

Abstract
Hypertension and associated inflammatory processes that accelerate cardiovascular damage are regulated by the innate immune system. Toll-like receptors (TLR) are major components of the innate immune system that recognize endogenous damage-associated molecular patterns to activate prominent inflammatory signaling including activation of nuclear factor-κB (NF-κB). However, the role of TLR in the etiology of hypertension is not well understood. TLR signaling is dependent on adaptor proteins that, along with the TLR expression patterns, confer specificity of the inflammatory response and its pathological targets. Here we review the conceptual framework of how TLR and their adaptor proteins may differentially affect hypertension and cardiac hypertrophy by different stimuli.
Keywords: hypertension, innate immune system, toll-like receptors, MyD88
innate immunity and the immune functions in which Toll-like receptors (TLR) play important roles are essential in our response to challenges by pathogens and in our autoimmune responses to cellular damage (3). The responses may include cytokine and chemokine production (e.g., TNF-α, interferons, interleukins) among others. The cytokine cascade initiated by TLR activation defines the transition from innate to adaptive immunity and T cell regulation and differentiation. In this minireview we focus on the role of immunity on hypertension (1, 7, 11, 13, 19, 26, 28, 35). The phenotypic manifestations of hypertension involve excessive sympathetic activation, renal glomerular injury, cardiac hypertrophy and arrhythmias, vascular end-organ damage, atherosclerosis, and stroke. The immune system plays a significant role in these events by inducing inflammatory processes in the central nervous system (CNS), the cardiovascular, and the renal systems (1, 7, 11–13, 18, 19, 21, 24, 26–28, 35).
Toll-Like Receptors and Adaptor Proteins
Since inflammation progresses even in the absence of pathogens as a result of endogenous “damage-associated molecular patterns” (DAMPs), a role of TLRs, which are activated by DAMPs has been proposed (20) as a link between inflammation and hypertension. TLRs are major components of the innate immune system that detect sterile injury and elicit cytokine and chemokine production primarily through activation of the pro-inflammatory transcription factor nuclear factor-κB (NF-κB). However, despite the known effects of NF-κB and inflammation in hypertension and end-organ damage, little is known about the mechanism of TLR involvement in hypertensive state.
A confounding factor in elucidation of this role of TLRs is their diversity. About a dozen TLRs have been identified in mammals (34). They are expressed in both immune cells and nonimmune cells in specific combinations (16). Different TLRs are expressed on the cell surface, and a few are in the intracellular endosomal compartments. They recognize specific agonists with distinct molecular patterns, yet all TLRs signal to activate NF-κB. They function as homo- or heterodimers in defined combinations and require adaptor proteins and a variety of kinases for downstream signaling (5).
At least five adaptor proteins are known to participate in propagation of downstream TLR signaling (22, 34). One adaptor protein in particular, the myeloid differentiation factor 88 (MyD88), is common to all TLRs except TLR-3. Based on the participation of adaptor proteins in TLR signaling, the pathways are defined as MyD88 dependent or MyD88 independent (15). Thus, despite having the commonality of NF-κB activation, the specificity of TLR signaling in a cell is achieved by its expression of a TLR type (e.g., TLR-2, -3, -4, etc.) and its ligand recognition as well as by expression and utilization of adaptor proteins. Consequently, this specificity of pathway selection in a given pathological state may yield different outcomes that define the phenotype of tissue injury and organ damage.
Role of TLRs in Different Types of Hypertension
Several studies have implicated the activation of various TLRs in the inflammatory responses and vascular or renal damage in different types of hypertension.
Preeclampsia.
In preeclampsia the placenta shows exaggerated necrosis of trophoblastic cells, which results in the release of mitochondrial DNA that becomes a ligand for TLR-9. The resulting activation of the innate immune cells produces significant maternal inflammation, vascular dysfunction that leads to hypertension, and intrauterine growth restriction (10). It is also proposed that viral mimetics, which are TLR-3 ligands, cause hypertension, endothelial dysfunction, and proteinuria in rats and mice only when pregnant and that this preeclampsia-like syndrome is exacerbated in mice by the absence of the anti-inflammatory IL-10 (6, 32).
