were typically 0.5–1.0 s, except sey1Δ dsl1ΔE and sey1Δ tip20-5 cells expressing Rtn1-GFP (Figures 4B and S1B) were imaged at 0.05–0.1 s to avoid pixel saturation.
ER–ER fusion assays
Cultures were grown overnight to early to mid–log phase in synthetic complete medium lacking leucine (SC −leu) at 30°C. Cells from each strain to be mated (0.01 OD600 unit) were added onto a pretreated 0.17-mm Delta T4 Culture Dish (Bioptechs, Butler, PA). Immediately before the cells were added, the Delta-T dishes were pretreated by coating with 25 μl of concanavalin A (0.1 mg/ml in 20 mM sodium acetate, pH 5.8) for 15 min and then being washed twice with 50 μl of 20 mM sodium acetate (pH 5.8). Cells were allowed to settle for 15 min before being washed with 200 μl of SC −leu to remove unstuck cells. Finally, 2 ml of SC −leu was added to the dish, and matings were imaged at room temperature. Mating pairs were imaged at 1-min intervals with minimal exposure times (usually 0.1 s) and at a single focal plane to reduce photobleaching and phototoxicity. After imaging, raw movies (not deconvolved) were scored for the time between cell fusion and ER–ER fusion. Cell fusion was identified by a rapid transfer of cytoplasmic GFP into the adjacent mating partner that usually equilibrated in less than 2 min. The first time point with cytoplasmic GFP in both cells, even if not yet equilibrated, was marked as the time of cell fusion. After cell fusion, mCherry-HDEL accumulated slowly in the mating partner's cytoplasm and ER, presumably due to cytoplasmic mCherry-HDEL and protein recycling. Eventually mCherry-HDEL transfer shifted to a rapid equilibration phase (see Figure 5A and Anwar et al., 2012). The first time point with this shift to fast equilibration was marked as the time of ER–ER fusion.
Fixed-cell assays (Figure 5C) were performed as previously described (Rogers et al., 2013). Briefly, cultures were grown at 30°C to mid–log phase, and 0.5 OD600 unit of cells was mixed and mated on a 0.45-μm nitrocellulose filter (EMD Millipore, Billerica, MA) for 3 h at 23°C. Cells were then washed into 900 μl of 1× phosphate-buffered saline (PBS), and 100 μl of 20% paraformaldehyde dissolved in distilled H2O was added. Cells were fixed at room temperature for 15 min; this was followed by one wash in 1× PBS, 4′,6-diamidino-2-phenylindole staining (2 μg/ml in PBS) for 15 min, two more washes in 1× PBS, and, finally, resuspension in 100–200 μl 1× PBS. Cells were imaged on the same day. GFP-HDEL was used in the fixed-cell assay rather than mCherry-HDEL, as GFP-HDEL intensity and localization is preserved better after fixation. Imaging on the same day ensures that the membranes stay intact and GFP-HDEL does not artifactually diffuse to equilibrium. ER was scored as unfused when GFP-HDEL appeared markedly brighter in one-half of the zygote than the other.
Electron microscopy
Cells were prepared for transmission electron microscopy as described in Gammie and Rose (2002). Briefly, the workflow included a glutaraldehyde fixation, potassium permanganate staining, sodium periodate treatment, uranyl acetate staining, and embedding in LR White resin. Specifically to our protocol, ∼5 OD600 units of mid–log phase sey1Δ dsl1ΔE cells (from diploid parent MY14907) were fixed in 2% glutaraldehyde for 30 min at room temperature. Cells were stained with 4% potassium permanganate for 4 h at 4°C. Ultrathin sections (∼80 nm) were placed on a nickel slotted-grid (Formvar film, FF-2010-Ni, Electron Microscopy Sciences, Hatfield, PA) and imaged directly, without lead citrate staining.
Colony size time-course analysis
Strain MY14454 was sporulated, and 100 tetrads were dissected on YEPD plates. The plates were incubated at 30°C and imaged after 2, 3, 4, 5, and 6 d of growth. Colony size was measured in ImageJ by applying a binary threshold mask to outline colonies. After 6 d, plates were replica plated to determine genotypes. Genotypes for lethal or extremely tiny colonies were inferred by assuming 2:2 segregation of genes. We excluded from our analysis tetrads that did not exhibit 2:2 segregation for all genes or tetrads that did not permit unambiguous determination of all four spores (e.g., only two cells were viable in the tetrad).
Supplementary Material
Acknowledgments
We thank members of the Rose, Gammie, Hughson, and Boone laboratories for helpful support and discussion. We also thank S. Ferro-Novick for providing parent strains used in this study and Bryan-Joseph San Luis for technical assistance. This work was supported by National Institutes of Health grants GM037739 (to M.D.R.) and GM071574 (to F.M.H.). J.V.R. was supported by National Institutes of Health training grant GM007388.
