

Abstract
20-Hydroxyeicosatetraenoic acid (20-HETE), Cyp4a-derived eicosanoid, is a lipid mediator that promotes tumor growth, as well as causing detrimental effects in cerebral circulation. We determined whether concurrent inhibition of cyclooxygenase-2 (COX-2) and 20-HETE affects colon tumor growth and ischemic stroke outcomes. The expression of Cyp4a and COXs and production of 20-HETE and PGE2 were determined in murine colon carcinoma (MC38) cells. We then examined the effects of combined treatment with rofecoxib, a potent COX-2 inhibitor, and HET0016, a potent Cyp4a inhibitor, on the growth and proliferation of MC38 cells. Subsequently, we tested the effects of HET0016 plus rofecoxib in MC38 tumor and ischemic stroke models. Cyp4a and COXs are highly expressed in MC38 cells. Respectively, HET0016 and rofecoxib inhibited 20-HETE and PGE2 formation in MC38 cells. Moreover, rofecoxib combined with HET0016 had greater inhibitory effects on the growth and proliferation of MC38 cells than did rofecoxib alone. Importantly, rofecoxib combined with HET0016 provided greater inhibition on tumor growth than did rofecoxib alone in MC38 tumor-bearing mice. Prolonged treatment with rofecoxib selectively induced circulating 20-HETE levels and caused cerebrovascular damage after ischemic stroke, whereas therapy with rofecoxib and HET0016 attenuated 20-HETE levels and reduced rofecoxib-induced cerebrovascular damage and stroke outcomes during anti-tumor therapy. Thus these results demonstrate that combination therapy with rofecoxib and HET0016 provides a new treatment of colon tumor, which can not only enhance the anti-tumor efficacy of rofecoxib, but also reduce rofecoxib-induced cerebrovascular damage and stroke outcomes.
Keywords: rofecoxib, HET0016, tumor growth, stroke
colorectal cancer (crc) is a major public health problem because it is estimated that 142,820 new cases and 50,830 deaths from CRC in the United States in 2013 (National Cancer Institute website). Early detection and chemoprevention of CRC are two major strategies against it. Moreover, early management of premalignant lesions can significantly reduce CRC-related mortality (20, 38). Coxibs are the potential chemopreventive agents in the clinical trials, because inflammation is associated with the development of CRC (41, 42).
Based on strong data that cyclooxygenase (COX)-2 is the major enzyme for the production of inflammatory PGE2 and PGI2, and that COX-1 is a key enzyme in the production of cytoprotective PGs in the stomach (9), the Food and Drug Administration has approved the use of three coxibs: rofecoxib, celecoxib, and valdecoxib. Clinically, nonsteroidal anti-inflammatory drugs (NSAIDs) are the primary choice for the treatment of inflammation (19). However, use of conventional NSAIDs is often associated with significant gastrointestinal complications, such as ulcers and bleeding. Coxibs were developed to reduce the side effects of NSAIDs (9). While substantial evidence from clinical trials [Adenoma Prevention with Celecoxib (APC), Adenomatous Polyp Prevention on Vioxx (APPROVe), and Prevention of Colorectal Sporadic Adenomatous Polyps (PRESAP) trial] demonstrates that coxibs reduce and prevent the incidence of CRC (3–5), long-term use of rofecoxib is also associated with an increased risk of side effects, including stroke and cardiovascular events (5). Consequently, rofecoxib (Vioxx) and valdecoxib were withdrawn from the market; clinical trials of the use of rofecoxib were stopped in 2004. Currently, celecoxib (Celebrex), which is less potent than rofecoxib, is the only coxib on the market. Thus the current unmet need in this field is to develop a new strategy that can not only enhance the efficacy of coxibs, but also reduce coxib-induced cerebrovascular damage.
20-Hydroxyeicosatetraenoic acid (20-HETE), the ω-hydroxylation product of arachidonic acid (AA), is a lipid mediator in cerebral microvasculature and has detrimental effects on cerebral circulation (8, 23, 29, 33, 43). In addition, several studies have demonstrated that 20-HETE acts as a mitogenic and angiogenic factor that promotes the growth of different types of tumors (1, 12, 45). In the present study, we determined whether concurrent inhibition of COX-2 and 20-HETE affects colon tumor growth and ischemic stroke outcomes.
METHODS
Animals.