Renal diseases.
There are also increasing data to support a major role of the innate immune receptors and particularly TLR-7 as a key regulator of immune complex-mediated glomerulonephritis and specifically murine models of systemic lupus erythematosus (8, 31).
Spontaneously hypertensive rat.
In the spontaneously hypertensive rat (SHR), Bomfim et al. (4) have identified increased expression of TLR-4 protein in mesenteric arteries and showed that treatment with anti-TLR-4 antibody for 15 days decreased blood pressure and decreased IL-6 levels in the serum and TLR-4 protein in arteries. Conversely, Eissler et al. (9) found that TLR-4 is overexpressed in cardiac myocytes of SHR and that treatment with the TLR-4 ligand heparan sulfate increased cytokines TNF-α and IL-6 mRNA in the heart. Others have reported that nuclear NF-κB p65 subunit was increased in the tissue of SHR indicating an inflammatory state and treatment with an α7-nicotinic acetylcholine receptor agonist PNU-282987 reduced the levels of inflammatory cytokines (17). It is of interest, however, that despite the suppression of inflammation in the end organs, the arterial pressure was not reduced suggesting an uncoupling of processes that lead to inflammation from those that elevate arterial pressure in the SHR.
In this genetic model of spontaneous hypertension, there is increased infiltration of T cells and macrophages and increased NF-κB activity in the kidney during the prehypertensive stage that progressively increase with age (25). Pharmacological inhibition of proinflammatory transcription factor NF-κB by a broad-spectrum inhibitor phyrrolidine dithiocarbamate normalized blood pressure in adult SHR and reduced immune cell infiltration in the kidney (23). Although it is not known that these inflammatory events are directly related to TLRs, defective TLR signaling in SHR immune cells may contribute to genetic hypertension. As mentioned earlier, neutralizing antibody to TLR-4 reduced the blood pressure in the adult SHR (4). Our laboratory has shown that the TLR signaling involving the neuro-immuno-sympathetic connection from the CNS to the immune system is altered in SHR (13). Treatment of splenocytes with nicotine, a ligand for anti-inflammatory α7-nicotinic acetylcholine receptors, inhibit TLR-activated cytokine production in normotensive Wistar-Kyoto rats (WKY) but increases cytokine production in SHR.
Angiotensin II hypertension.
ANG II-induced hypertension has a major adaptive immune component dependent on the presence of T cells and their migration to end organs including the kidney (11). Ahn et al. (2) reported that ANG II also plays a pivotal role in activating the innate immune response in chronic cyclosporine nephropathy. Infusion of ANG II for 14 days increased mRNA for TNF-α and TLR-2 and the number of dendritic cells in the kidney, whereas losartan decreased these responses as well as the cyclosporine A-induced renal injury. ANG II also upregulated TLR-4 expression and increased TNF-α in rat vascular smooth muscle cells, which appear to be mediated by AT-1 receptors and ERK activation (14). An increase in superoxide resulting from a reduction in extracellular superoxide dismutase (ecSOD) is abrogated in TLR4−/− mice.
ANG II-induced hypertension and organ damage also depend on specific adaptor protein molecules based in the signaling pathway of TLR. Our preliminary results suggest that different TLR adaptors may be operational in the cardiac hypertrophy from myocardial infarction (MI) than in the ANG II-induced cardiac hypertrophy (29).
