

Abstract
Hypoglycemia poses a serious threat to the integrity of the brain, owing to its reliance on blood glucose as a fuel. Protecting against hypoglycemia is an extended network of glucose sensors located within the brain and in the periphery that serve to mediate responses restoring euglycemia, i.e., counterregulatory responses. This review examines the various glucose sensory loci involved in hypoglycemic detection, with a particular emphasis on peripheral glucose sensory loci and their contribution to hypoglycemic counterregulation.
The brain normally relies on circulating blood glucose as a primary source of energy and accounts for 60–80% of total body glucose uptake under postabsorbtive conditions (112, 116). Maintaining plasma glucose levels high enough to support brain glucose uptake, while avoiding the deleterious effects of long-term hyperglycemia (i.e., diabetes), requires that plasma levels be tightly regulated, normally between 4 and 6 mM. Although we can and do tolerate transient elevations beyond these normal limits with no apparent harm, acute decrements in blood glucose below 3.0 mM can have a profound impact. Beginning with various neurogenic symptoms, e.g., nervousness, irritability, etc., hypoglycemia if left unchecked eventually leads to impaired cognitive function, impaired motor function, coma, and even death. Unlike the complications of hyperglycemia, which require years to develop, the impact of hypoglycemia can be felt within minutes, and severe consequences can manifest in a few hours. Thus it is not surprising that the body has evolved a number of defenses to counter the fall in blood glucose.
Even as glucose levels fall within the normal range, insulin secretion is decreased, thereby reducing uptake by nonneuronal tissues (20). However, as blood glucose falls below 4 mM, a number of counterregulatory measures are initiated to restore blood glucose levels. Primary among these are the secretion of glucagon and epinephrine, which collectively stimulate hepatic glucose production and decrease glucose uptake, leading to the restoration of blood glucose levels. Normally, glucagon and epinephrine are sufficient to restore blood glucose, but norepinephrine and cortisol may play important roles when hypoglycemia is prolonged or when glucagon secretion is defective, e.g., diabetes. All counterregulatory hormone responses (CRR) are initiated at blood glucose levels just below the normal range, i.e., 3.6–3.8 mM (20). This narrow range is rather remarkable, given the disparate origins of these hormones, i.e., pancreatic alpha-cell, adrenal medulla, sympathetic nerve endings, and adrenal cortex, as well as the various signaling mechanisms involved. Toward this end, the body retains a widely distributed network of glucose sensing neurons, which, together with integrative, premotor, and motor elements, serve to detect and defend against hypoglycemia (113). Whereas a number of recent reviews have focused on the role of the brain, the present review examines peripheral glucose sensors and their interaction with the CNS in mediating the counterregulatory response to hypoglycemia.
Peripheral Glucose Sensors
Peripheral glucose-sensing neurons are found in a number of locations throughout the body, including the oral cavity (93), gastrointestinal tract (85), portal-mesenteric vein (28), and carotid body (77). Those found in the oral cavity and gastrointestinal tract are associated with the detection of ingested glucose and convey information relevant to elevations in blood glucose and satiety (85, 108). Although the portal-mesenteric vein (PMV) may also retain glucose-sensing neurons capable of detecting elevations in blood glucose, PMV glucose sensors have been implicated in the detection of hypoglycemia (28), as have the polymodal chemosensors of the carotid body (52).
Portal-Mesenteric Vein Glucose Sensors
The existence of portohepatic glucose sensors was first proposed by Russek (96) over 50 years ago, followed shortly thereafter by Niijima's discovery of hepatic vagal afferents sensitive to portal glucose infusions (69). Although considerable evidence would accrue over the next two decades regarding the ability of portohepatic glucose sensors to respond to elevations in glycemia, evidence for their role in hypoglycemic detection was not forthcoming until the early 1990s. In the ensuing 15 years, several different approaches would be employed, each establishing the significance of portohepatic glucose sensing in hypoglycemic detection. Primary among these was the “local irrigation technique,” which utilizes a local portal vein glucose infusion to establish a euglycemic clamp across the portohepatic circulation, while systemic hypoglycemia is maintained via a hyperinsulinemic-hypoglycemic clamp (25). The fundamental premise of this approach is that if the portoheptic region contains critical glucose sensors, then normalizing glycemia across that region during general systemic hypoglycemia should impair the detection of hypoglycemia, blunting any counterregulatory responses. Normalizing portohepatic glucose in this manner resulted in a 42% suppression of the epinephrine response to moderate systemic hypoglycemia of 3.25 mM and with an even greater suppression of up to 73% when glycemia was lowered to 2.5 mM (26, 27, 42). A similar effect was observed for norepinephrine with a 42–67% suppression when portohepatic glycemia was normalized during deep systemic hypoglycemia. This same approach was subsequently used to establish the portal vein as the locus for the “portohepatic glucose sensors,” not the liver as had long been believed (43). By selectively adjusting the point of infusion, it was shown that normalizing liver glycemia alone was ineffective in blunting the sympathoadrenal response to hypoglycemia and that normalization of portal vein glycemia was essential to that response. Consistent with this finding, it was shown that normalizing liver glycemia via the hepatic artery during systemic hypoglycemia was also ineffective in suppressing the sympathoadrenal response (45). Denervation studies have provided added support for both the locus and importance of portal vein glucose sensors in mediating the sympathoadrenal response to hypoglycemia. Thus chemically stripping the nerves of the portal vein via topical phenol yielded a 50% suppression of the sympathoadrenal response to deep hypoglycemia (2.5 mM), comparable to that observed with the local irrigation technique (44). A similar result was obtained with the topical application of capsaicin, a chemical agent that selectively removes afferent nerves (37). The efficacy of the capsaicin treatment was confirmed by the extensive loss of CGRP immunoreactive fibers in the portal vein of treated animals. Interestingly, CGRP immunoreactivity extends beyond the portal vein proper to include the superior mesenteric vein, suggesting that the locus for hypoglycemic detection might extend to the mesenteric vein, an observation that was confirmed by Saberi et al. (97). Although denervating the portal vein alone was shown to suppress the epinephrine response by ∼60%, extending the denervation to include both the portal and superior mesenteric vein resulted in a 91% suppression. Although denervation of portal-messenteric vein substantially suppressed the epinephrine response, it effectively eliminated the norepinephrine response to slow-onset hypoglycemia. Consistent with these observations in animals, Perseghin et al. (83) reported that humans having undergone liver transplantation, i.e., with denervated livers, also demonstrate impaired hypoglycemic counterregulation. The impaired counterregulation in these subjects was evidenced by suppressed endogenous glucose production during a hyperinsulinemic-hypoglycemic clamp, accompanied by a 53% suppression in the sympathoadrenal response. Additional evidence for hypoglycemic detection at the portal vein derives from the study of Lamarche et al. (54). Utilizing a complex cross-perfusion design, they were able to establish hypoglycemia across the portohepatic circulation during systemic euglycemia. Despite anesthetic conditions known to blunt the sympathoadrenal response, they demonstrated an approximately threefold increase in the catecholamine output from the adrenal gland with selective portal vein hypoglycemia. Collectively, these studies not only served to establish the portal-mesenteric vein as a locus for hypoglycemic detection but suggest a quantitative contribution in mediating the counterregulatory response comparable to that of the CNS glucose sensors.
