

Bim protein mediates neuronal apoptosis under certain stress conditions. In neurons exposed to oxidative stress, Hsp27 prevents neuronal death, repressing bim mRNA translation through its binding to the 3′-untranslated region. This neuroprotective mechanism may provide novel therapeutic approaches in diverse neurodegenerative conditions.
Abstract
Neurons face a changeable microenvironment and therefore need mechanisms that allow rapid switch on/off of their cytoprotective and apoptosis-inducing signaling pathways. Cellular mechanisms that control apoptosis activation include the regulation of pro/antiapoptotic mRNAs through their 3′-untranslated region (UTR). This region holds binding elements for RNA-binding proteins, which can control mRNA translation. Here we demonstrate that heat shock protein 27 (Hsp27) prevents oxidative stress–induced cell death in cerebellar granule neurons by specific regulation of the mRNA for the proapoptotic BH3-only protein, Bim. Hsp27 depletion induced by oxidative stress using hydrogen peroxide (H2O2) correlated with bim gene activation and subsequent neuronal death, whereas enhanced Hsp27 expression prevented these. This effect could not be explained by proteasomal degradation of Bim or bim promoter inhibition; however, it was associated with a specific increase in the levels of bim mRNA and with its binding to Hsp27. Finally, we determined that enhanced Hsp27 expression in neurons exposed to H2O2 or glutamate prevented the translation of a reporter plasmid where bim-3′UTR mRNA sequence was cloned downstream of a luciferase gene. These results suggest that repression of bim mRNA translation through binding to the 3′UTR constitutes a novel cytoprotective mechanism of Hsp27 during stress in neurons.
INTRODUCTION
Bcl-2 homology domain (BH) 3–containing proteins (BH3-only proteins) couple stress signals to the intrinsic mitochondrial pathway of apoptosis. Accordingly, their levels within cells are tightly regulated to avoid inappropriate activation of the apoptosis program. The BH3-only protein Bim (Bcl-2 interacting mediator of cell death) mediates apoptosis in different neuronal types under diverse stress situations (Gilley et al., 2003; Butts et al., 2005; Zimmermann et al., 2005; Concannon et al., 2010; Xie et al., 2011). Bim promoter activation depends on the simultaneous binding of transcription factors such as FOXO3 and AP1 (Whitfield et al., 2001; Gilley et al., 2003; Biswas et al., 2007). Bim can also be posttranscriptionally regulated by phosphorylation and subsequent degradation via the proteasome (Ley et al., 2003; Luciano et al., 2003; Meller et al., 2006). Furthermore, bim mRNA can be regulated through the 3′-untranslated region (3′UTR; Matsui et al., 2007; Terasawa et al., 2009). This region binds miRNAs and RNA-binding proteins (RBPs), which are able to regulate mRNA stability and/or translation (de Moor et al., 2005; Bolognani et al., 2008).
The heat shock proteins (Hsps) are stress-inducible proteins that exert cytoprotective and antiapoptotic actions (Xanthoudakis and Nicholson, 2000; Lanneau et al., 2008). The Hsp27 isoform (Hsp25 in rodents) has an important neuroprotective role (Latchman et al., 2005), whose exact cellular mechanisms have not yet been fully elucidated. Hsp27 is able to prevent neuronal apoptosis (Wagstaff et al., 1999; Benn et al., 2002; King et al., 2009) by its direct binding to proapoptotic proteins such as cytochrome C (Bruey et al., 2000); however, evidence indicates that Hsp27 can also interfere with apoptotic signaling upstream of mitochondrial cytochrome C release (Paul et al., 2002; Havasi et al., 2008; Stetler et al., 2008). Hsp effects are associated with their chaperone activity, binding misfolded, oxidized, or proapoptotic proteins and facilitating their degradation by the proteasome (Garrido et al., 2006; Arrigo 2007; Kalmar and Greensmith, 2009). Hsps also display the capacity to act as RNA-binding proteins (Henics et al., 1999; Matsui et al., 2007; Sinsimer et al., 2008). Here we describe how enhanced Hsp27 levels during oxidative stress in cerebellar granule neurons (CGNs) prevent Bim protein up-regulation and subsequent neuronal death. Hsp27 effects depend on binding to the bim mRNA transcript and the regulation of its 3′UTR to repress its translation. Together these results identify a novel posttranscriptional mechanism by which Hsp27 opposes neuronal death.
RESULTS
Bim mediates oxidative stress–induced cell death in cerebellar granule neurons
We induced oxidative stress in CGN cultures using hydrogen peroxide (H2O2; 25–50 μM) addition. This treatment induces an abrupt increase in reactive oxygen species (ROS) and oxidative stress in CGNs (Davila and Torres-Aleman, 2008). ROS can act as signaling molecules, and their harmful effects include the activation of redox-sensitive proapoptotic pathways (Ueada et al., 2002; Handy and Loscalzo, 2012). H2O2 treatment induced cell death in CGNs with typical features of apoptosis, including nuclear pyknosis, caspase activation, and DNA fragmentation (Figure 1A).
FIGURE 1:

Bim mediates oxidative stress–induced cell death in CGNs. (A) CGNs treated with H2O2 (37.5 μM), showing typical features of apoptotic cell death 4–6 h after addition. Nuclear pyknosis was detected by Hoechst staining, caspase activation was detected by in situ staining with the fluorescence marker CaspACE FITC-VAD-FMK, and DNA fragmentation was detected by TUNEL staining. (B) Bim mRNA levels were quantified by qPCR. CGNs treated with H2O2 (37.5 or 50 μM) showed significant up-regulation of bim (*p < 0.05; n = 3). (C) Western blot analysis revealed a significant increase in Bim protein levels 4–6 h after H2O2 (37.5 or 50 μM) addition (*p < 0.05; n = 5). This up-regulation correlated with a decrease of pAKT (Ser-473), pFOXO3 (Thr-32), and Hsp25 (rodent homologue of Hsp27) levels. β-Actin served as loading control. (D) CGNs were transfected with a vector containing a 0.8-kb fragment of the bim promoter. Bim promoter activation was significantly increased after H2O2 (37.5 μM) addition (*p < 0.05; n = 3). Mutations of the FOXO3-binding site significantly reduced this activation (*p < 0.05; n = 3). (E) CGNs from bim−/− mice and wild-type (WT) controls were treated with H2O2 (37.5 μM) or sham conditions. At 4–6 h posttreatment, the neurons were stained live with Hoechst. Bar, 2.5 μm. (F) Pyknotic nuclei were scored (*p < 0.05; n = 3).