Eight-week-old male C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). All mice were maintained on a 12:12-h light-dark cycle and were housed five mice to a cage. All animal protocols were approved by the Institutional Animal Care and Use Committee and were in accord with the requirements of the National Research Council Guide for the Care and Use of Laboratory Animals.
Cell culture of MC38 cells, HCMECL, RBMEC, and NCM460.
Murine colon carcinoma (MC38) cells were obtained from Dr. Michael R. Shurin (University of Pittsburgh, Pittsburgh, PA). MC38 cells were maintained in complete medium consisting of DMEM supplemented with 10% heat-inactivated FBS, 100 mg/ml streptomycin, and 100 U/ml penicillin in a humidified incubator at 37°C with 5% CO2. Subconfluent cultured cells were harvested by washing T-75 flasks two times with Ca2+- and Mg2+-free Earl's balanced salts, followed by 2-min incubation with 0.25% trypsin-EDTA (Life Technologies, Grand Island, NY) at 37°C. A human cerebral microvascular endothelial cell line (HCMECL) was obtained from Dr. Jason Zastre (University of Georgia, Athens, GA). Primary rat brain microvascular endothelial cells (RBMEC) were prepared as described previously (30). HCMECL and RBMEC were maintained in MCDB-131 complete medium (VEC Technologies, Rensselaer, NY). NCM460, a normal human colon mucosal epithelial cell line, cells were obtained from Dr. Kebin Liu (Georgia Regents University) and were maintained in RPMI-1640 medium (Corning Cellgro, Manassas, VA).
Measurement of 20-HETE and PGE2 levels in the media and cell lysates of MC38 cells.
MC38 cells were collected using 0.25% trypsin-EDTA. After centrifugation, a cell pallet was resuspended in 300 μl of PBS. Cell lysates were generated by brief sonication. The concentrations of 20-HETE in media and cell lysates were determined by liquid chromatography–tandem mass spectrometry (LC/MS/MS) using 15(S)-HETE-d8 as an internal standard, as described previously (7). The media and cell lysates were collected for PGE2 measurement. The concentrations of PGE2 in media and cell lysates were determined by an ELISA kit (Cayman Chemical, Ann Arbor, MI), according to the manufacturer's instructions.
Western blot analysis.
Expression of Cyp4a, COXs, β-actin, and GADPH was analyzed by Western blot analysis using the lysates of MC38 cells, HCMECL, and RBMEC, as well as homogenates of mouse brain microvessels (MBM) and normal mouse colon samples. Identical amounts of protein samples were separated by NuPAGE 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA) at 125 V for 3 h. Our laboratory has previously described the detailed procedures for transfer, blocking, and washing the samples (7). The membranes were incubated with antibody against Cyp4a polyclonal antibody [1:2,000; Abcam (catalog no.: ab3573), Cambridge, MA], COX-1 [1:500; Cayman Chemical (catalog no.: 160110)], COX-2 [1:500; Cayman Chemical (catalog no.: 160106)], β-actin (1:5,000; Sigma, St. Louis, MO), or GAPDH [1:10,000; Abcam (catalog no.: ab8245)]. According to the manufacturer's instruction, this Cyp4a antibody can recognize human, rat, and mouse Cyp4a isoforms. Similarly, the COX-1 and COX-2 antibody can recognize human, rat, and mouse COX-1 and COX-2, respectively. The membranes were incubated with secondary antibody for Cyp4a, COX-1, COX-2, β-actin, or GAPDH. We developed the immunoblots using an ECL detection kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Measurement of cell proliferation of MC38 and NCM460 cells by counting cell numbers.
For cell growth experiments, cells were seeded onto six-well plates at a density of 2.5 × 105 cells/well. Cell growth of MC38 and NCM460 was followed 48 h after seeding with serum-free medium containing epidermal growth factor (EGF) (200 ng/ml) in the presence or absence of various concentrations of HET0016 (Cayman Chemical), rofecoxib (Toronto Research Chemicals), or HET0016 + rofecoxib. HET0016 and rofecoxib were dissolved in EtOH; an equal volume of EtOH was used as vehicle control. After 48 h of treatment, cells were collected using 0.25% trypsin-EDTA and counted using a hemocytometer.
Measurement of cell proliferation of MC38 and NCM460 cells by bromodeoxyuridine ELISA assay.