It has long been believed that all glucose sensory information from the portohepatic region is conveyed to the CNS via hepatic vagal afferents. This was based primarily on the extensive work of Niijima (69–72, 75), who first elucidated the existence of glucose-inhibited vagal afferents within the hepatic branch of the vagus nerve (69). He subsequently demonstrated that the firing rate of these vagal afferents was inversely related to the glucose concentration in the portal vein over a broad range of portal glucose concentrations (5–27 mM) (70). It was further shown that these vagal afferents were specific for glucose, failing to respond to other hexoses, e.g., fructose, mannose, and galactose, and that the suppression in firing rate appeared to be mediated by an ATP-dependent sodium pump (70, 74). Niijima and colleagues would further demonstrate that elevations in portal vein glucose led to a suppression in the efferent firing rate of the adrenal nerve, while increasing the efferent firing rate for the pancreatic branch of the vagus (73). Sectioning of the hepatic branch of the vagus eliminated both of these responses. These observations were consistent with the prevailing perception that visceral chemosensory information was communicated by vagal afferents, while spinal afferents transmitted mechanosensory and noicioceptive inputs. Although these findings clearly established their existence, glucose-sensitive vagal afferents appear to play no role in mediating hypoglycemic detection at the portal-mesenteric vein. Sectioning the hepatic vagus nerve, i.e., the nerve containing the glucose-sensitive afferents identified by Niijima, was shown to have no impact on the sympathoadrenal response to hypoglycemia (38) (FIGURE 1). Furthermore, when all vagal transmission below the diaphragm was eliminated via bilateral subdiaphragmatic vagotomy, the sympathoadrenal response to hypoglycemia remained fully intact (38). The counterregulatory response to hypoglycemia was also observed to be normal in animals undergoing acute vagal blockade via cooling (2°C) despite the loss of vagal transmission below the neck (46, 47). Thus the vagal afferents, as originally suggested by Niijima, may primarily serve to detect elevations in portal vein glucose levels subsequent to feeding (74, 75). This hypothesis is consistent with the observation that these afferents demonstrate a linear suppression of their firing rate over a wide range of portal glucose concentrations ranging from euglycemia to hyperglycemia, i.e., 5.5–27 mM (68).
FIGURE 1.
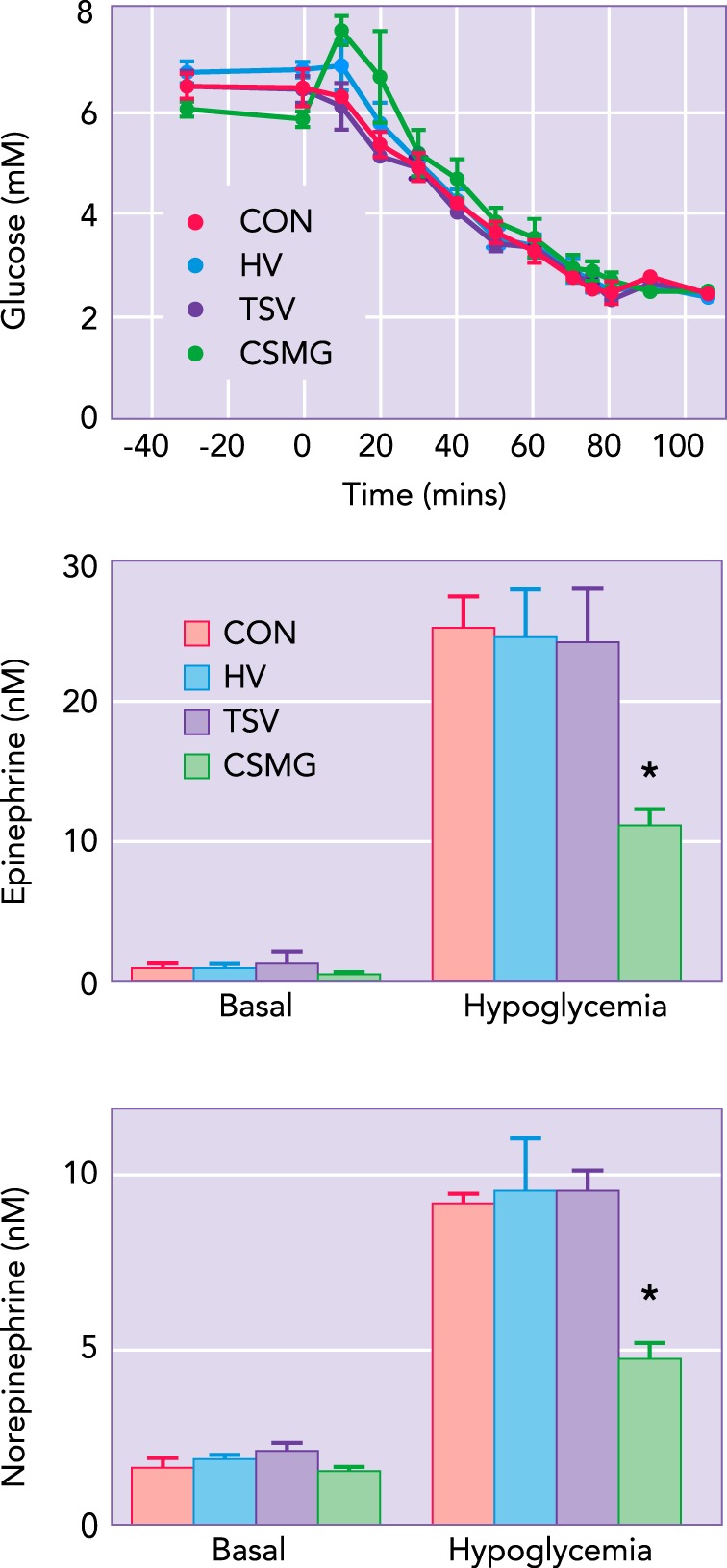
The effect of lesioning visceral vagal vs. spinal afferents on the sympathoadrenal response to hypoglycemia
The peak epinephrine and norepinephrine responses to matched hyperinsulinemic-hypoglycemic clamps in nerve-intact controls (CON), hepatic vagotomized (HV), total subdiphragmatic vagotomized (TSV), and celiac-superior mesenteric ganglionectomized (CSMG) animals. Basal values (means ± SE) were not significantly different between groups, nor were any differences observed between control and vagotomized animals during hypoglycemia. However, animals in which the spinal afferent innervation of the portal-mesenteric vein was removed via celiac-superior mesenteric ganglionectomy demonstrate a substantial suppression in the sympathoadrenal response to hypoglycemia. Figure adapted from Ref. 38.
All evidence to date indicates that hypoglycemic detection at the portal-mesenteric vein is mediated by glucose-sensitive spinal afferents. Where vagotomy proved ineffective, sectioning the celiac-superior mesenteric ganglion (CSMG) was shown to induce marked blunting of the sympathoadrenal response to hypoglycemia (38). The portal vein is richly innervated by spinal afferents, as denoted by CGRP and substance P immunoreactivity (4, 37), which traverse the celiac-superior mesenteric ganglion entering the spinal cord at T8–T13 (4, 6). Although some vagal afferents originating from the celiac branch pass through the CSMG, this branching occurs caudal to the subdiaphragmatic sectioning of the vagus nerve (84), a procedure shown to have no effect on the sympathoadrenal response to hypoglycemia (38). Both CSMG and dorsal root rhizotomy (T8–T13) are effective in eliminating CGRP and SP immunoreactivity in the portal-mesenteric vein (4, 40). Vagotomy by contrast was ineffective in decreasing the content of either CGRP or SP in the portal vein (4, 40), consistent with a spinal origin for those portal-mesenteric vein glucose sensors detecting hypoglycemia. As with CSMG, topical application of capsaicin substantially reduces CGRP reactivity in the portal-mesenteric vein, and with it the ability for hypoglycemic detection (37, 97). Capsaicin acts through the TRPV1 receptor, which in the abdominal viscera is found primarily on spinal afferents (82, 111). That glucose sensing at the portal vein might be mediated by spinal afferents was suggested some years ago by Schmitt (102). Having identified select neurons in the lateral hypothalamus responsive to portal glucose infusion, she demonstrated that sectioning either the splanchnic nerve or spinal cord (T3) was effective in eliminating this response. In contrast, sectioning both branches of the vagus failed to impact the firing rate of these LH neurons in response to portal glucose infusion. These findings were supported by the observation that CSMG was significantly more effective than hepatic vagotomy in eliminating the feeding response induced by portal vein infusion of the glucopenic agent 2-deoxyglucose (103).