Four hours after H2O2 addition, we detected a significant up-regulation of the proapoptotic BH3-only protein Bim at both mRNA and protein levels (Figure 1, B and C). These results correlated with the activation of the bim promoter. Thus neurons transfected with a luciferase reporter plasmid bearing the bim promoter sequence showed higher luciferase activity after H2O2 addition (37.5 μM) compared with control neurons treated with vehicle (Figure 1D). Mutation of the FOXO3-binding sites on the bim promoter prevented its activation (Figure 1D). Moreover, we also found that H2O2 addition decreased activation of the kinase AKT, as assessed by Western blot analysis of pAKT (Ser-473) levels, and also the phosphorylation of the AKT target, FOXO3 (Thr-32) (Figure 1C). This step is necessary to allow FOXO3 nuclear accumulation and transcriptional activity (Brunet et al., 1999). Parallel to Bim up-regulation, we also detected a decrease of the total levels of Hsp25 (rodent homologue of Hsp27) induced by H2O2 treatment (Figure 1C).
Finally, to determine whether Bim has a causal role in H2O2-induced cell death, we compared CGN survival between bim−/− and wild-type (wt) control cultures treated with H2O2 (37.5 μM). Bim-deficient neurons showed resistance to H2O2, displaying a lower percentage of cells with nuclear pyknosis compared with wt neurons after treatment (Figure 1, E and F).
Hsp27 regulates Bim protein levels and oxidative stress–induced cell death
Having observed the down-regulation of Hsp25 levels induced by H2O2 treatment, we analyzed its possible relation with the expression of Bim. For this purpose, we transfected CGNs with a construct expressing the wild-type form of the human Hsp27 (pNEO-Hsp27). Despite the fact that we also observed a decrease in levels of Hsp27 in H2O2-treated CGNs, transfection of Hsp27 prevented the up-regulation of Bim protein levels (Figure 2A) and subsequent apoptotic events such as cytosolic accumulation of cytochrome C (Figure 2B) or accumulation of the active caspase 3 subunit (Supplemental Figure S1A). Hsp27 transfection in CGNs also prevented the nuclear pyknosis induced by H2O2 treatment (Figure 2C). On the other hand, depletion of the endogenous Hsp25 levels in CGNs by expression of a specific small interfering RNA (siRNA) targeting Hsp25 (200 pmol) was able to mimic the effect of H2O2 treatment, up-regulating significantly Bim protein levels and inducing nuclear pyknosis (Figure 2, D and E).
FIGURE 2:

Hsp27 regulates Bim protein levels and oxidative stress–induced cell death. (A) CGNs were transfected with pNEO-Hsp27 or a control construct before H2O2 (37.5 μM) addition. pNEO-Hsp27 neurons displayed significantly lower Bim protein levels than did control neurons after H2O2 treatment (*p < 0.05; n = 3). β-Actin served as loading control. (B) At 4 h after H2O2 (37.5 μM) treatment, pNEO-Hsp27 neurons displayed lower cytosolic cytochrome C protein levels than in control neurons. Mitochondrial membrane levels of cytochrome C were also higher in pNEO-Hsp27 neurons than in control neurons. IκB-α protein served as marker of cytosolic fraction and LAMP1 as marker of the membrane fraction, including mitochondrial membranes. (C) CGNs transfected with pNEO-Hsp27 or a control construct were treated with H2O2 (37.5 μM) or sham conditions and live-stained with Hoechst 4–6 h later. pNEO-Hsp27 neurons showed a significantly lower percentage of cells with pyknotic nuclei than control neurons after H2O2 addition (*p < 0.05; n = 3). Bar, 2.5 μm. (D) CGNs were electroporated with Hsp25 siRNA (rodent homologue of Hsp27; 100/200 pmol) or control siRNA (200 pmol), and Bim and Hsp25 protein levels were analyzed after 72 h by Western blot. Hsp25 levels were depleted in Hsp25 siRNA (200 pmol) neurons, which also displayed significantly higher levels of Bim than control siRNA neurons (*p < 0.05; n = 3). β-Actin served as loading control. (E) CGNs electroporated with Hsp25 siRNA or control siRNA were live-stained with Hoechst. Hsp25 siRNA neurons presented a significantly higher percentage of cells with pyknotic nuclei than control neurons (*p < 0.05; n = 3). Bar, 2.5 μm.
Hsp27 effects depend on a posttranscriptional mechanism
We next sought to determine the mechanism used by Hsp27 to prevent Bim induction during oxidative stress–induced cell death. First, we analyzed a possible effect of Hsp27 on bim promoter activation by the AKT/FOXO3 pathway. Overexpression of the pNEO-Hsp27 construct in CGNs did not prevent the down-regulation of pAKT (Ser-473) and pFOXO3 (Thr-32) levels induced by H2O2 (37.5 μM) treatment (Figure 3A). We also examined the JNK/AP1 signaling pathway, which is activated by oxidative stress and involved in bim promoter activation (Torres and Forman, 2003; Biswas et al., 2007). Treatment of CGNs with H2O2 (37.5 μM) increased JNK activation, as detected by the up-regulation of pJNK (Thr-183/Tyr-185) levels (Figure 3A). However, overexpression of the pNEO-Hsp27 did not prevent this effect. Finally, we cotransfected CGNs with the luciferase reporter plasmid bearing the bim promoter sequence and the pNEO-Hsp27 construct or alternatively its control vector. Hsp27 overexpression did not prevent the up-regulation of the bim promoter after H2O2 addition (Figure 3B). These results indicate that the effect of Hsp27 on Bim expression does not depend on the regulation of its promoter.
FIGURE 3:

Hsp27 effects depend on a posttranscriptional mechanism. (A) CGNs were transfected with pNEO-Hsp27 or a control construct before H2O2 (37.5 μM) addition. Western blot analysis showed that H2O2 treatment decreased pAKT (Ser-473) and pFOXO3 (Thr-32) levels and increased pJNK (Thr-183/Tyr-185) levels both in pNEO-Hsp27 neurons and in control neurons. β-Actin served as loading control. (B) CGNs were cotransfected with the luciferase reporter containing the bim promoter sequence and either pNEO-Hsp27 or a control construct. Bim promoter activation was significantly increased after H2O2 (37.5 μM) addition both in pNEO-Hsp27 neurons and in control neurons (*p < 0.05; n = 3). (C) CGNs were pretreated with the proteasome inhibitor epoxomicin (50 nM) or vehicle 45 min before H2O2 (37.5/50 μM) addition. Western blot analysis revealed that H2O2 treatment down-regulated Hsp25 protein level (*p < 0.05; n = 3) and up-regulated Bim levels. Epoxomicin pretreatment prevented Hsp25 down-regulation and increased Bim protein levels induced by H2O2. β-Actin served as loading control. (D) CGNs were transfected with pNEO-Hsp27 or a control construct and pretreated with epoxomicin (50 nM) or vehicle 45 min before H2O2 (37.5 μM) addition. pNEO-Hsp27 neurons presented lower Bim protein levels after H2O2 addition than control neurons in the presence of epoxomicin or vehicle (*p < 0.05; n = 3). β-Actin served as loading control. (E) CGNs were transfected with pNEO-Hsp27 or a control construct before H2O2 (37.5 μM) addition. qPCR analysis showed that H2O2 treatment up-regulated bim mRNA levels (*p < 0.05; n = 3). pNEO-Hsp27 neurons treated with H2O2 showed significant higher levels of bim mRNA than H2O2-treated control neurons (*p < 0.05; n = 3). (F) ATF4 mRNA levels were also quantified by qPCR. H2O2 (37.5 μM) addition increased ATF4 mRNA levels (*p < 0.05; n = 3); however pNEO-Hsp27 transfection did not have any significant effect on these levels.