MC38 cells were seeded at a density of 5 × 103 cells/well in 10% FBS/DMEM in 96-well plates, and NCM460 cells were maintained in 10% FBS/RPMI-1640 in 96-well plates. The next day, 10% FBS medium was removed and replaced with serum-free medium containing EGF (200 ng/ml) in the presence or absence of various concentrations of HET0016, rofecoxib, or HET0016 + rofecoxib for 48 h. Bromodeoxyuridine (BrdU) ELISA assay was done using an ELISA kit (CycLex, Nagano, Japan). Briefly, 3 h before assay, BrdU (10 μM final concentration) was added to the 96-well plates containing MC38 cells or NCM460 cells given different incubation conditions. After 3 h of incubation, the culture medium was removed and fixed with fixing/denaturing solution (200 μl/well) at room temperature for 30 min. After removing fixing/denaturing solution, plates were incubated with monoclonal anti-BrdU antibody (50 μl/well) at room temperature for 1 h. Plates were washed five times with wash buffer (200 μl/well) and then incubated with the secondary antibody (50 μl/well) at room temperature for 1 h. After removing the secondary antibody and doing a second wash procedure, plates were incubated with substrate reagent (50 μl/well) at room temperature for 15 min in the dark. The reaction was then stopped by adding stop solution (50 μl/well). The absorbance in each well was measured using a Tecan GENios Plus microplate reader at dual wavelengths of 450/540 nm.
Measurement of tumor growth in vivo.
We subcutaneously injected 8-wk-old male C57BL/6J mice with MC38 cells (1.4 × 106 cells/mouse) to induce colon tumor. MC38 colon tumor-bearing mice were divided into vehicle, HET0016 (5 mg·kg−1·day−1 or 10 mg·kg−1·day−1 ip), rofecoxib (50 mg/l in drinking water), or HET0016 + rofecoxib. All treatments were done for 21 days. Rofecoxib (50 mg/l) was suspended in drinking water containing 1% (vol/vol) PEG 400 (Sigma-Aldrich) and 0.5% (wt/vol) 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich). The formula and dose of rofecoxib were prepared according to a previous publication (21). Tumor growth rates were monitored by measuring tumor volume with a sliding caliper on day 9 or day 18 after cell implantation. The volumes of tumors were calculated from the major dimension (L) and minor dimension (S), using the equation: tumor volume = L × (S)2/2, as described previously (16).
Measurement of 20-HETE and other eicosanoids in plasma samples by LC/MS/MS analysis.
MC38 tumor mice were divided into three treatment groups and treated for 3 wk with the following: rofecoxib (50 mg/l), rofecoxib (50 mg/l) plus HET0016 (5 mg·kg−1·day−1 ip), or vehicle. The formula and doses of rofecoxib and HET0016 were based on previous publications (21, 25).
After 3 wk of different treatments, MC38 tumor mice were anesthetized with 2% isoflurane delivered by an anesthesia apparatus. About 0.6 ml of venous blood was collected from mice using a 1-ml syringe containing sodium citrate. The samples were centrifuged at 2,400 g for 15 min to obtain platelet-free plasma. To stabilize eicosanoids in plasma samples, 10-μl combinations of antioxidants [EDTA (0.2 mg/l), butylated hydroxytoluene (0.2 mg/l), and triphenyphosphine (2 mg/l)] were added to each plasma sample. The plasma samples were stored at −80°C until LC/MS/MS analysis. Plasma samples were spiked with 10 ng of 15(S)-HETE-d8, applied to preconditioned SEP-Pak C18 cartridges (100 mg adsorbent, Waters), and washed with water followed by hexane. Eicosanoids were eluted with 500 μl of ethyl acetate-hexane (3:1). The eluate was dried under nitrogen and reconstituted in methanol-aqueous ammonium acetate (25 mM) (7:3). The extracted and reconstituted sample was subjected to HPLC (Shimadzu Prominence XR system) on a Max-RP C18 column (2 × 150 mm, 3 μm, Phenomenex) isocratically eluted with methanol-aqueous ammonium acetate (13 mM) (8:2) at a flow rate of 0.4 ml/min. The eluent was monitored for 20-HETE, epoxyeicosatrienoic acids (EETs) (5,6-EET, 8,9-EET, 11,12-EET, 14,15-EET), dihydroxyeicosatrienoic acids (DHETs) (5,6-DHET, 8,9-DHET, 11,12-DHET, 14,15-DHET), 5-HETE, 8-HETE, 11-HETE, 15-HETE, PGJ2, PGE2, thromboxane (Tx) B2, and 15-keto-PGE2 by mass spectrometer (QTRAP5500, ABSCIEX) in the negative ion mode using multiple reaction monitoring under optimized conditions. 15(S)-HETE-d8 was used as the internal standard for recovery and quantitation. The concentrations of eicosanoids were determined by comparing the ratio of ion intensity of each eicosanoid vs. that of 15(S)-HETE-d8, as described previously (7, 22).