The mechanism, by which portal-mesenteric vein glucose sensors detect hypoglycemia remains to be fully elucidated but appears to involve the uptake and subsequent metabolism of glucose. Thus mice deficient in the glucose transporter GLUT2 lack the ability to detect changes in portal vein glucose levels following direct portal glucose infusion (14). This is true even when GLUT2 levels are restored in the liver, indicating an essential role for glucose transport in portal vein glucose sensing. However, much like the pancreatic β-cell and ventromedial hypothalamic glucose-sensing neurons, the subsequent metabolism of glucose appears critical for hypoglycemic detection at the portal vein. This was shown by selectively elevating portal vein lactate (5 mM) or pyruvate (2 mM) levels during insulin-induced hypoglycemia, which led to a suppression in the sympathoadrenal response comparable to that observed when glucose was normalized across the portal vein (58). In contrast, portal infusion of the ketone β-hydroxybutyrate failed to impact the sympathoadrenal response under the same conditions. In all cases, the elevation of lactate, pyruvate, or β-hydroxybutyrate was restricted to the portal vein, i.e., arterial levels were normal, constraining the site of action to the portal vein. Interestingly, as has been shown for the CNS (109), glucose sensing at the portal-mesenteric vein may involve more than one mechanism. Delaere et al. (23) recently provided evidence for a metabolism-independent mechanism of portal vein glucose sensing involving the Na+-dependent glucose transporter SGLT3. Although not addressing hypoglycemia per se, their findings suggest a mechanism sensitive to relatively small changes in blood glucose that appears mediated by portal-mesenteric spinal afferents. Portal glucose sensing may also be subject to the influence of other metabolic signals, as suggested by the observation that an intact GLP-1 receptor is essential for portal glucose sensing (13). The role of the GLP-1 receptor in this case does not require an elevation in GLP-1 levels and therefore may be relevant to the hypoglycemic condition. It should be noted that, in contrast to central glucose sensing neurons, all known peripheral glucose sensory mechanisms involve transduction of the glucose signal by a primary sensory cell (76, 85, 93). Although no such primary sensory cell has been identified for the portal-mesenteric glucose sensors, the patterns of innervation for the portal vein demonstrate similarities to other peripheral glucose sensory loci (e.g., oral cavity, gut, and carotid body), i.e., innervation of the portal vein appears restricted largely to the adventitia surrounding the vein, with some limited projections to the bilayer of smooth muscle below (4, 6). There are no reports of any afferents projecting into the lumen of the portal vein and thus positioned to directly detect portal glucose concentrations.
Carotid Body
That the carotid body might be involved in glucose homeostasis was proposed over 20 years ago by Alvarez-Byulla et al. (1) when they demonstrated a hyperglycemic response following treatment of the carotid body with NaCN. Since that time, evidence has accumulated regarding the ability of the carotid body to sense low glucose, although controversy persists as to whether glucose sensing by the carotid body is direct or indirect (9, 77). The controversy appears in part to derive from the type of in vitro preparation employed in these studies. Experiments employing either the carotid body slice or cocultured (type 1 cell and sensory neuron) preparations indicate a direct effect of low glucose on the secretion of ACh and ATP by the type 1 cell (76). The mechanism appears to share elements with other proposed models of glucose sensing. That is, low glucose induces K+ channel inhibition, resulting in depolarization of the type I cell membrane, leading to an influx of Ca2+ and ultimately neurotransmitter release. Acetylcholine and ATP then bind to nicotinic acetylcholine and purinergic receptors, respectively, on the adjacent petrosal (sensory) neuron, leading to a postsynaptic potential. In contrast to these findings, studies utilizing the whole intact carotid body preparation have failed to observe any direct impact of low glucose on neurotransmitter discharge (9). These authors have suggested that the in vivo findings for altered ventilation with hypoglycemia might best be explained by the sensitizing of the carotid body to Pco2 with low glucose. Although putative explanations for the disparity in these results have been offered (53, 77), compelling evidence resolving this controversy remains to be provided.
As with in vitro studies, in vivo experiments have also proven equivocal with respect to the role of the carotid body in hypoglycemic detection. Koyama et al. (52) reported impaired hypoglycemic counterregulation following sectioning of the carotid bodies, as evidenced by a suppressed glucagon response, decreased endogenous glucose production, and elevated exogenous glucose infusion rate (GINF). However, carotid body resected animals also demonstrated increased glucose disposal rates, suppressed glucagon levels before the onset of hypoglycemia, and normal intact sympathoadrenal responses to hypoglycemia. Interestingly, carotid body resection leads to a similar suppression in the exercise-induced glucagon response that is apparently unrelated to glucose levels (51). Although carotid body resected animals clearly demonstrate impaired counterregulation, it is unclear whether this derives from impaired glucose sensing. Other in vivo studies have attempted to exploit the polymodal characteristics of the carotid body by employing elevated Po2 to suppress glucose sensing by the carotid body. Employing this approach with human subjects, Wehrwein et al. (114) reported a substantial suppression in the counterregulatory hormone response to insulin-induced hypoglycemia for subjects whose arterial Po2 was elevated to 345 Torr. Glucagon, epinephrine, and norepinephrine were all suppressed by 49–63% during hypoglycemia under hyperoxic conditions, leading to an increase in the required GINF of 51%. However, Ward et al. (110), employing a very similar protocol, found no significant suppression of the counterregulatory response to hypoglycemia with hyperoxia. Although an intriguing site for hypoglycemic detection and its integration with other metabolic signals, e.g., hypoxia, the carotid body's role in hypoglycemic sensing and counterregulation remains unclear.
Central Glucose-Sensing Neurons
The concept that the brain plays an important role in glucose homeostasis dates back over 150 years to the seminal work by Claude Bernard (5). Subsequent studies would identify the hypothalamus and hindbrain as important regions for glycemic control (65, 98). That the brain actually detects blood glucose levels gained traction in the 1950s with the “glucostatic hypothesis” put forward by Jean Mayer (59).