Bim protein levels are tightly controlled by ubiquitin-dependent proteasomal degradation. Pretreatment of CGNs with an inhibitor of the proteasome, epoxomicin (50 nM), 45 min before H2O2 addition prevented down-regulation of Hsp25 and increased Bim protein levels. In addition, we also observed an increase in phosphorylation of Bim on Ser-69, a necessary step for its proteasomal degradation (Luciano et al., 2003). These results suggest the partial degradation of both proteins at the proteasome during oxidative stress (Figure 3C). Considering this, we examined whether enhanced levels of Hsp27 could stimulate Bim protein degradation during oxidative stress. To this end, we pretreated CGNs transfected with pNEO-Hsp27 or its control vector with epoxomicin and 45 min later exposed them to H2O2 (37.5 μM) or vehicle. Epoxomicin induced a slight increase in Bim protein levels in all the experimental groups; however, this was not statistically significant. Epoxomicin was also not able to prevent the significant decrease in Bim proteins levels induced in H2O2-treated cells by pNEO-Hsp27 overexpression (Figure 3D). These results indicate that Hsp27 modulates Bim expression during oxidative stress by a mechanism independent of its proteasomal degradation.
Finally, we analyzed by quantitative real-time reverse transcription (RT)-PCR analysis (qPCR) the effect of Hsp27 on bim mRNA levels during oxidative stress. CGNs transfected with a control vector showed up-regulation of bim mRNA levels after H2O2 (37.5 μM) treatment. This up-regulation was significantly higher in CGNs treated with H2O2 but transfected with the pNEO-Hsp27 construct (Figure 3E). To determine whether this effect was specific for the bim mRNA transcript or was generic for every mRNA transcript, we also quantified the mRNA levels of ATF4, an oxidative stress–inducible transcription factor in neurons (Lange et al., 2008). H2O2 (37.5 μM) treatment also up-regulated ATF4 mRNA levels in CGNs; however, in this case, pNEO-Hsp27 overexpression did not affect ATF4 mRNA levels (Figure 3F). These results suggest that during oxidative stress, Hsp27 overexpression specifically represses bim mRNA translation.
Hsp27 regulates the bim-3′UTR during oxidative stress
To determine whether Hsp27 was able to inhibit bim mRNA translation, we analyzed the amount of bim RNA present at the polysomes (the ribonucleoprotein particles where translation occurs) using the translating ribosome affinity purification (TRAP) methodology (Heiman et al., 2008; Kapeli and Yeo, 2012). Specifically, we determined by coimmunoprecipitation and subsequent qPCR analysis the levels of bim mRNA associated with the ribosomal protein RPL10a present at the polysome. In CGNs treated with H2O2 (37.5 μM), overexpression of Hsp27 significantly reduced the levels of bim mRNA associated with RPL10a and therefore present at the polysome (Figure 4A).
FIGURE 4:

Hsp27 regulates bim-3′UTR sequence during oxidative stress. (A) Quantification of bim mRNA levels bound to the ribosomal protein RPL10a present at the polysome. CGNs were treated with H2O2 (37.5 μM). Hsp27 overexpression reduced bim mRNA levels coimmunoprecipitated with RPL10a when compared with controls (*p < 0.05; n = 3). (B) CGNs were cotransfected with a luciferase reporter plasmid containing the bim-3′UTR (divided into two sequences, named 1 and 2) and either pNEO-Hsp27 or a control construct. Bim-3′UTR (sequence 1) neurons showed a significant increase in the luciferase activity after H2O2 (37.5 μM) addition (*p < 0.05; n = 3). Coexpression of pNEO-Hsp27 prevented this effect, significantly reducing the luciferase activity (*p < 0.05; n = 3). (C) Bim-3′UTR (sequence 2) neurons did not show any modification of the luciferase activity induced by H2O2 treatment or pNEO-Hsp27 coexpression. (D) CGNs were coelectroporated with the luciferase reporter plasmid containing the bim-3′UTR (sequence 1) and either Hsp25 siRNA or control siRNA. Hsp25 siRNA neurons displayed significantly higher luciferase activity than control siRNA neurons (*p < 0.05; n = 3). (E) Quantification of bim mRNA levels bound to the endogenous Hsp25 protein and to the wild-type form of the human Hsp27 (pNEO-Hsp27 construct) overexpressed in CGNs. H2O2 (37.5 μM) treatment significantly increased bim mRNA levels coimmunoprecipitated with the Hsp25/Hsp27 compared to control neurons (*p < 0.05; n = 4). The coimmunoprecipitated bim mRNA levels were significantly increased in H2O2-treated cells when Hsp27 was overexpressed (*p < 0.05; n = 4). As positive and negative controls for the immunoprecipitation, a specific antibody for Argonaute-2 protein and an unspecific serum were used, respectively. Argonaute-2 levels were detected by Western blot. bim mRNA levels were not detected in the negative control.
Next we analyzed whether Hsp27 was able to regulate bim mRNA translation via its 3′UTR. This region contains microRNA sites and cis-acting sequences within AU-rich sequence elements (AREs), which can bind RBPs. These elements are critical for the regulation of bim mRNA stability and/or translation (Matsui et al., 2007; Terasawa et al., 2009). To this end, this region was divided into two overlapping sequences (named here 1 and 2), which were cloned downstream of a luciferase gene in a chimeric reporter plasmid whose expression was controlled by a constitutively active promoter. Therefore the luciferase activity of these reporter plasmids could only be posttranscriptionally regulated by the modulation of the bim-3′UTR. Exposure of CGNs to H2O2 (37.5 μM) resulted in up-regulation of the luciferase activity of the reporter plasmid containing sequence 1 (Figure 4B). This effect was abrogated by coexpression of the pNEO-Hsp27 construct (Figure 4B). As a control for the specificity of this effect, we observed that Hsp27 coexpression did not have an effect on the luciferase activity of the reporter plasmid containing sequence 2 (Figure 4C). Next we demonstrated that depletion of endogenous Hsp25 levels in CGNs by expression of a specific siRNA targeting Hsp25 was able to mimic the effect of H2O2 treatment, up-regulating the luciferase activity of the reporter plasmid containing sequence 1 (Figure 4D). These results indicated that enhanced levels of Hsp27 during oxidative stress was able to repress bim mRNA translation by a mechanism dependent on the bim-3′UTR.