Contrasting Pathways in Cardiac Hypertrophy
TLR-4 has been implicated to play a role in hypertension (4, 9, 20). It is unique among TLRs as it signals through both MyD88-dependent and MyD88-independent pathways. We have previously reported that the MyD88-dependent pathway is involved in post-MI myocardial inflammation and cardiac remodeling. MyD88-deficient mice had a remarkably high survival after an MI (82% in MyD88−/− vs. 50% in wild type) (30). In addition, expression of proinflammatory genes, fibrosis and cardiac hypertrophy were attenuated in MyD88−/− mice. When we directly tested the role of MyD88 in ANG II-induced hypertension, we found that chronic ANG II infusion increased blood pressure and caused cardiac hypertrophy equally in wild-type (WT) and in MyD88−/− mice (29). Thus, whereas the MyD88-dependent pathway contributed to MI-induced cardiac hypertrophy, it did not play a significant role in ANG II-induced cardiac hypertrophy, which must be mediated by a MyD88-independent pathway. In fact, expression of proinflammatory and cardiac hypertrophy marker genes tended to be enhanced with ANG II infusion in MyD88−/− mice. Interestingly, ANG II-induced cardiac hypertrophy was reduced in TLR-4 deficient mice, whereas ANG II-mediated hypertension was sustained (29).
These results suggest that cardiac hypertrophy may be induced by two different innate immune inflammatory pathways, one being MyD88 dependent (in myocardial infarction) and the other MyD88 independent (ANG II infusion). Furthermore, the ANG II-induced hypertension may be uncoupled from ANG II-cardiac hypertrophy with the latter being dependent on TLR-4 activation and an alternative adaptor protein, while hypertension may require an alternative TLR and adaptor protein (Fig. 1).
Fig. 1.
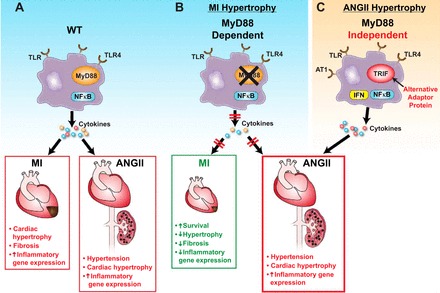
Pathways of Toll-like receptors (TLR) in hypertension and cardiac hypertrophy. A: myocardial infarction (MI)-induced cardiac hypertrophy and ANG II-induced hypertension and cardiac hypertrophy through intact TLR pathways in the wild type (WT). B: MI-induced cardiac hypertrophy is dependent on the major TLR adaptor protein myeloid differentiation factor 88 (MyD88) but ANG II-induced hypertension and hypertrophy are not. C: MyD88-independent pathways may regulate ANG II-induced cardiac hypertrophy and hypertension.
In conclusion, available data clearly suggest an association between inflammation and hypertension and related end-organ damage. The innate immune system, especially the TLRs, appears to play an important role in the hypertensive state. However, the link between TLRs and hypertension has not been fully explored. Activation of TLRs in CNS neurons and glia may evoke a sympathetic drive that causes and sustains hypertension. Activation of cholinergic, adrenergic, or AT1 receptors on immune cells may in turn significantly modulate their cytokine responses to TLR activation and the resulting induction of inflammatory genes in end organs. Given the diversity of TLRs and their adaptor proteins, it is likely that several distinct signaling pathways might be involved in the pathology of hypertension in various tissues, e.g., brain, kindeys, blood vessels, as well as in different hypertension phenotypes. Further studies in the role of specific combinations of TLRs and their adaptors will unravel these important signaling paradigms.
Perspectives and Significance
Increased immune cell infiltration and cytokine production are clearly associated with hypertension and end-organ damage. However, the molecular mechanisms underlying these events are now being elucidated. A role of the innate immune system and TLRs is emerging as a determinant of the pathology. These exciting findings allow us to set further goals to understand the specific molecular mechanisms involved in bridging inflammation, hypertension, and end-organ damage to the TLRs. Among the important results will be the identification of specific TLRs and their endogenous ligands activating the innate immune pathways. In addition, adaptor proteins may also be important targets of therapeutic interventions since they define the specificity of the pathological processes (33).
GRANTS
The work of the authors referred to in this review was supported by research grant from the National Institutes of Health (HL-14388).