Abbreviations used:
- CATCHR
complex associated with tethering containing helical rods
- COPI
cytosolic coat protein I
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- ORF
open reading frame
- PBS
phosphate-buffered saline
- SGA
synthetic genetic array
- SM
Sec1/Munc18
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- YEPD
yeast extract/peptone/dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www .molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1220) on September 3, 2014.
*These authors contributed equally to this work.
REFERENCES
- Amberg D, Burke J, Strathern J. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Andag U, Neumann T, Schmitt HD. The coatomer-interacting protein Dsl1p is required for Golgi-to-endoplasmic reticulum retrieval in yeast. J Biol Chem. 2001;276:39150–39160. doi: 10.1074/jbc.M105833200. [DOI] [PubMed] [Google Scholar]
- Andag U, Schmitt HD. Dsl1p, an essential component of the Golgi-endoplasmic reticulum retrieval system in yeast, uses the same sequence motif to interact with different subunits of the COPI vesicle coat. J Biol Chem. 2003;278:51722–51734. doi: 10.1074/jbc.M308740200. [DOI] [PubMed] [Google Scholar]
- Anwar K, Klemm RW, Condon A, Severin KN, Zhang M, Ghirlando R, Hu J, Rapoport TA, Prinz WA. The dynamin-like GTPase Sey1p mediates homotypic ER fusion in S. cerevisiae. J Cell Biol. 2012;197:209–217. doi: 10.1083/jcb.201111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ, Myers CL, Andrews B, Boone C. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Methods Enzymol. 2010;470:145–179. doi: 10.1016/S0076-6879(10)70007-0. [DOI] [PubMed] [Google Scholar]
- Burri L, Lithgow T. A complete set of SNAREs in yeast. Traffic. 2004;5:45–52. doi: 10.1046/j.1600-0854.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- Casey AK, Dawson TR, Chen J, Friederichs JM, Jaspersen SL, Wente SR. Integrity and function of the Saccharomyces cerevisiae spindle pole body depends on connections between the membrane proteins Ndc1, Rtn1, and Yop1. Genetics. 2012;192:441–455. doi: 10.1534/genetics.112.141465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadrin A, Hess B, San Roman M, Gatti X, Lombard B, Loew D, Barral Y, Palancade B, Doye V. Pom33, a novel transmembrane nucleoporin required for proper nuclear pore complex distribution. J Cell Biol. 2010;189:795–811. doi: 10.1083/jcb.200910043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Novick P, Ferro-Novick S. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat Cell Biol. 2012;14:707–716. doi: 10.1038/ncb2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Novick P, Ferro-Novick S. ER structure and function. Curr Opin Cell Biol. 2013;25:428–433. doi: 10.1016/j.ceb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TR, Lazarus MD, Hetzer MW, Wente SR. ER membrane-bending proteins are necessary for de novo nuclear pore formation. J Cell Biol. 2009;184:659–675. doi: 10.1083/jcb.200806174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene J-O, Coleman J, Estrada de Martin P, Pypaert M, Anderson S, Yates JR, Ferro-Novick S, Novick P. Rtn1p is involved in structuring the cortical endoplasmic reticulum. Mol Biol Cell. 2006;17:3009–3020. doi: 10.1091/mbc.E06-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delic M, Valli M, Graf AB, Pfeffer M, Mattanovich D, Gasser B. The secretory pathway: exploring yeast diversity. FEMS Microbiol Rev. 2013;37:872–914. doi: 10.1111/1574-6976.12020. [DOI] [PubMed] [Google Scholar]
- Diefenbacher M, Thorsteinsdottir H, Spang A. The Dsl1 tethering complex actively participates in soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor (SNARE) complex assembly at the endoplasmic reticulum in Saccharomyces cerevisiae. J Biol Chem. 2011;286:25027–25038. doi: 10.1074/jbc.M110.215657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AR, Voeltz GK. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol. 2013;5:a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada de Martin P, Du Y, Novick P, Ferro-Novick S. Ice2p is important for the distribution and structure of the cortical ER network in Saccharomyces cerevisiae. J Cell Sci. 2005;118:65–77. doi: 10.1242/jcs.01583. [DOI] [PubMed] [Google Scholar]
- Gammie AE, Rose MD. Assays of cell and nuclear fusion. Methods Enzymol. 2002;351:477–498. doi: 10.1016/s0076-6879(02)51866-8. [DOI] [PubMed] [Google Scholar]
- Hong W, Lev S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014;24:35–43. doi: 10.1016/j.tcb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- Hosaka K, Nikawa J, Kodaki T, Ishizu H, Yamashita S. Cloning and sequence of the SCS3 gene which is required for inositol prototrophy in Saccharomyces cerevisiae. J Biochem. 1994;116:1317–1321. doi: 10.1093/oxfordjournals.jbchem.a124681. [DOI] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Voss C, Shemesh T, Li Z, Coughlin M, Kozlov MM, Rapoport T A, Prinz W A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 2008;319:1247–1250. doi: 10.1126/science.1153634. [DOI] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu P-P, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedd G, Richardson C, Litt R, Segev N. The Ypt1 GTPase is essential for the first two steps of the yeast secretory pathway. J Cell Biol. 1995;131:583–590. doi: 10.1083/jcb.131.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamena F, Diefenbacher M, Kilchert C, Schwarz H, Spang A. Ypt1p is essential for retrograde Golgi-ER transport and for Golgi maintenance in S. cerevisiae. J Cell Sci. 2008;121:1293–1302. doi: 10.1242/jcs.016998. [DOI] [PubMed] [Google Scholar]