Finally, we analyzed whether the effect of Hsp27 on bim mRNA translation depended on the regulation of other RBPs or was a direct effect, since several members of the heat-shock protein family, including Hsp27, show affinity for AREs and can act as RBPs (Henics et al., 1999; Sinsimer et al., 2008). To this end, we immunoprecipitated the endogenous Hsp25 protein and the wild-type form of the human Hsp27 (pNEO-Hsp27 construct) overexpressed in CGNs and quantified by qPCR the levels of bound bim mRNA. CGNs treatment with H2O2 (37.5 μM) significantly increased bim mRNA levels that were coimmunoprecipitated with endogenous Hsp25 when compared with control neurons (Figure 4E). The coimmunoprecipitated bim mRNA levels were significantly increased in H2O2-treated cells when Hsp27 was overexpressed (Figure 4E). These results demonstrated binding of Hsp25/27 to bim mRNA during oxidative stress and suggest that overexpression of Hsp27 stimulates this binding.
Hsp27 also regulates the bim-3′UTR during excitotoxicity
Previous results from our group showed that excitotoxic apoptosis in two different types of neurons—CGNs and cortical neurons—required Bim induction (Concannon et al., 2010). We therefore explored whether the described regulation of the bim-3′UTR sequence by Hsp27 was also evident during excitotoxic cell death. First, we observed that overexpression of the pNEO-Hsp27 construct was able to prevent Bim protein up-regulation in CGNs exposed to glutamate (100 μM)/glycine (10 μM; Figure 5A). Next we explored whether Hsp27 could also modify the luciferase activity of the reporter plasmid containing the bim-3′UTR (sequence 1) during excitotoxicity. CGNs exposed to glutamate (100 μM)/glycine (10 μM) showed a decrease of the luciferase activity compared with control CGNs. This effect was in contrast to the increase in luciferase activity observed in H2O2-treated cells, suggesting that additional mechanisms regulate bim-3′UTR during excitotoxicity. However, the decrease was significantly enhanced by overexpression of Hsp27 (Figure 5B). This effect was also observed in cortical neurons exposed to N-methyl-d-aspartate (NMDA; 100 μM)/glycine (10 μM), where Hsp27 overexpression decreased luciferase activity (Figure 5C). These results demonstrate that Hsp27 overexpression also represses bim mRNA translation during excitotoxicity.
FIGURE 5:
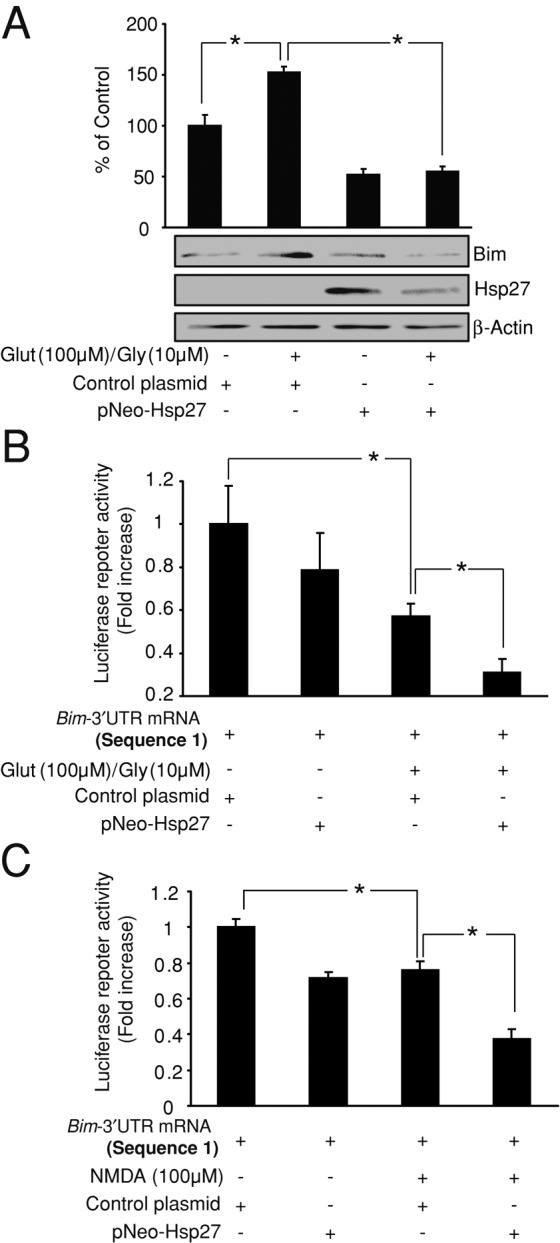
Hsp27 also regulates bim-3′UTR sequence during excitotoxicity. (A) CGNs were transfected with pNEO-Hsp27 or a control construct before glutamate (100 μM)/glycine (10 μM) exposure. pNEO-Hsp27–transfected neurons exhibited significantly lower Bim protein levels than control neurons after H2O2 treatment (*p < 0.05; n = 3). β-Actin served as loading control. (B) CGNs were cotransfected with a luciferase reporter plasmid containing the bim-3′UTR (sequence 1) and either pNEO-Hsp27 or a control construct. Glutamate/glycine (100/10 μM) exposure decreased the luciferase activity in control neurons (*p < 0.05; n = 3), whereas expression of the pNEO-Hsp27 construct significantly enhanced this effect (*p < 0.05; n = 3). (C) Cortical neurons were cotransfected with a luciferase reporter plasmid containing the bim-3′UTR (sequences 1) and either pNEO-Hsp27 or a control construct. NMDA (100 μM)/glycine (10 μM) exposure decreased the luciferase activity in control neurons (*p < 0.05; n = 3), and expression of the pNEO-Hsp27 construct significantly enhanced this effect (*p < 0.05; n = 3).
DISCUSSION
The present study describes the regulation of the proapoptotic BH3-only protein Bim by Hsp27 in neurons undergoing oxidative stress. In an in vitro model, we observed that H2O2 addition to CGNs induced the up-regulation of bim mRNA and protein levels, which was necessary for CGN cell death. In parallel, we also detected a down-regulation of Hsp25 levels induced by H2O2. We report that enhancement of Hsp27 levels during oxidative stress is able to prevent Bim protein up-regulation and downstream events such as cytochrome C release, caspase-3 activation, and neuronal death despite the accumulation of bim mRNA. Finally, we show that the main mechanism that mediates the effect of Hsp27 is regulation of the 3′UTR of bim mRNA, repressing its translation. Together these studies reveal a novel posttranscriptional mechanism controlling oxidative stress–induced neuronal death and describe a novel cytoprotective mechanism of Hsp27.