Evidence that the CNS plays a direct role in hypoglycemic detection and subsequent counterregulation derives from two general types of investigations. The first of these relies on the induction of glucopenia via a glucose analog, e.g., 2-deoxyglucose (2DG), that effectively blocks glycolytic flux. Although not strictly hypoglycemia, an intravenous injection of 2DG does induce many similar responses, including a strong counterregulatory hormone response resulting in systemic hyperglycemia, increased feeding, and Fos expression in several areas of the CNS (21, 38, 88, 99). Injection of 2-deoxyglucose directly into the lateral or third cerebral ventricles was shown to induce both hyperglycemia and feeding (7, 31, 64). This was ascribed to known glucose-sensing neurons in the hypothalamus, and subsequent work has shown that 2-DG applied directly to the ventromedial hypothalamus via microdialysis elicits a strong counterregulatory hormonal response (12). However, Ritter et al. (86) reported that, when the cerebral aqueduct was blocked, injection of the 5-thioglucose was effective in inducing hyperglycemia only when injected into the fourth ventricle, not the lateral ventricles, suggesting the critical glucose sensors resided in the hindbrain. Consistent with this observation, it was shown that nanoliter injections of 5-thio-d-glucose were effective in eliciting a hyperglycemic response when applied to specific loci in the hindbrain but not the in the hypothalamus (87). This same group had previously identified neurons in the same hindbrain regions that responded to 2DG injection with increased cFos expression (90). That the hindbrain retains the essential counterregulatory elements to hypoglycemia is supported by the observation that decerebrate animals demonstrate a robust hyperglycemic response to an infusion of 2-deoxyglucose (24).
The second approach to establishing the CNS as a critical locus for glucose sensing in hypoglycemia involves the normalization of brain glucose during the induction of systemic hypoglycemia. Toward this end, Borg et al. (11) demonstrated suppression of the counterregulatory response to insulin-induced hypoglycemia when glucose was applied to the ventromedial hypothalamus via microdialysis. This observation was subsequently confirmed, although the glucose concentration of the infusate required to achieve this suppression was substantial (i.e., 100 mM) (115). A similar approach has been employed for the hindbrain, where it has been shown that lactate, a substrate readily used by neurons, when infused into the fourth ventricle will suppress the restoration of euglycemia following insulin-induced hypoglycemia (81). In contrast to studies utilizing direct infusions into the brain, studies employing local irrigation to normalize glucose delivery to the brain via the circulation suggest that the critical hypoglycemic sensors are widely distributed, providing redundant sensory input. Thus normalizing brain glycemia proved effective in suppressing the counterregulatory response to systemic hypoglycemia only when delivered via both the carotid and the vertebral arteries (8). Attempts to normalize brain glycemia via the carotid artery alone have repeatedly failed in this respect, even though this approach should normalize glycemia to the hypothalamus (18, 19, 35). Interestingly, glucose injections into the carotid artery resulting in hyperglycemia lead to a strong Fos response in the hypothalamus (29, 32, 41). In similar fashion, normalizing brain glycemia via the vertebral arteries, which should normalize hindbrain glycemia, also failed to impact hypoglycemic counterregulation (25, 35). Although this discrepancy between local brain glucose infusion and brain “glycemic clamps” remains to be resolved, both approaches support the existence of CNS glucose sensors critical to the detection and modulation of the counterregulatory response to hypoglycemia.
Since it was first reported that select neurons in the hypothalamus altered their firing rates in response to the direct application of glucose (3, 78), glucose-sensing neurons have been identified in numerous regions of the brain, including the ventromedial nucleus of the hypothalamus (VMH), tuberal nucleus, and arcuate nucleus (ARH), amygdala, nucleus of the solitary tract (NTS), dorsal motor nucleus of the vagus, and area postrema (55, 79). Recent evidence suggests that there are perhaps as many as seven different types of glucose-sensing neurons in the brain, utilizing half a dozen different sensing mechanisms (32, 105, 109). An in-depth review of the various glucose-sensing neurons and mechanisms involved is beyond the scope of this short review, and the reader is directed to several excellent recent reviews for a more comprehensive discussion of this literature (16, 89, 95, 109). However, it is worth noting that the interstitial glucose concentration of the brain, to which most of the identified glucose sensors are exposed, is ∼20–30% of that found in the plasma (62, 104), and under hypoglycemic conditions this gradient may increase (22, 61). Thus only glucose-sensing neurons responding to glucose concentrations below 1.5 mM are plausible candidates for hypoglycemic detection within the brain. The exception would be glucose-sensing neurons located in sensory circumventricular organs (CVOs), which may be exposed to much higher glucose concentrations, given the absence of an intact blood-brain barrier. Of the sensory CVOs, only the area postrema (AP) and subfornical organ (SFO) have been shown to contain glucose-sensing neurons (39, 63). Although little is known regarding the potential contribution of the SFO in hypoglycemic detection, neurons in the AP do demonstrate robust Fos expression in response to systemic hypoglycemia and 2-deoxyglucose (10, 80, 100). There has been some suggestion that neurons of the arcuate nucleus may also be exposed to higher glucose concentrations, given their proximity to the median eminence (60). Although this remains an area of some debate (36, 92), the measured glucose concentrations in the ARH were shown to be similar to those found in the VMH (30).
Although the low interstitial glucose levels in the brain appear to exclude certain types of glucosensing neurons, e.g., the high glucose-excited (HGE) and -inhibited (HGI) (32), at least two types of neurons appear capable of hypoglycemic detection in this environment. Both the glucose-excited (GE) and glucose-inhibited (GI) neurons of the VMH demonstrate a relatively linear response in firing rate to changes in glucose concentrations between 0.1 and 1.5 mM (50, 106). The GE neurons, which increase their firing rate in response to elevations in glucose, are purported to employ a glucose-sensing mechanism similar to that of the pancreatic beta cell (57, 94). In this model, glucose “sensing” is dependent on the uptake and subsequent metabolism of glucose, which dictates the adenylate charge for the cell (57). The increase in glucose flux purportedly leads to an elevation in ATP, resulting in the closure of an ATP-dependent potassium channel, thereby eliciting an action potential. Critical components of this pathway, i.e., the GLUT2 glucose transporter, glucokinase, and Kir6.2 subunit of the ATP dependent K+ channel, have all been identified in the VMH GE neurons. Furthermore, there is strong evidence that glucokinase plays a primary regulatory role in mediating the glucose sensing of GE neurons as it does in the pancreatic beta cell (49). That glucokinase, the Kir6.2 subunit, and GLUT2 are not uniformly expressed in VMH GE neurons (50) suggests that the GE neurons may employ additional mechanisms for glucose sensing (106). However, these may simply reflect variations in the model, e.g., GLUT4 more abundantly expressed in GE neurons may augment or supplant the role of GLUT2 in mediating glucose uptake by some GE neurons (95).
Although the underlying mechanism for glucose detection by GE neurons appears somewhat conserved, multiple mechanisms have been reported for GI neurons (16). How many of these GI neurons may be involved in hypoglycemic counterregulation remains unclear, but those of the VMH have been linked to the CRR (33, 107), and thus their mechanism is discussed herein. In contrast to GE neurons, depolarization of the VMH GI neuron is dependent on inhibition of chloride conductance (105), which recent evidence suggests maybe mediated by the cystic fibrosis transmembrane regulator (CFTR) (66). Furthermore, glucose sensing by the GI neurons appears to be modulated by AMP-activated protein kinase (AMPK) (17, 67). AMPK inhibition of the CFTR Cl− channel in turn appears dependent on its interaction with nitric oxide (NO) in a feed-forward augmentation of the AMPK activity (66). In this model, a decrease in ambient glucose leads to a reduction in glucose uptake and catabolism, resulting in an elevation of the AMP-to-ATP ratio, thereby allosterically activating AMPK. However, this activation of AMPK alone appears insufficient to sustain closure of the Cl− channel. Rather, AMPK activity appears to require further augmentation through a cyclical process in which AMPK phosphorylates neuronal NO synthase (nNOS), resulting in increased NO production. NO then binds to soluble guanylyl cyclase (sGC), increasing cyclic guanosine monophosphate (cGMP) production, presumably increasing AMPK kinase activity. AMPK kinase in turn potentiates the allosteric effect of the elevated AMP on AMPK activity, ultimately leading to inhibition of the CFTR Cl− channel (34). The central role of AMPK in this model provides for a mechanism that may explain the ability of other substrates, e.g., lactate and ketones, to suppress the counterregulatory respone to hypoglycemia.