Assessment of neurovascular injury and functional outcome after ischemic stroke.
MC38 tumor mice were divided into three treatment groups and treated for 3 wk with the following: rofecoxib (50 mg/l), rofecoxib (50 mg/l) plus HET0016 (5 mg·kg−1·day−1 ip), or vehicle. We also used age-matched non-tumor mice as control. At the end of the treatment period, mice were subjected to thromboembolic stroke. Under isoflurane anesthesia, the right common carotid artery, the right external carotid artery, and the right internal carotid artery were exposed through a middle incision on the ventral side of the neck. A PE10 catheter containing a fibrin-rich clot was gently inserted from the external carotid artery stump into the distal internal carotid artery just proximal to the origin of the middle cerebral artery. Each clot (∼1 cm), prepared from a donor mouse, was injected with 75 μl of sterile PBS; after 2 min, the catheter was withdrawn as described previously (14). Occlusion was confirmed by a ≥70% drop in cerebral blood flow compared with the preischemic value. The success rate of thromboembolic middle cerebral artery occlusion was 95% based on changes in cerebral blood flow.
At day 3 after surgery, mice were first evaluated for functional outcome and then killed for evaluation of neurovascular injury, including infarct size and bleeding. Extracted brains were analyzed for infarct size in coronal slices of 2 mm thickness, labeled A-G, front to back. Hemorrhagic transformation (HT), secondary bleeding into the brain after ischemic stroke, was evaluated by the presence of visible macroscopic bleeding and measured in a binary fashion: yes or no. Number of animals that had HT was reported per group. 2,3,5-Triphenyltetrazolium chloride, a mitochondria stain, was used to outline the infarct area. The captured images were numerically labeled and analyzed using specialized KS300 software. Infarct size was expressed as a percentage of the contralateral hemisphere. Neurological deficits in mice given different treatments were assessed at 24 and 72 h after stroke, using a 5-point scale for scoring: 0, no deficit; 1, forelimb flexion deficit on contralateral side; 2, decreased resistance to lateral push and torso turning to the ipsilateral side when held by tail; 3, significant circling to the affected side and reduced capability to bear weight on the affected side; and 4, rarely moved spontaneously and preferred to stay at rest.
Statistical analysis.
All values are expressed as means ± SE. All data were analyzed by GraphPad Instat Software (LaJolla, CA). We used one-way ANOVA and Tukey-Kramer tests for multiple comparisons or independent Student's t-test for unpaired groups. Bleeding was analyzed by Fisher's exact test. Statistical significance was set at P < 0.05 or 0.01.
RESULTS
Cyp4a expression and 20-HETE production in MC38 cells.
It is well established that Cyp4a isoforms are important for the production of 20-HETE (34). To determine whether MC38, the murine colon carcinoma, cells have the capacity to synthesize 20-HETE, cell lysates were prepared from MC38 cells for Western blot analysis. We used normal mouse colon tissue homogenates as a control. Intriguingly, the expression of Cyp4a is absent in normal colon tissue, whereas its expression is upregulated in MC38 cells (Fig. 1A).
Fig. 1.
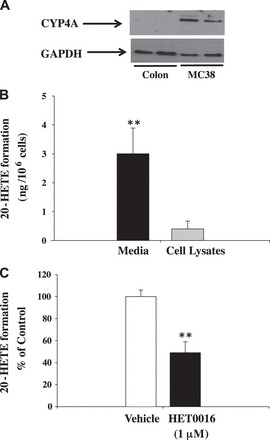
A: representative Western blot analysis of Cyp4a in samples of normal mouse colon and murine colon carcinoma (MC38) cell lysate. Western blot analysis showed that Cyp4a is highly expressed in murine colon carcinoma cells. Based on protein standards, the size of Cyp4a isoforms is ∼52 kDa. B: 20-hydroxyeicosatetraenoic acid (20-HETE) levels were higher in media than in cell lysates of MC38 cells. C: effect of HET0016 on 20-HETE production in MC38 cells, and results are expressed as %control. 20-HETE levels were determined by liquid chromatography–tandem mass spectrometry (LC/MS/MS). Values are means ± SE; n = 4. **P < 0.01 vs. control.