Protein levels of endogenous Hsp25 were down-regulated in CGNs treated with H2O2, in spite of its described stress-dependent expression (Xanthoudakis and Nicholson, 2000; Lanneau et al., 2008). Hsp25/27 is able to act as a chaperone of oxidized proteins, facilitating their ubiquitination and proteasomal degradation (Arrigo, 2001, 2007), and this function could be related to its down-regulation after H2O2 treatment. Supporting this concept, it has been described that Hsp27 can be ubiquitinated and degraded (Sun et al., 2011). Indeed, our results show that pretreatment with the proteasome inhibitor epoxomicin abrogated H2O2-induced down-regulation of Hsp25. Both H2O2 treatment and transfection with a siRNA-Hsp25 up-regulated Bim protein levels and induced neuronal apoptosis, which was prevented in bim−/− CGNs. On the other hand, Hsp27 overexpression prevented Bim protein expression and subsequent cytochrome C release, caspase-3 activation, and neuronal death. These results indicate that Hsp25/27 down-regulation could be a key process to allow Bim expression and neuronal apoptosis during oxidative stress conditions, confirming previous studies that correlate Hsp27 levels with cell survival/death during diverse stress conditions (Wagstaff et al., 1999; Stetler et al., 2008; King et al., 2009; Gibert et al., 2012). Expression of another BH3-only protein, PUMA, has been described as necessary for oxidative stress–induced apoptosis in cortical neurons (Steckley et al., 2007). However, puma-deficient CGNs were not protected against H2O2 in our experimental setting (Supplemental Figure S1B), suggesting a cell type– and stimulus-dependent role of BH3-only proteins.
Apoptosis is a terminal cellular decision, and the proteins that control its activation, including Bim, require multiple levels of regulation. We observed that epoxomicin pretreatment significantly increased Bim protein levels and its phosphorylation (Ser-69) in H2O2-treated CGNs but did not prevent the effect of Hsp27 overexpression on Bim protein levels. These results confirm the regulation of Bim by proteasomal degradation (Luciano et al., 2003) but also indicate a proteasome-independent effect of Hsp27. Hsp27 overexpression in H2O2-treated cells altered neither bim promoter activation nor the signaling pathways involved. However, Hsp27 overexpression increased bim mRNA levels without modifying bim promoter activity. This stabilization and up-regulation of bim mRNA levels could be associated with the repression of mRNA translation (Kawai et al., 2004). This hypothesis was confirmed by determining bim mRNA levels at the polysome using TRAP methodology. Epoxomicin also increased Hsp25 protein levels in H2O2-treated cells, but in contrast to Hsp27 overexpression, this increase did not affect Bim protein levels. These results can be explained by Hsp25 proteosomal degradation occurring after bim mRNA translation or nondegraded Hsp25 protein being trapped at the proteasome. On the contrary, Hsp27 overexpression before oxidative stress and bim expression would still exert an effect on Bim translation. Finally, Hsp27 overexpression did not affect other stress response genes, such as ATF4, also up-regulated during oxidative stress (Lange et al., 2008). This result excludes general inhibition of translation as a main mechanism of the effects observed (Cuesta et al., 2000) but instead indicates specific repression of the bim mRNA translation by Hsp27.
Our reporter assays revealed the involvement of the bim-3′UTR in the repression of bim mRNA translation by Hsp25/27. Hsp25 depletion induced by H2O2 treatment or siRNA-mediated Hsp25 knockdown allowed the translation of a luciferase reporter plasmid bearing half of the bim-3′UTR (sequence 1), whereas Hsp27 overexpression in H2O2-treated cells prevented this translation. Hsp27 overexpression also increased the levels of bim mRNA coimmunoprecipitated in H2O2-treated cells, indicating that oxidative stress stimulates Hsp27 binding to the bim-3′UTR. In agreement with this concept, previous studies showed that oxidative stress stimulates the antiapoptotic activity of Hsp27 (Huot et al., 1996). Furthermore, these results also confirm Hsp25/27 as a RBP that binds to 3′UTRs (Sinsimer et al., 2008). The binding of Hsp27 to the 3′UTR of bim could be a result of the affinity of Hsp27 for AREs (Sinsimer et al., 2008), which stimulate mRNA degradation but also stabilization and translational repression (Mukhopadhyay et al., 2003; Barreau et al., 2005). A small sequence of 1 kb that holds phylogenetically conserved AREs (able to bind other heat shock proteins; Matsui et al., 2007) is present in the reporter plasmid bearing the first half of the bim-3′UTR. The presence of these AREs could explain the effect of Hsp25/27 on the reporter plasmid bearing bim-3′UTR (sequence 1) but not on the reporter that contains the second half of the bim-3′UTR (sequence 2). However, multiple AREs are present along the bim-3′UTR, and additional studies are required to map specific interactions with Hsp25/27.
Our results demonstrate how Hsp25/27 levels during oxidative stress situations are crucial in regulating bim mRNA translation and neuronal apoptosis and also underscore the existence of multiple control points between transcription and protein production in the neuroprotective mechanisms of Hsps. The observed bim-mediated, antiapoptotic mechanism would occur before the irreversible step of cytochrome C release and thus may explain the known upstream role of Hsp27 in regulating the mitochondrial pathway of apoptosis (Paul et al., 2002; Havasi et al., 2008). Other mechanisms by which Hsp27 prevents neuronal apoptosis have also been identified, including direct inhibition of apoptosis signal–regulating kinase 1 (Stetler et al., 2008).
Hsp27 overexpression also prevented bim mRNA translation and Bim protein up-regulation during excitotoxic injury in cortical and cerebellar granule neurons. These results suggest the possible importance of Bim regulation by Hsp27 as a broad neuroprotective mechanism, since oxidative stress and excitotoxicity are implicated in multiple neurologic insults and neurodegenerative disorders. Of note, transient and sublethal ischemia (preconditioning) is able to up-regulate Hsps without inducing neuronal death, contributing to tolerance against ischemic brain injury (Liu et al., 2009). The described mechanism by which enhanced Hsp27 levels represses bim mRNA translation potentially expands the number and type of posttranscriptional mechanisms by which ischemic preconditioning reduces Bim protein levels and protects against neuronal death (Meller et al., 2006).
In conclusion, our study describes a new role for Hsp27 levels in the posttranscriptional regulation of Bim during neuronal death induced by oxidative stress (summarized in Figure 6). This involves repression of bim translation by Hsp27 binding to the bim-3′UTR. This mechanism contributes a novel mechanism explaining the neuroprotective activity of Hsp27 and may provide novel therapeutic approaches to prevent neuronal death in diverse neurodegenerative conditions.
FIGURE 6:

Hsp27 posttranscriptionally regulates Bim levels during neuronal death induced by oxidative stress. H2O2 treatment induces Bim transcription and neuronal death by the modulation of redox-sensitive pathways such as AKT and JNKs, which leads to the activation of transcription factors that regulate the bim promoter such as FOXO3. H2O2 also reduces the levels of the antiapoptotic protein Hsp27. Up-regulation of Hsp27 levels during oxidative stress does not affect bim mRNA transcription or processing; however, it represses its translation by modulation of its 3′UTR. This mechanism contributes a novel mechanism for the neuroprotective activity of Hsp27.