Integration of Peripheral and Central Glucose Sensing
The multitude of glucose-sensing loci for hypoglycemic detection, peripheral and central, has frequently been explained as a redundancy essential to protecting blood glucose levels and brain function. Although there is undoubtedly some redundancy as suggested by the “brain clamp” experiments (8), most attempts to block the counterregulatory response by normalizing glycemia at specific loci or lesioning specific afferents would have failed were redundancy an overriding factor. A more plausible explanation is that the distributed glucose sensors provide critical inputs to an integrative network, allowing for a more finely tuned response to glycemic challenges. For example, it has been known for some time that the rate of fall in glycemia has no effect on the counterregulatory response in normal nerve-intact individuals (2, 101). However, when peripheral portal-mesenteric vein glucose sensors are eliminated, the counterregulatory response to slow onset hypoglycemia, i.e., ≤1 mg·dl−1·min−1, is severely diminished (FIGURE 2) (97). Concomitant with the suppression in the CRR is a failure for hindbrain neuronal activation, as denoted by Fos expression under these conditions (10). Interestingly, lesioning catecholaminergic afferents projecting from the hindbrain to the hypothalamus results in a similar impaired response to slow-onset hypoglycemia (48). Together, these data suggest an extended network of peripheral and hindbrain afferents critical for the detection of physiological decrements in blood glucose. Alternatively, the brain appears fully capable of responding to very rapid decrements in blood glucose, i.e., ≥2mg·dl−1·min−1, generating a full-blown CRR in the absence of PMV glucose sensors. For these rapid responses, the local brain glycemia, particularly in the ventromedial hypothalamus and hindbrain, appears most critical (11, 81, 115). Thus infusing glucose or an alternative fuel, e.g., lactate, into these areas of the brain during rapid-onset hypoglycemia severely blunts the counterregulatory hormonal responses. Furthermore, with rapid onset hypoglycemia, the catecholaminergic neurons projecting from the hindbrain to the hypothalamus are essential for the corticosterone and feeding responses but not the sympathoadrenal response (91).
FIGURE 2.

The disparate response to slow- vs. rapid-onset hypoglycemia with portal-mesenteric denervation
A and B: matched hyperinsulinemic-hypoglycemic clamps in which glucose is allowed to fall either slowly (−0.05 mM/min; A) or rapidly (−0.18 mM/min; B) for nerve-intact controls (CON) or animals undergoing portal-mesenteric vein sensory denervation via topical capsaicin (PMV-DN). C and D: the epinephrine response to hypoglycemia, which is severely blunted in PMV-DN animals with slow-onset hypoglycemia (C) but is largely unaffected by PMV-DN during rapid-onset hypoglycemia (D). E and F: the norepinephrine response to hypoglycemia, which is essentially eliminated in PMV-DN animals with slow-onset hypoglycemia (E) but is unaffected by PMV-DN during rapid-onset hypoglycemia (F). Figure adapted from Ref. 97.
Given their lack of sensitivity to a slow decline or small decrements in blood glucose, it is tempting to posit the CNS glucose-sensing neurons as an emergency mechanism responding only to catastrophic declines in blood glucose. Although they clearly appear to serve in this capacity, this may substantially underestimate the contribution of CNS glucose sensors to hypoglycemic counterregulation. Distributing specific glucose sensitivity to peripheral afferents may in fact be essential to allowing central glucose-sensing neurons to serve as metabolic sensors monitoring the overall energy status of the organism during periods of deficit (15, 56). Decreasing their sensitivity to any single metabolite may allow these neurons to better detect and integrate a variety of incoming signals to help achieve the appropriate motor response. This distributed network would also allow for the comparison and integration of inputs regarding the glucose status of the entire organism, i.e., the CNS and periphery. Such information would be vital to allowing for the proper efferent response during hypoglycemia in the presence of multiple substrates, peripheral inputs from the ingestion of food, as well as reestablishment of euglycemia.
Combining the above functional data with known anatomical connections as elucidated by various tracing techniques, we recently proposed an extended glucose-sensing network (FIGURE 3) (113). In this model, sensory inputs regarding glucose status derive from a number of loci, including the portal-mesenteric vein, carotid body, hindbrain, and hypothalamus (LHA, ARH, VMH). Those inputs are then processed in one of two integrative networks, i.e., hypothalamic and hindbrain, which may in turn exchange critical information. Information from the integrative networks is then projected, directly or indirectly, to premotor networks located in the hypothalamus (LHA, PVH, RCH), hindbrain, and spinal cord. The premotor networks allow one more potential site of modulation, before projecting onto motoneurons generating the counterregulatory autonomic and neuroendocrine responses. Although this model is not comprehensive in terms of accounting for all known behavioral and neurogenic symptoms of hypoglycemia, it does provide a framework from which we can begin to intelligently describe, interpret, and even predict interactions between the various peripheral and central glucose-sensing elements.
FIGURE 3.

A schematic diagram illustrating the extended neural network mediating hypoglycemic counterregulation
The model provides for peripheral (PMV and carotid body) and central (hindbrain and hypothalamic) glucose sensory inputs that feed into integrative networks located in the hindbrain and hypothalamus. Utilizing neural pathways, these networks can integrate glucosensory information with other inputs to generate the appropriate output to premotor networks and ultimately the motoneurons generating the autonomic and neuroendocrine responses. Figure adapted from Ref. 113.
Footnotes
This work was supported by grants from the National Institutes of Health, DK-062471 (C.M.D.) and NS-029728 (A.G.W.), and the Juvenile Diabetes Research Foundation, 1-2007-605 (C.M.D.) and 1-2008-710 (A.G.W.).
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: C.M.D. and A.G.W. interpreted results of experiments; C.M.D. and A.G.W. prepared figures; C.M.D. drafted manuscript; C.M.D. and A.G.W. edited and revised manuscript; C.M.D. and A.G.W. approved final version of manuscript.