To test whether 20-HETE production occurs in MC38 cells, we determined 20-HETE levels in the lysates and media of cultured MC38 cells. We detected 20-HETE predominantly in the cell media and found much lower amounts in cell lysates (Fig. 1B). To determine the effect of HET0016, the selective inhibitor of Cyp4a enzymes, on 20-HETE production, we incubated MC38 cells with HET0016 (1 μM) or vehicle. We found that HET0016 was effectively to block 20-HETE production in the media of cultured MC38 cells (Fig. 1C).
COX-2 expression and PGE2 production in MC38 cells.
To determine whether MC38 cells have the capacity to generate PGs, we did Western blot analysis of COX enzymes in the lysates of MC38 cells. MC38 cell lysates contained a 72-kDa protein that cross-reacted with COX-2 antibody (Fig. 2A), which is in agreement with a previous report that COX-2 is expressed in MC38 cells (17). Similarly, COX-1 is also expressed in MC38 cells. Experiments were also done to compare PGE2 production in MC38 cells. PGE2 was detected mainly in the media of cultured MC38 cells (72 ng/106 cells in the media vs. 0.8 ng/106 cells in cell lysates) (Fig. 2B). Moreover, we found that rofecoxib dose dependently inhibited PGE2 production in MC38 cells (Fig. 2C), indicating that rofecoxib effectively blocks PGs synthesis in these cells.
Fig. 2.
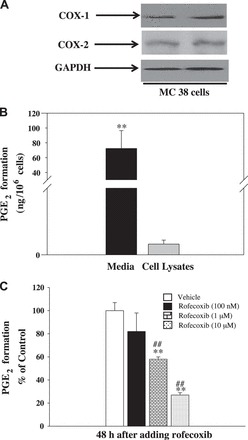
A: representative Western blot analysis of cyclooxygenases (COXs) in the lysates of MC38 cells. Western blot analysis showed that COX-1 and COX-2 are highly expressed in MC38 cells. Based on protein standards, the size of COXs is ∼72 kDa. B: PGE2 levels were higher in media than in cell lysates of MC38 cells. C: effect of rofecoxib on PGE2 production in MC38 cells, and results are expressed as %control. PGE2 production was estimated by a PGE2 ELISA. Values are means ± SE; n = 5. **P < 0.01 vs. cell lysates. ##P < 0.01 vs. other groups.
Combination of rofecoxib with HET0016 causes an additive inhibitory effect on the growth and proliferation of MC38 cells.
It is well known that HET0016 avidly binds to serum proteins (13). To determine whether HET0016 alters the inhibitory effects of rofecoxib on the proliferation of colon cancer cells, MC38 cells were cultured in serum-free medium. The growth of these cells was stimulated with EGF, a mitogen of many cells, at a concentration of 200 ng/ml, as described previously (13). Using the same culture conditions as those described earlier, both HET0016 alone (Fig. 3A) and rofecoxib alone (Fig. 3B) inhibited cell growth. Interestingly, combining rofecoxib (1 μM) with HET0016 (1 μM) increased the inhibitory effect on the growth of MC38 cells compared with rofecoxib alone (Fig. 3C).
Fig. 3.
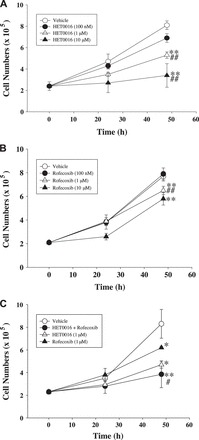
Effects of HET0016, rofecoxib, and rofecoxib + HET0016 on MC38 cell proliferation. MC38 cells growing in six-well plates were treated with HET0016 (100 nM to 10 μM; A), rofecoxib (100 nM to 10 μM; B), or HET0016 (1 μM) + rofecoxib (1 μM) (C) for 48 h. Cells were harvested by trypsinization and counted using a hemacytometer. Results are expressed as cell number/well and are means ± SE; n = 5. *P < 0.05, **P < 0.01 vs. vehicle. #P < 0.05, ##P < 0.01 vs. other groups.