MATERIALS AND METHODS
Materials
Fetal calf serum and MEM were obtained from Invitrogen (Bio Sciences, Dublin, Ireland). Hydrogen peroxide, glutamate, glycine, Hoechst 33258, and propidium iodide (PI) were from Fluka (Sigma-Aldrich, Arklow, Ireland). Epoxomicin was obtained from Calbiochem (Merck Biosciences, Nottingham, United Kingdom). FITC-VAD-FMK was obtained from Promega (Medical Supply, Dublin, Ireland).
Preparation of primary neuron cultures
CGNs were isolated from postnatal day 7 rat (Sprague Dawley) or mouse (C57BL/6) pups as described previously (Ward et al., 2000). CGNs were plated on poly-l-lysine–coated glass coverslips, glass Willco dishes, and six- and 24-well plates at 1 × 106 cells/ml and maintained at 37°C in a humidified atmosphere of 5% CO2/95% air. CGNs were used for oxidative stress experiments after 6–7 d in culture. Oxidative stress was induced by H2O2 treatment (final concentration 25–50 μM). H2O2 is an endogenously produced ROS, and previous evidence showed that its exogenous H2O2 administration is a model of oxidative stress–induced neuronal death (Davila and Torres-Aleman, 2008). For excitotoxicity experiments, CGNs were used after 9 d in culture and treated for 10 min with glutamate (100 μM)/glycine (10 μM) in experimental buffer (120 mM NaCl, 3.5 mM KCl, 0.4 mM KH2PO4, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 5 mM NaHCO3, 1.2 mM Na2SO4, 1.2 mM CaCl2, 1.2 mM MgCl2, and 15 mM glucose; pH 7.4). As a control, cells were treated with experimental buffer only. Mouse neocortical neurons were used for specific excitotoxicity experiments as outlined in Results and isolated from embryonic day 16 pups as described previously (Concannon et al., 2010). They were used after 9 d in culture and treated 5 min with NMDA (100 μM)/glycine (10 μM) in experimental buffer. All animal work was performed with ethical approval from the Royal College of Surgeons in Ireland under licenses granted to the authors by the Irish Department of Health and Children.
Plasmids, siRNA, and transfection
The firefly luciferase reporter plasmid containing 0.8 kb of the bim promoter sequence along with similar constructs with mutated FOXO3-binding sites were kindly provided by Eric Lam (Imperial College London, London, United Kingdom). The pmax–green fluorescent protein (GFP) construct was obtained from Lonza (Basel, Switzerland). The pRL-TK (TK-Renilla luciferase) was a kind gift of B. M. Burgering (University Medical Center, Utrecht, Netherlands). pNeo-Hsp27 and pNeo-Control were kindly provided by Caoimhin Concannon (Royal College of Surgeons in Ireland, Dublin, Ireland). The bim-3′UTR mRNA sequence was divided into two different luciferase reporter plasmids (termed here 1 and 2). These constructs were obtained from GeneCopoeia (LabOmics, Nivelles, Belgium). The siRNAs targeting the homologue of Hsp27 in rat (Hsp25) and the control sequence were obtained from Sigma-Aldrich. For luciferase assays, CGNs were transfected using the calcium phosphate technique at 7 d in vitro (Weisova et al., 2009). For coimmunoprecipitation and siRNA transfection experiments, CGNs were electroporated before plating using an electroporator (Rat Neuron Nucleofector Kit; Lonza) and treated 5 d after electroporation. Mouse neocortical neurons (Figure 5C) were transfected at 5 d in vitro using Lipofectamine 2000 (Invitrogen) as per manufacturer's instructions.
Western blotting
Western blotting was performed as described (Weisova et al., 2009). Neurons were removed from the plates by washing once with ice-cold phosphate-buffered saline (PBS) and lysed with PIK buffer (1% NP-40, 150 mM NaCl, 20 mM Tris, pH 7.4, 10% glycerol, 1 mM Cl2Ca, 1 mM Cl2Mg, 400 μM sodium vanadate, 0.2 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 0.1% phosphatase inhibitor cocktails I and II [Sigma-Aldrich]). Blots were probed with rabbit polyclonal antibodies to Bim (H-191; 1:500) and IκB-α (C-21; 1:2000) from Santa Cruz Biotechnology (Dublin, Ireland); phospho (Thr-183/Thr-185) JNK (1:1000), total JNK (1:2000), phospho (Ser-473) AKT (1:2000), total AKT (1:2000), phospho (Thr-32) FOXO3 (1:1000), and cleaved caspase-3 (Asp-175; 1:1000), all from Cell Signaling Technology (Danvers, MA); total FOXO3 (1:2000) from Millipore (Carrigtwohill, Ireland); Hsp25 (1:3000) from Enzo Life Science (Exeter, United Kingdom); and Argonaute-2 (1:1000) and LAMP1 (1:1000) from Abcam (Cambridge, United Kingdom). Blots were probed also with mouse monoclonal antibodies to Hsp27 (1:3000) from Enzo Life Science, β-actin (clone DM 1A; 1:10000) from Sigma-Aldrich, cytochrome C (A-8; 1:2000) and RPL10a (JK-16; 1:1000) from Santa Cruz Biotechnology, and Neu-N (clone A60; 1:2000) from Millipore. Horseradish peroxidase–conjugated secondary antibodies (1:10,000) from Thermo Scientific (Fisher, Dublin, Ireland) were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and imaged using a FujiFilm LAS-3000 imaging system (FujiFilm, Dublin, Ireland). To normalize for protein load, membranes were reblotted (Re-Blot; Millipore) and incubated with an appropriate control antibody. Levels of the protein under study were expressed relative to protein load in each lane as determined by appropriate control protein content. Different exposures of each blot were collected to ensure linearity and to match control levels for quantification. Densitometric analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD). A representative blot is shown from a total of at least three independent experiments except when indicated.
Cytochrome C release detection
CGNs cultured on six-well plates were lysated 4 h after H2O2 (37.5μM) or vehicle treatment in plasma membrane permeabilization buffer (50 μg/ml digitonin, 80 mM KCl in PBS), and cytochrome C mitochondrial release was detected as described previously (Waterhouse et al., 2004). IκB-α protein was used as marker of the cytosolic fraction, and LAMP1 protein was used as marker of the membrane fraction, including mitochondrial membranes.
Hoechst staining of nuclear chromatin
Neurons cultured on 24-well plates were stained live with Hoechst 33258 and PI at a final concentration of 0.1 μg/ml, respectively. After incubation for 20 min, nuclear morphology was observed in living cells (PI−) using an Eclipse TE 300 inverted microscope (Nikon, Amstelveen, Netherlands) and a 20× dry objective. For each treatment, cells were analyzed for apoptotic morphology in three subfields of each culture. Condensed nuclei were scored as pyknotic nuclei and expressed as a percentage of the total population. All experiments were performed in triplicate dishes in at least three independent experiments. Neurons transfected with pNeo-Hsp27 or siRNA-Hsp27 were also cotransfected with a pMax-GFP construct in a 5:1 ratio, with the apoptotic morphology analyzed only in GFP+ cells.