References
- 1.Alvarez-Buylla R, de Alvarez-Buylla ER. Carotid sinus receptors participate in glucose homeostasis. Respir Physiol 72: 347–359, 1988 [DOI] [PubMed] [Google Scholar]
- 2.Amiel SA, Simonson DC, Tamborlane WV, DeFronzo RA, Sherwin RS. Rate of glucose fall does not affect counterregulatory hormone responses to hypoglycemia in normal and diabetic humans. Diabetes 36: 518–522, 1987 [DOI] [PubMed] [Google Scholar]
- 3.Anand BK, China GS, Sharma KN, Dua S, Singh B. Activity of single neurons in the hypothalamic feeeding centers: effect of glucose. Am J Physiol 207: 1146–1154, 1964 [DOI] [PubMed] [Google Scholar]
- 4.Barja F, Mathison R. Sensory innervation of the rat portal vein and the hepatic artery. J Auton Nerv Syst 10: 117–125, 1984 [DOI] [PubMed] [Google Scholar]
- 5.Bernard C. Magendie annonce à l'Académie des Sciences que Bernard a achevé une augmentation de glucose dans le sang par une blessure d'un certain point du cerveau. C Rhebd Acad Sci 28: 393–394, 1849 [Google Scholar]
- 6.Berthoud HR. Anatomy and function of sensory hepatic nerves. Anat Rec Part A Disc Mol Cell Evol Biol 280: 827–835, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Berthoud HR, Mogenson GJ. Ingestive behavior after intracerebral and intracerebroventricular infusions of glucose and 2-deoxy-d-glucose. Am J Physiol Regul Integr Comp Physiol 233: R127–R133, 1977 [DOI] [PubMed] [Google Scholar]
- 8.Biggers DWD, Myers SRS, Neal DD, Stinson RR, Cooper NBN, Jaspan JBJ, Williams PEP, Cherrington ADA, Frizzell RTR. Role of brain in counterregulation of insulin-induced hypoglycemia in dogs. Diabetes 38: 7–16, 1989 [DOI] [PubMed] [Google Scholar]
- 9.Bin-Jaliah I, Maskell PD, Kumar P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J Physiol 556: 255–266, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bohland MA, Matveyenko AV, Saberi M, Khan AM, Watts AG, Donovan CM. Activation of hindbrain neurons is mediated by portal-mesenteric vein glucosensors during slow-onset hypoglycemia. Diabetes. First published April 11, 2014; 10.2337/db13-1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest 99: 361–365, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44: 180–184, 1995 [DOI] [PubMed] [Google Scholar]
- 13.Burcelin R, Da Costa A, Drucker D, Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 50: 1720–1728, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes 49: 1643–1648, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Burdakov D, Karnani MM, Gonzalez A. Lateral hypothalamus as a sensor-regulator in respiratory and metabolic control. Physiol Behav 121: 117–124, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burdakov D, Lesage F. Glucose-induced inhibition: how many ionic mechanisms? Acta Physiol (Oxf) 198: 295–301, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canabal DD, Song Z, Potian JG, Beuve A, McArdle JJ, Routh VH. Glucose, insulin, and leptin signaling pathways modulate nitric oxide synthesis in glucose-inhibited neurons in the ventromedial hypothalamus. Am J Physiol Regul Integr Comp Physiol 292: R1418–R1428, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Cane P, Artal R, Bergman RN. Putative hypothalamic glucoreceptors play no essential role in the response to moderate hypoglycemia. Diabetes 35: 268–277, 1986 [DOI] [PubMed] [Google Scholar]
- 19.Cane PP, Haun CKC, Evered JJ, Youn JHJ, Bergman RNR. Response to deep hypoglycemia does not involve glucoreceptors in carotid perfused tissue. Am J Physiol Endocrinol Metab 255: E680–E687, 1988 [DOI] [PubMed] [Google Scholar]
- 20.Cryer PE. Hierarchy of physiological responses to hypoglycemia: relevance to clinical hypoglycemia in type I (insulin dependent) diabetes mellitus. Horm Metab Res 29: 92–96, 1997 [DOI] [PubMed] [Google Scholar]
- 21.Darling RA, Ritter S. 2-Deoxy-d-glucose, but not mercaptoacetate, increases food intake in decerebrate rats. Am J Physiol Regul Integr Comp Physiol 297: R382–R386, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 52: 2767–2773, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Delaere F, Duchampt A, Mounien L, Seyer P, Duraffourd C, Zitoun C, Thorens B, Mithieux G. The role of sodium-coupled glucose co-transporter 3 in the satiety effect of portal glucose sensing. Mol Metab 2: 47–53, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiRocco RJR, Grill HJH. The forebrain is not essential for sympathoadrenal hyperglycemic response to glucoprivation. Science 204: 1112–1114, 1979 [DOI] [PubMed] [Google Scholar]
- 25.Donovan CM, Cane P, Bergman RN. Search for the hypoglycemia receptor using the local irrigation approach. Adv Exp Med Biol 291: 185–196, 1991 [DOI] [PubMed] [Google Scholar]
- 26.Donovan CM, Halter JB, Bergman RN. Importance of hepatic glucoreceptors in sympathoadrenal response to hypoglycemia. Diabetes 40: 155–158, 1991 [DOI] [PubMed] [Google Scholar]
- 27.Donovan CM, Hamilton-Wessler M, Halter JB, Bergman RN. Primacy of liver glucosensors in the sympathetic response to progressive hypoglycemia. Proc Natl Acad Sci USA 91: 2863–2867, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donovan CM. Portal vein glucose sensing. Diabetes Nutr Metab 15: 308–312, 2002 [PubMed] [Google Scholar]
- 29.Dunn-Meynell AA, Govek E, Levin BE. Intracarotid glucose selectively increases Fos-like immunoreactivity in paraventricular, ventromedial and dorsomedial nuclei neurons. Brain Res 748: 100–106, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Dunn-Meynell AA, Sanders NM, Compton D, Becker TC, Eiki JI, Zhang BB, Levin BE. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J Neurosci 29: 7015–7022, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engeset RMR, Ritter RCR. Intracerebroventricular 2-DG causes feeding in the absence of other signs of glucoprivation. Brain Res 202: 229–233, 1980 [PubMed] [Google Scholar]
- 32.Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes 56: 1219–1227, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Fioramonti X, Marsollier N, Song Z, Fakira KA, Patel RM, Brown S, Duparc T, Pica-Mendez A, Sanders NM, Knauf C, Valet P, McCrimmon RJ, Beuve A, Magnan C, Routh VH. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 59: 519–528, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fioramonti X, Song Z, Vazirani RP, Beuve A, Routh VH. Hypothalamic nitric oxide in hypoglycemia detection and counterregulation: a two-edged sword. Antioxid Redox Signal 14: 505–517, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frizzell RT, Jones EM, Davis SN, Biggers DW, Myers SR, Connolly CC, Neal DW, Jaspan JB, Cherrington AD. Counterregulation during hypoglycemia is directed by widespread brain regions. Diabetes 42: 1253–1261, 1993 [DOI] [PubMed] [Google Scholar]
- 36.Fry M, Hoyda TD, Ferguson AV. Making sense of it: roles of the sensory circumventricular organs in feeding and regulation of energy homeostasis. Exp Biol Med (Maywood) 232: 14–26, 2007 [PubMed] [Google Scholar]
- 37.Fujita S, Bohland M, Sanchez-Watts G, Watts AG, Donovan CM. Hypoglycemic detection at the portal vein is mediated by capsaicin-sensitive primary sensory neurons. Am J Physiol Endocrinol Metab 293: E96–E101, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Fujita S, Donovan CM. Celiac-superior mesenteric ganglionectomy, but not vagotomy, suppresses the sympathoadrenal response to insulin-induced hypoglycemia. Diabetes 54: 3258–3264, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Funahashi M, Adachi A. Glucose-responsive neurons exist within the area postrema of the rat: in vitro study on the isolated slice preparation. Brain Res Bull 32: 531–535, 1993 [DOI] [PubMed] [Google Scholar]