To confirm the effects of rofecoxib and HET0016 on the proliferation of colon cancer cells, MC38 cells were cultured in serum-free medium with added EGF (200 ng/ml). The effects of HET0016 alone, rofecoxib alone, and HET0016 + rofecoxib on the proliferation of MC38 cells were determined by assessing the incorporation of a thymidine analog, BrdU. HET0016 alone (Fig. 4A) concentration-dependently inhibited the incorporation of BrdU. Rofecoxib at 1 μM significantly inhibited BrdU incorporation (Fig. 4B). Notably, HET0016 (1 μM) + rofecoxib (1 μM), compared with rofecoxib alone, displayed an enhanced inhibitory effect on BrdU incorporation (Fig. 4C). These results demonstrate that inhibition of 20-HETE enhances the inhibitory effect of rofecoxib on the proliferation of MC38 cells.
Fig. 4.
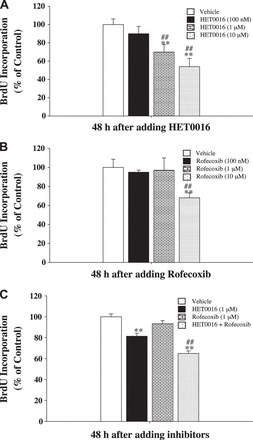
Effects of HET0016, rofecoxib, and rofecoxib + HET0016 on bromodeoxyuridine (BrdU) incorporation into MC38 cells. MC38 cells growing in 96-well plates were treated with HET0016 (100 nM to 10 μM; A), rofecoxib (100 nM to 10 μM; B), or HET0016 (1 μM) + rofecoxib (1 μM) (C) for 48 h. In the last 3 h, BrdU was added (10 μM final concentration). Incorporation of BrdU was determined using an ELISA kit. Results are expressed as %control and are means ± SE; n = 5. **P < 0.01 vs. vehicle. ##P < 0.01 vs. other groups.
In complementary experiments, we determined whether HET0016, rofecoxib, or HET0016 + rofecoxib affect the proliferation of a normal human colon mucosal epithelial cell line, NCM460 cells. We found that neither HET0016 or rofecobix alone nor HET0016 + rofecoxib affected cell growth (Fig. 5A) and BrdU incorporation (Fig. 5B) in NCM460 cells. These results indicate that blockade of COX-2 and 20-HETE does not affect the proliferation of normal colon epithelial cells.
Fig. 5.
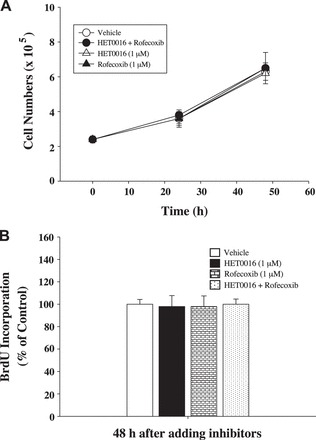
Effects of HET0016, rofecoxib, and rofecoxib + HET0016 on cell numbers (A) and BrdU incorporation in NCM460 cells (B). NCM460 cells were treated with HET0016 (1 μM), rofecoxib (1 μM), HET0016 (1 μM) + rofecoxib (1 μM), or vehicle for 48 h. A: cell proliferation by counting cell numbers was determined as described in Fig. 3. B: BrdU incorporation was determined using an ELISA kit as described in Fig. 4. Values are means ± SE; n = 4 (A), n = 5 (B).
Inhibition of 20-HETE enhances anti-tumor effects of rofecoxib in MC38 colon tumor-bearing mice.
To study the effects of combination therapy with rofecoxib and HET0016 on colon tumor, we subcutaneously injected 8-wk-old male C57BL/6J mice with MC38 cells (1.4 × 106 cells/mouse) to induce colon tumor. The effects of HET0016 alone, rofecoxib alone, and HET0016 + rofecoxib on tumor growth were determined by assessing tumor volumes at 9 days and 18 days after the implantation of MC38 cells. Treatment with HET0016 did not affect tumor size on day 9 postimplantation, but dose dependently reduced tumor size on day 18 postimplantation (Fig. 6A). Treatment with rofecoxib significantly reduced tumor size on both day 9 and day 18 postimplantation (Fig. 6B). Interestingly, on days 9 and 18 postimplantation, treatment with HET0016 + rofecoxib increased the inhibition of tumor growth achieved with rofecoxib alone (Fig. 6B), clearly demonstrating that inhibition of 20-HETE enhances the anti-tumor efficacy of rofecoxib in MC38 tumor-bearing mice.
Fig. 6.