- Kraynack BA, Chan A, Rosenthal E, Essid M, Umansky B, Waters MG, Schmitt HD. Dsl1p, Tip20p, and the novel Dsl3(Sec39) protein are required for the stability of the Q/t-SNARE complex at the endoplasmic reticulum in yeast. Mol Biol Cell. 2005;16:3963–3977. doi: 10.1091/mbc.E05-01-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Chen LB. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Gallwitz D, Peng R. Structure-based functional analysis reveals a role for the SM protein Sly1p in retrograde transport to the endoplasmic reticulum. Mol Biol Cell. 2005;16:3951–3962. doi: 10.1091/mbc.E05-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Barlowe C. Analysis of Sec22p in endoplasmic reticulum/Golgi transport reveals cellular redundancy in SNARE protein function. Mol Biol Cell. 2002;13:3314–3324. doi: 10.1091/mbc.E02-04-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Gross DA, Silver DL, Willis IM. SCS3 and YFT2 link transcription of phospholipid biosynthetic genes to ER stress and the UPR. PLoS Genet. 2012;8:e1002890. doi: 10.1371/journal.pgen.1002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orso G, Pendin D, Liu S, Tosetto J, Moss TJ, Faust JE, Micaroni M, Egorova A, Martinuzzi A, McNew JA, Daga A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- Ossig R, Dascher C, Trepte HH, Schmitt HD, Gallwitz D. The yeast SLY gene products, suppressors of defects in the essential GTP-binding Ypt1 protein, may act in endoplasmic reticulum-to-Golgi transport. Mol Cell Biol. 1991;11:2980–2993. doi: 10.1128/mcb.11.6.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SK, Indig FE, Olivieri N, Levine ND, Latterich M. Organelle membrane fusion: a novel function for the syntaxin homolog Ufe1p in ER membrane fusion. Cell. 1998;92:611–620. doi: 10.1016/s0092-8674(00)81129-0. [DOI] [PubMed] [Google Scholar]
- Ren Y, Yip CK, Tripathi A, Huie D, Jeffrey PD, Walz T, Hughson FM. A structure-based mechanism for vesicle capture by the multisubunit tethering complex Dsl1. Cell. 2009;139:1119–1129. doi: 10.1016/j.cell.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JV, Arlow T, Inkellis ER, Koo TS, Rose MD. ER-associated SNAREs and Sey1p mediate nuclear fusion at two distinct steps during yeast mating. Mol Biol Cell. 2013;24:3896–3908. doi: 10.1091/mbc.E13-08-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrock TM, O'Dell JL, Adams AE. Allele-specific suppression by formation of new protein-protein interactions in yeast. Genetics. 1997;147:1635–1642. doi: 10.1093/genetics/147.4.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt HD. Dsl1p/Zw10: common mechanisms behind tethering vesicles and microtubules. Trends Cell Biol. 2010;20:257–268. doi: 10.1016/j.tcb.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Weissman JS. The contribution of systematic approaches to characterizing the proteins and functions of the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5:a013284. doi: 10.1101/cshperspect.a013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, Voeltz GK. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem. 2008;283:18892–18904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A. Retrograde traffic from the Golgi to the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5:a013391. doi: 10.1101/cshperspect.a013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tong AHY, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Tripathi A, Ren Y, Jeffrey PD, Hughson FM. Structural characterization of Tip20p and Dsl1p, subunits of the Dsl1p vesicle tethering complex. Nat Struct Mol Biol. 2009;16:114–123. doi: 10.1038/nsmb.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen SM, Reilly BA, Chamberlain SJ, Waters MG. Dsl1p, an essential protein required for membrane traffic at the endoplasmic reticulum/Golgi interface in yeast. Traffic. 2001;2:212–231. doi: 10.1034/j.1600-0854.2001.020307.x. [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport T A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman B V, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- West M, Zurek N, Hoenger A, Voeltz GK. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol. 2011;193:333–346. doi: 10.1083/jcb.201011039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink S, Wenzel D, Wurm Ca, Schmitt HD. A link between ER tethering and COP-I vesicle uncoating. Dev Cell. 2009;17:403–416. doi: 10.1016/j.devcel.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Zurek N, Sparks L, Voeltz G. Reticulon short hairpin transmembrane domains are used to shape ER tubules. Traffic. 2011;12:28–41. doi: 10.1111/j.1600-0854.2010.01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.