REFERENCES
- 1.Abboud FM, Harwani SC, Chapleau MW. Autonomic neural regulation of the immune system: implications for hypertension and cardiovascular disease. Hypertension 59: 755–762, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn KO, Lim SW, Li C, Yang HJ, Ghee JY, Kim JY, Kim SH, Kim J, Yang CW. Influence of angiotensin II on expression of toll-like receptor 2 and maturation of dendritic cells in chronic cyclosporine nephropathy. Transplantation 83: 938–947, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430: 257–263, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 122: 535–543, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure 19: 447–459, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee P, Chiasson VL, Kopriva SE, Young KJ, Chatterjee V, Jones KA, Mitchell BM. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension 58: 489–496, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27: 801–810, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eissler R, Schmaderer C, Rusai K, Kuhne L, Sollinger D, Lahmer T, Witzke O, Lutz J, Heemann U, Baumann M. Hypertension augments cardiac Toll-like receptor 4 expression and activity. Hypertens Res 34: 551–558, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Goulopoulou S, Matsumoto T, Bomfim GF, Webb RC. Toll-like receptor 9 activation: a novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin Sci (Lond) 123: 429–435, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, hypertension. Hypertension 57: 132–140, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res 111: 1190–1197, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji Y, Liu J, Wang Z, Liu N. Angiotensin II induces inflammatory response partly via toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem 23: 265–276, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol 167: 5887–5894, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun 388: 621–625, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Li DJ, Evans RG, Yang ZW, Song SW, Wang P, Ma XJ, Liu C, Xi T, Su DF, Shen FM. Dysfunction of the cholinergic anti-inflammatory pathway mediates organ damage in hypertension. Hypertension 57: 298–307, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Luft FC, Dechend R, Muller DN. Immune mechanisms in angiotensin II-induced target-organ damage. Ann Med 44, Suppl 1: S49–S54, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, Iwakura Y, Blinder Y, Rahman A, Quyyumi AA, Harrison DG. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 31: 1565–1572, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 306: H184–H196, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mian MO, Paradis P, Schiffrin EL. Innate immunity in hypertension. Current Hypertens Reports 16: 413, 2014 [DOI] [PubMed] [Google Scholar]
- 22.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Rev Immunol 7: 353–364, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Rodríguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor-κB prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exptl Ther 315: 51–57, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nature Rev Nephrol 10: 56–62, 2014 [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND. Evolution of renal interstitial inflammation and NF-kappaB activation in spontaneously hypertensive rats. Am J Nephrol 24: 587–594, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Ryan MJ. Does interleukin 6 contribute to renal hemodynamic changes during angiotensin II-dependent hypertension? Hypertension 56: 819–821, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryan MJ. An update on immune system activation in the pathogenesis of hypertension. Hypertension 62: 226–230, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh M, Chapleau M, Harwani S, Abboud F, The immune system and hypertension. Immunol Res, 2014, May 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh MV, Cicha MZ, Chapleau MW, Abboud FM. Selective pathways of innate immune responses of heart and kidney during angiotensin II hypertension. In: Experimental Biology 2014. San Diego, CA: 2014, p. A54–A857.851 [Google Scholar]
- 30.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol 52: 1135–1144, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith KD. Toll-like receptors in kidney disease. Curr Opin Nephrol Hypertensi 18: 189–196, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tinsley JH, Chiasson VL, Mahajan A, Young KJ, Mitchell BM. Toll-like receptor 3 activation during pregnancy elicits preeclampsia-like symptoms in rats. Am J Hypertens 22: 1314–1319, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Ve T, Gay NJ, Mansell A, Kobe B, Kellie S. Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr Drug Targets 13: 1360–1374, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Watters TM, Kenny EF, O'Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol 85: 411–419, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Zubcevic J, Waki H, Raizada MK, Paton JF. Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension 57: 1026–1033, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]