- 40.Goehler LE, Sternini C. Calcitonin gene-related peptide innervation of the rat hepatobiliary system. Peptides 17: 209–217, 1996 [DOI] [PubMed] [Google Scholar]
- 41.Guillod-Maximin E, Lorsignol A, Alquier T, Pénicaud L. Acute intracarotid glucose injection towards the brain induces specific c-fos activation in hypothalamic nuclei: involvement of astrocytes in cerebral glucose-sensing in rats. J Neuroendocrinol 16: 464–471, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Hamilton-Wessler M, Bergman RN, Halter JB, Watanabe RM, Donovan CM. The role of liver glucosensors in the integrated sympathetic response induced by deep hypoglycemia in dogs. Diabetes 43: 1052–1060, 1994 [DOI] [PubMed] [Google Scholar]
- 43.Hevener AL, Bergman RN, Donovan CM. Novel glucosensor for hypoglycemic detection localized to the portal vein. Diabetes 46: 1521–1525, 1997 [DOI] [PubMed] [Google Scholar]
- 44.Hevener AL, Bergman RN, Donovan CM. Portal vein afferents are critical for the sympathoadrenal response to hypoglycemia. Diabetes 49: 8–12, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Hevener AL, Bergman RN, Donovan CM. Hypoglycemic detection does not occur in the hepatic artery or liver: findings consistent with a portal vein glucosensor locus. Diabetes 50: 399–403, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Jackson PA, Cardin S, Coffey CS, Neal DW, Allen EJ, Penaloza AR, Snead WL, Cherrington AD. Effect of hepatic denervation on the counterregulatory response to insulin-induced hypoglycemia in the dog. Am J Physiol Endocrinol Metab 279: E1249–E1257, 2000 [DOI] [PubMed] [Google Scholar]
- 47.Jackson PA, Pagliassotti MJ, Shiota M, Neal DW, Cardin S, Cherrington AD. Effects of vagal blockade on the counterregulatory response to insulin-induced hypoglycemia in the dog. Am J Physiol Endocrinol Metab 273: E1178–E1188, 1997 [DOI] [PubMed] [Google Scholar]
- 48.Jokiaho A, Donovan CM, Watts AG. The rate of fall of blood glucose determines the necessity of forebrain-projecting catecholaminergic neurons for male rat adrenomedullary responses. Diabetes. First published April 16, 2014; 10.2337/db13-1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, Eiki J, Zhang BB, Levin BE. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 55: 412–420, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 53: 549–559, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Koyama Y, Coker RH, Denny JC, Lacy DB, Jabbour K, Williams PE, Wasserman DH. Role of carotid bodies in control of the neuroendocrine response to exercise. Am J Physiol Endocrinol Metab 281: E742–E748, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Koyama Y, Coker RH, Stone EE, Lacy DB, Jabbour K, Williams PE, Wasserman DH. Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes 49: 1434–1442, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Kumar P. How sweet it is: sensing low glucose in the carotid body. J Physiol 578: 627–627, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamarche L, Yamaguchi N, Péronnet F. Selective hypoglycemia in the liver induces adrenomedullary counterregulatory response. Am J Physiol Regul Integr Comp Physiol 270: R1307–R1316, 1996 [DOI] [PubMed] [Google Scholar]
- 55.Levin BE, Kang L, Sanders NM, Dunn-Meynell AA. Role of neuronal glucosensing in the regulation of energy homeostasis. Diabetes 55: S122–S130, 2006 [Google Scholar]
- 56.Levin BE, Magnan C, Dunn-Meynell A, Le Foll C. Metabolic sensing and the brain: who, what, where, and how? Endocrinology 152: 2552–2557, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levin BE. Metabolic sensors: viewing glucosensing neurons from a broader perspective. Physiol Behav 76: 397–401, 2002 [DOI] [PubMed] [Google Scholar]
- 58.Matveyenko AV, Donovan CM. Metabolic sensors mediate hypoglycemic detection at the portal vein. Diabetes 55: 1276–1282, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Mayer J. Regulation of energy intake and the body weight: the glucostatic theory and the lipostatic hypothesis. Ann NY Acad Sci 63: 15–43, 1955 [DOI] [PubMed] [Google Scholar]
- 60.McCrimmon R. The mechanisms that underlie glucose sensing during hypoglycaemia in diabetes. Diabet Med 25: 513–522, 2008 [DOI] [PubMed] [Google Scholar]
- 61.Mccrimmon RJ, Jacob RJ, Fan X, Mcnay EC, Sherwin RS. Effects of recurrent antecedent hypoglycaemia and chronic hyperglycaemia on brainstem extra-cellular glucose concentrations during acute hypoglycaemia in conscious diabetic BB rats. Diabetologia 46: 1658–1661, 2003 [DOI] [PubMed] [Google Scholar]
- 62.Mcnay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem 72: 785–790, 1999 [DOI] [PubMed] [Google Scholar]
- 63.Medeiros N, Dai L, Ferguson AV. Glucose-responsive neurons in the subfornical organ of the rat-a novel site for direct CNS monitoring of circulating glucose. Neurosci 201: 157–165, 2012 [DOI] [PubMed] [Google Scholar]
- 64.Miselis RR, Epstein AN. Feeding induced by intracerebroventricular 2-deoxy-d-glucose in the rat. Am J Physiol 229: 1438–1447, 1975 [DOI] [PubMed] [Google Scholar]
- 65.Morgan LO, Johnson CA. Experimental lesions in the tuber cinereum of the dog: followed by epileptiform convulsions and changes in blood chemistry. Arch Neurol Psychiatry 24: 696, 1930 [Google Scholar]
- 66.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol 297: C750–C758, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy BA, Fioramonti X, Jochnowitz N, Fakira K, Gagen K, Contie S, Lorsignol A, Penicaud L, Martin WJ, Routh VH. Fasting enhances the response of arcuate neuropeptide Y (NPY)-glucose-inhibited (GI) neurons to decreased extracellular glucose. Am J Physiol Cell Physiol 296: C746–C756, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niijima A, Fukuda A, Taguchi T, Okuda J. Suppression of afferent activity of the hepatic vagus nerve by anomers of D-glucose. Am J Physiol Regul Integr Comp Physiol 244: R611–R614, 1983 [DOI] [PubMed] [Google Scholar]
- 69.Niijima A. Afferent impulse discharges from glucoreceptors in the liver of the guinea pig. Ann NY Acad Sci 157: 690–700, 1969 [DOI] [PubMed] [Google Scholar]
- 70.Niijima A. Glucose-sensitive afferent nerve fibres in the hepatic branch of the vagus nerve in the guinea-pig. J Physiol 332: 315–323, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niijima A. Glucose-sensitive afferent nerve fibers in the liver and their role in food intake and blood glucose regulation. J Auton Nerv Syst 9: 207–220, 1983 [DOI] [PubMed] [Google Scholar]
- 72.Niijima A. The effect of D-glucose on the firing rate of glucose-sensitive vagal afferents in the liver in comparison with the effect of 2-deoxy-D-glucose. J Auton Nerv Syst 10: 255–260, 1984 [DOI] [PubMed] [Google Scholar]
- 73.Niijima A. Reflex control of the autonomic nervous system activity from the glucose sensors in the liver in normal and midpontine-transected animals. J Auton Nerv Syst 10: 279–285, 1984 [DOI] [PubMed] [Google Scholar]
- 74.Niijima A. Nervous regulation of metabolism. Prog Neurobiol 33: 135–147, 1989 [DOI] [PubMed] [Google Scholar]
- 75.Niijima A. Neural mechanisms in the control of blood glucose concentration. J Nutr 119: 833–840, 1989 [DOI] [PubMed] [Google Scholar]