Terminal deoxynucleotide transferase–mediated dUTP nick-end labeling and caspase activation
Terminal deoxynucleotide transferase–mediated dUTP nick-end labeling (TUNEL) of CGN cultures was performed using the Dead End Fluorometric TUNEL kit (Promega) as per manufacturer's instructions. Detection of activated caspases in CGN cultures was performed with an in situ fluorescent marker, CaspACE FITC-VAD-FMK (Promega), as per manufacturer's instructions. Photographs were taken using an Eclipse TE 300 inverted microscope (Nikon) and a 20× dry objective.
Luciferase assays
Neurons were transfected with a reporter construct in which the expression of the luciferase firefly was regulated by the wild-type bim promoter or promoter with mutated FOXO3-binding sites (see Plasmids, siRNA, and transfection). In other experiments, neurons were transfected with a reporter plasmid in which the bim-3′UTR mRNA sequence was cloned downstream of the luciferase gene, whose expression is controlled by a constitutively active promoter. For luciferase assays, cells were lysed in passive lysis buffer, and luciferase activity was analyzed using a luminometer and dual luciferase assay kit, according to the manufacturer (Promega). Transfections were performed in triplicate dishes, and luciferase counts (RLUs) were normalized using coexpression of Renilla luciferase. Background luminescence was subtracted, and luciferase activity was expressed as fold increase with respect to the control.
Quantitative real-time RT-PCR analysis
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, West Sussex, United Kingdom). First-strand cDNA synthesis was performed using 2 μg of total RNA as template and reverse transcribed using Superscript II (Invitrogen) primed with 50 pmol of random hexamers. Real-time qPCR was performed using the Light-Cycler 2.0 (Roche, West Sussex, United Kingdom) and the QuantiTech SYBR green PCR kit (Qiagen). Sense and antisense primers were as follows: GCAGTCTCAGGAGGAACCTG and AGTGCCTTCTCCAGACCAGA for Bim; AGAATGGCTGGCTATGGATG and GCCAATTGGGTTCACTGTCT for ATF-4, and GGGAAATCGTGCGTGACATT and TGCCACAGGATTCCATACCC for β-actin. The data were analyzed using the LightCycler software 4.0 (Roche), with all samples normalized to β-actin.
RNA coimmunoprecipitation
Neurons cultured on Petri dishes (100 mm) were transfected or electroporated with the plasmid pNeo-Hsp27 or its control vector and 3 or 4 d later treated with H2O2 (30–37.5 μM). After 4 h, cells were homogenized in buffer A (50 mM Tris HCl, pH 7.4, 300 mM NaCl, 5 mM MgCl2, 0.1% NP-40, and protease and recombinant RNase inhibitors) for Hsp27/Hsp25 immunoprecipitation or buffer B (10 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl2, 1% NP-40, 0.5 mM dithiothreitol, 100 mg/ml cycloheximide, and protease and recombinant RNase inhibitors) for RPL10a immunoprecipitation and specific buffer for the polysome extraction (Heiman et al., 2008). In both cases, lysates were centrifuged, and 400 μl of the supernatants was incubated overnight at 4°C with 5 μg of the corresponding antibodies against Hsp27/Hsp25 (both from Enzo Life Science) or RPL10. Supernatants were also immunoprecipitated with Argonaute-2 or an unspecific serum as positive and negative controls for the immunoprecipitation, respectively. Protein A agarose beads (Santa Cruz Biotechnology) were added, mixed, and incubated for 1 h at 4°C and then centrifuged, and the supernatant was removed. The pellet was washed and processed with TRIzol (Invitrogen) for RNA and protein extraction. First-strand c-DNA synthesis was performed using 2 μg of the RNA, and real-time qPCR was performed using bim primers (as described previously). bim mRNA levels were expressed as fold increase with respect to the control. bim mRNA levels were below detection levels in the control experiments.
Gene-targeted mice
The generation and genotyping of puma−/− and bim−/− mice were previously described. (Bouillet et al., 1999; Villunger et al., 2003). The puma−/− mice were generated on an inbred C57BL/6 background, using C57BL/6-derived embryonic stem cells. The bim−/− mice were originally generated on a mixed C57BL/6 × 129SV genetic background, using 129SV-derived embryonic stem cells, but had been backcrossed for >12 generations onto the C57BL/6 background. Wild-type animals of the same genetic background (C57BL/6) were used in this study
Statistical analysis
Data are expressed as mean ± SE. Differences among groups were analyzed by one-way analysis of variance followed by Tukey test. Comparison between two groups was performed with the t test. p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This study was supported by a Marie Curie Intra-European Fellowship (PIEF-GA-2009-237765) and the Science Foundation Ireland (08/IN1/B1949; 08/IN1/B1875; 12/COEN/18). We thank Ina Woods and Sarah Cannon for excellent technical assistance and Andreas Strasser (Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia) for bim- and puma-deficient mice.
Abbreviations used:
- ARE
AU-rich sequence element
- BH
Bcl-2 homology domain
- BH3-only proteins
BH 3–containing proteins
- CGN
cerebellar granule neuron
- Hsp27
heat shock protein 27
- RBP
RNA-binding protein
- ROS
reactive oxygen species
- TRAP
translating ribosome affinity purification
- RLU
relative light unit
- UTR
untranslated region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www .molbiolcell.org/cgi/doi/10.1091/mbc.E13-08-0495) on September 3, 2014.