- 76.Nurse C. Neurotransmission and neuromodulation in the chemosensory carotid body. Auton Neurosci 120: 1–9, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Nurse CA. Carotid body: new stimuli and new preparations: invited article. Adv Exp Med Biol 648: 29–38, 2009 [DOI] [PubMed] [Google Scholar]
- 78.Oomura Y, Ooyama H, Sugimori M, Nakamura T, Yamada Y. Glucose inhibition of the glucose-sensitive neurone in the rat lateral hypothalamus. Nature 247: 284–286, 1974 [DOI] [PubMed] [Google Scholar]
- 79.Oomura Y, Yoshimatsu H. Neural network of glucose monitoring system. J Auton Nerv Syst 10: 359–372, 1984 [DOI] [PubMed] [Google Scholar]
- 80.Paranjape SA, Briski KP. Recurrent insulin-induced hypoglycemia causes site-specific patterns of habituation or amplification of CNS neuronal genomic activation. Neurosci 130: 957–970, 2005 [DOI] [PubMed] [Google Scholar]
- 81.Patil GD, Briski KP. Lactate is a critical “sensed” variable in caudal hindbrain monitoring of CNS metabolic stasis. Am J Physiol Regul Integr Comp Physiol 289: R1777–R1786, 2005 [DOI] [PubMed] [Google Scholar]
- 82.Patterson LM, Zheng H, Ward SM, Berthoud HR. Vanilloid receptor (VR1) expression in vagal afferent neurons innervating the gastrointestinal tract. Cell Tissue Res 311: 277–287, 2003 [DOI] [PubMed] [Google Scholar]
- 83.Perseghin G, Regalia E, Battezzati A, Vergani S, Pulvirenti A, Terruzzi I, Baratti D, Bozzetti F, Mazzaferro V, Luzi L. Regulation of glucose homeostasis in humans with denervated livers. J Clin Invest 100: 931–941, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prechtl JC, Powley TL. Organization and distribution of the rat subdiaphragmatic vagus and associated paraganglia. J Comp Neurol 235: 182–195, 1985 [DOI] [PubMed] [Google Scholar]
- 85.Raybould HE. Gut chemosensing: Interactions between gut endocrine cells and visceral afferents. Auton Neurosci: Basic Clin 153: 41–46, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritter RC, Slusser PG, Stone S. Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213: 451–452, 1981 [DOI] [PubMed] [Google Scholar]
- 87.Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856: 37–47, 2000 [DOI] [PubMed] [Google Scholar]
- 88.Ritter S, Dinh TT. 2-Mercaptoacetate and 2-deoxy-D-glucose induce Fos-like immunoreactivity in rat brain. Brain Res 641: 111–120, 1994 [DOI] [PubMed] [Google Scholar]
- 89.Ritter S, Li AJ, Wang Q, Dinh TT. Minireview: the value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology 152: 4019–4032, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ritter S, Llewellyn-Smith I, Dinh TT. Subgroups of hindbrain catecholamine neurons are selectively activated by 2-deoxy-D-glucose induced metabolic challenge. Brain Res 805: 41–54, 1998 [DOI] [PubMed] [Google Scholar]
- 91.Ritter S, Watts AG, Dinh TT, Sanchez-Watts G, Pedrow C. Immunotoxin lesion of hypothalamically projecting norepinephrine and epinephrine neurons differentially affects circadian and stressor-stimulated corticosterone secretion. Endocrinology 144: 1357–1367, 2003 [DOI] [PubMed] [Google Scholar]
- 92.Rodríguez EM, Blázquez JL, Guerra M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: the former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 31: 757–776, 2010 [DOI] [PubMed] [Google Scholar]
- 93.Roper SD. Signal transduction and information processing in mammalian taste buds. Pflügers Arch 454: 759–776, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Routh VH, McArdle JJ, Levin BE. Phosphorylation modulates the activity of the ATP-sensitive K+ channel in the ventromedial hypothalamic nucleus. Brain Res 778: 107–119, 1997 [DOI] [PubMed] [Google Scholar]
- 95.Routh VH. Glucose sensing neurons in the ventromedial hypothalamus. Sensors 10: 9002–9025, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Russek M. Participation of hepatic glucoreceptors in the control of intake of food. Nature 197: 79–80, 1963 [DOI] [PubMed] [Google Scholar]
- 97.Saberi M, Bohland M, Donovan CM. The locus for hypoglycemic detection shifts with the rate of fall in glycemia: the role of portal-superior mesenteric vein glucose sensing. Diabetes 57: 1380–1386, 2008 [DOI] [PubMed] [Google Scholar]
- 98.Sachs E, Macdonald ME. Blood sugar studies in experimental pituitary and hypothalamic lesions. Arch Neurol Psychiatry 13: 335–368, 1925 [Google Scholar]
- 99.Salter-Venzon D, Watts AG. The role of hypothalamic ingestive behavior controllers in generating dehydration anorexia: a Fos mapping study. Am J Physiol Regul Integr Comp Physiol 295: R1009–R1019, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanders NM, Ritter S. Repeated 2-deoxy-D-glucose-induced glucoprivation attenuates Fos expression and glucoregulatory responses during subsequent glucoprivation. Diabetes 49: 1865–1874, 2000 [DOI] [PubMed] [Google Scholar]
- 101.Santiago JV, Clarke WL, Shah SD, Cryer PE. Epinephrine, norepinephrine, glucagon, and growth hormone release in association with physiological decrements in the plasma glucose concentration in normal and diabetic man. J Clin Endocrinol Metab 51: 877–883, 1980 [DOI] [PubMed] [Google Scholar]
- 102.Schmitt M. Influences of hepatic portal receptors on hypothalamic feeding and satiety centers. Am J Physiol 225: 1089–1095, 1973 [DOI] [PubMed] [Google Scholar]
- 103.Schneider K, Rezek M, Novin D. Effects of visceral sympathectomy on 2-deoxy-d-glucose induced eating. Physiol Behav 16: 55–58, 1976 [DOI] [PubMed] [Google Scholar]
- 104.Silver IA, Erecińska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14: 5068–5076, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50: 2673–2681, 2001 [DOI] [PubMed] [Google Scholar]
- 106.Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 54: 15–22, 2005 [DOI] [PubMed] [Google Scholar]
- 107.Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus. Am J Physiol Regul Integr Comp Physiol 291: R1283–R1287, 2006 [DOI] [PubMed] [Google Scholar]
- 108.Teff KL. How neural mediation of anticipatory and compensatory insulin release helps us tolerate food. Physiol Behav 103: 44–50, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thorens B. Sensing of Glucose in the Brain. Berlin, Heidelberg: Springer-Verlag, 2011 [Google Scholar]
- 110.Ward DS, Voter WA, Karan S. The role of the carotid bodies in the counter-regulatory response to hypoglycemia. Adv Exp Med Biol 648: 273–280, 2009 [DOI] [PubMed] [Google Scholar]
- 111.Ward SM, Bayguinov J, Won KJ, Grundy D, Berthoud HR. Distribution of the vanilloid receptor (VR1) in the gastrointestinal tract. J Comp Neurol 465: 121–135, 2003 [DOI] [PubMed] [Google Scholar]
- 112.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab 296: E11–E21, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watts AG, Donovan CM. Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendocrinol 31: 32–43, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wehrwein EA, Basu R, Basu A, Curry TB, Rizza RA, Joyner MJ. Hyperoxia blunts counterregulation during hypoglycaemia in humans: possible role for the carotid bodies? J Physiol 588: 4593–4601, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhu W, Czyzyk D, Paranjape SA, Zhou L, Horblitt A, Szabó G, Seashore MR, Sherwin RS, Chan O. Glucose prevents the fall in ventromedial hypothalamic GABA that is required for full activation of glucose counterregulatory responses during hypoglycemia. Am J Physiol Endocrinol Metab 298: E971–E977, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zierler K. Whole body glucose metabolism. Am J Physiol Endocrinol Metab 276: E409–E426, 1999 [DOI] [PubMed] [Google Scholar]