REFERENCES
- Arrigo AP. Hsp27: novel regulator of intracellular redox state. IUBMB Life. 2001;52:303–307. doi: 10.1080/152165401317291156. [DOI] [PubMed] [Google Scholar]
- Arrigo AP. The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv Exp Med Biol. 2007;594:14–26. doi: 10.1007/978-0-387-39975-1_2. [DOI] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles. Nucleic Acid Res. 2005;33:7130–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron. 2002;36:45–56. doi: 10.1016/s0896-6273(02)00941-8. [DOI] [PubMed] [Google Scholar]
- Biswas SC, Shi Y, Sproul A, Greene LA. Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J Biol Chem. 2007;282:29368–29374. doi: 10.1074/jbc.M702634200. [DOI] [PubMed] [Google Scholar]
- Bolognani F, Perrone-Bizzozero NI. RNA-protein interactions and control of mRNA stability in neurons. J Neurosci Res. 2008;86:481–489. doi: 10.1002/jnr.21473. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Butts BD, Hudson HR, Linseman DA, Le SS, Ryan KR, Bouchard RJ, Heidenreich KA. Proteasome inhibition elicits a biphasic effect on neuronal apoptosis via differential regulation of pro-survival and pro-apoptotic transcription factors. Mol Cell Neurosci. 2005;30:279–289. doi: 10.1016/j.mcn.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Tuffy LP, Weisova P, Bonner HP, Davila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JH. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta R, Laroia G, Schneider RJ. Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000;14:1460–1470. [PMC free article] [PubMed] [Google Scholar]
- Davila D, Torres-Aleman I. Neuronal death by oxidative stress involves activation of FOXO3 through a two-arm pathway that activates stress kinases and attenuates insulin-like growth factor I signaling. Mol Biol Cell. 2008;19:2014–2025. doi: 10.1091/mbc.E07-08-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor CH, Meijer H, Lissenden S. Mechanisms of translational control by the 3'UTR in development and differentiation. Semin Cell Dev Biol. 2005;16:49–58. doi: 10.1016/j.semcdb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–2601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- Gibert B, Eckel B, Fasquelle L, Moulin M, Bouhallier F, Gonin V, Mellier G, Simon S, Kretz-Remy C, Arrigo AP, et al. Knock down of heat shock protein 27 (HspB1) induces degradation of several putative client proteins. PLoS One. 2012;7:e29719. doi: 10.1371/journal.pone.0029719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havasi A, Li Z, Wang Z, Martin JL, Botla V, Ruchalski K, Schwartz JH, Borkan SC. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2008;283:12305–12313. doi: 10.1074/jbc.M801291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suaréz-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A translational profiling approach for the molecular characterization of cns cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henics T, Nagy E, Oh HJ, Csermely P, von Gabain A, Subjeck JR. Mammalian Hsp70 and Hsp110 proteins bind to RNA motifs involved in mRNA stability. J Biol Chem. 1999;274:17318–17324. doi: 10.1074/jbc.274.24.17318. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Spitz DR, Landry J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996;56:273–279. [PubMed] [Google Scholar]
- Kalmar B, Greensmith L. Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev. 2009;61:310–318. doi: 10.1016/j.addr.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Kapeli K, Yeo GW. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front Neurosci. 2012;6:144. doi: 10.3389/fnins.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Fan J, Mazan-Mamczarz K, Gorospe M. Global mRNA stabilization preferentially linked to translational repression during the endoplasmic reticulum stress response. Mol Cell Biol. 2004;24:6773–6787. doi: 10.1128/MCB.24.15.6773-6787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M, Nafar F, Clarke J, Mearow K. The small heat shock protein Hsp27 protects cortical neurons against the toxic effects of beta-amyloid peptide. J Neurosci Res. 2009;87:3161–3175. doi: 10.1002/jnr.22145. [DOI] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. 2008;12:743–761. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman DS. Hsp27 and cell survival in neurons. Int J Hyperthermia. 2005;21:393–402. doi: 10.1080/02656730400023664. [DOI] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- Liu XQ, Sheng R, Qin ZH. The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol Sin. 2009;30:1071–1080. doi: 10.1038/aps.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- Matsui H, Asou H, Inaba T. Cytokines direct the regulation of bim mRNA stability by heat-shock cognate protein 70. Mol Cell. 2007;25:99–112. doi: 10.1016/j.molcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Meller R, Cameron JA, Torrey DJ, Clayton CE, Ordonez AN, Henshall DC, Minami M, Schindler CK, Saugstad JA, Simon RP. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J Biol Chem. 2006;281:7429–7436. doi: 10.1074/jbc.M512138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S. Coupled mRNA stabilization and translational silencing of cyclooxygenase-2 by a novel RNA binding protein, CUGBP2. Mol Cell. 2003;11:113–126. doi: 10.1016/s1097-2765(03)00012-1. [DOI] [PubMed] [Google Scholar]
- Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol. 2002;22:816–834. doi: 10.1128/MCB.22.3.816-834.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinsimer KS, Gratacos FM, Knapinska AM, Lu J, Krause CD, Wierzbowski AV, Maher LR, Scrudato S, Rivera YM, Gupta S, et al. Chaperone Hsp27, a novel subunit of AUF1 protein complexes, functions in AU-rich element-mediated mRNA decay. Mol Cell Biol. 2008;28:5223–5237. doi: 10.1128/MCB.00431-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckley D, Karajgikar M, Dale LB, Fuerth B, Swan P, Drummond-Main C, Poulter MO, Ferguson SS, Strasser A, Cregan SP. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J Neurosci. 2007;27:12989–12999. doi: 10.1523/JNEUROSCI.3400-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Cao G, Gao Y, Zhang F, Wang S, Weng Z, Vosler P, Zhang L, Signore A, Graham SH, et al. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci. 2008;28:13038–13055. doi: 10.1523/JNEUROSCI.4407-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhou M, Fu D, Xu B, Fang T, Ma Y, Chen J, Zhang J. Ubiquitination of heat shock protein 27 is mediated by its interaction with Smad ubiquitination regulatory factor 2 in A549 cells. Exp Lung Res. 2011;37:568–573. doi: 10.3109/01902148.2011.619627. [DOI] [PubMed] [Google Scholar]
- Terasawa K, Ichimura A, Sato F, Shimizu K, Tsujimoto G. Sustained activation of ERK1/2 by NGF induces microRNA-221 and 222 in PC12 cells. FEBS J. 2009;276:3269–3276. doi: 10.1111/j.1742-4658.2009.07041.x. [DOI] [PubMed] [Google Scholar]
- Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxid Redox Signal. 2002;4:405–414. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Wagstaff MJ, Collaco-Moraes Y, Smith J, de Belleroche JS, Coffin RS, Latchman DS. Protection of neuronal cells from apoptosis by Hsp27 delivered with a herpes simplex virus-based vector. J Biol Chem. 1999;274:5061–5069. doi: 10.1074/jbc.274.8.5061. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse N, Steel R, Kluck R, Trapani J. Assaying cytochrome C translocation during apoptosis. Methods Mol Biol. 2004;284:307–313. doi: 10.1385/1-59259-816-1:307. [DOI] [PubMed] [Google Scholar]
- Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009;29:2997–3008. doi: 10.1523/JNEUROSCI.0354-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing Bim expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Nicholson DW. Heat-shock proteins as death determinants. Nat Cell Biol. 2000;2:163–165. doi: 10.1038/35023643. [DOI] [PubMed] [Google Scholar]
- Xie B, Wang C, Zheng Z, Song B, Ma C, Thiel G, Li M. Egr-1 transactivates bim gene expression to promote neuronal apoptosis. J Neurosci. 2011;31:5032–5044. doi: 10.1523/JNEUROSCI.5504-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann AK, Loucks FA, Le SS, Butts BD, Florez-McClure ML, Bouchard RJ, Heidenreich KA, Linseman DA. Distinct mechanisms of neuronal apoptosis are triggered by antagonism of Bcl-2/Bcl-x(L) versus induction of the BH3-only protein Bim. J Neurochem. 2005;94:22–36. doi: 10.1111/j.1471-4159.2005.03156.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.