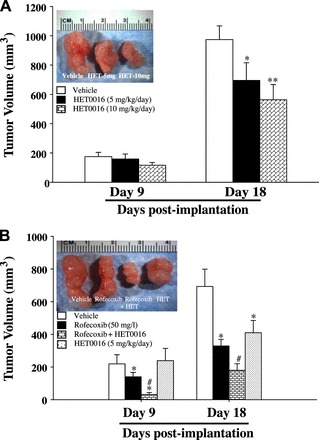
A: dose effects of HET0016 treatment on subcutaneous MC38 tumors in C57B6 mice. MC38 colon tumor-bearing mice (1.4 × 106 cells/mouse, injected subcutaneously) were divided into vehicle, HET0016 (5 mg·kg−1·day−1 ip), or HET0016 (10 mg·kg−1·day−1 ip) treatment groups. After 9 or 18 days postimplantation, tumor volumes were determined using a sliding caliper. B: effects of rofecoxib + HET0016 on tumor growth in MC38 tumor mice. MC38 tumor mice were divided into vehicle, rofecoxib (50 mg/l), HET0016 (5 mg·kg−1·day−1 ip), or rofecoxib + HET0016 treatment groups. After 9 and 18 days postimplantation, tumor volumes were determined. Values are means ± SE; n = 6. *P < 0.05, **P < 0.01 vs. vehicle. #P < 0.05 vs. other groups.
In a complementary experiment, we determined the water intake and body weight of mice treated with water, vehicle, and rofecoxib (50 mg/l). We found that neither vehicle (3.02 ± 0.17 vs. 3.16 ± 0.16 ml·24 h−1·mouse−1, n = 5) nor rofecoxib (3.07 ± 0.1 vs. 3.16 ± 0.16 ml·24 h−1·mouse−1, n = 5) treatment affected water intake. Likewise, there is no significant change of body weight among water (24 ± 1.63 g/mouse, n = 5), vehicle (23.8 ± 1.1 g/mouse, n = 5), and rofecoxib (24.5 ± 1.0 g/mouse, n = 5) groups. These results indicate that neither vehicle nor rofecoxib treatment has significant impact on eating/drinking habits.
Prolonged treatment with rofecoxib selectively increases circulating 20-HETE levels in MC38 colon tumor-bearing mice.
To determine whether treatment with rofecoxib and rofecoxib + HET0016 affects circulating 20-HETE levels during anti-tumor therapy, we measured the levels of plasma 20-HETE and other eicosanoids in MC38 tumor mice given different treatments. As shown in Fig. 7, rofecoxib selectively increased 20-HETE levels in MC38 tumor mice. It is possible that the increased 20-HETE levels were caused by elevation of the substrate, AA, after blocking COX-2. However, we found no significant changes in levels of EETs/DHETs, 5-HETE, 8-HETE, 11-HETE, or 15-HETE after rofecoxib treatment (Fig. 7A). Notably, HET0016 selectively reduced plasma 20-HETE levels without affecting EETs/DHETs, 5-HETE, 8-HETE, 11-HETE, or 15-HETE in MC38 tumor mice (Fig. 7A). To exclude that HET0016 affects COX pathway, we determined plasma PGs levels after HET0016 treatment. HET0016 did not affect plasma PGJ2, PGE2, TxB2, and 15-keto-PGE2 levels (Fig. 7B), suggesting that HET0016 does not alter COX pathway in vivo. These results demonstrate that prolonged treatment with rofecoxib selectively induces circulating 20-HETE levels. To exclude the possibility that rofecoxib affects the expression of 20-HETE synthesizing enzyme, we determined the expression of Cyp4a in the liver and kidney. Rofecoxib had no effect on Cyp4a expression (Fig. 7C), suggesting that rofecoxib does not affect 20-HETE levels through altering the expression of Cyp4a levels.
Fig. 7.
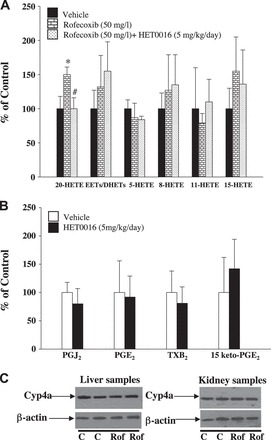
A: MC38 colon tumor-bearing mice were divided into 3 treatment groups: vehicle, rofecoxib (50 mg/l), or rofecoxib + HET0016 (5 mg·kg−1·day−1 ip). After 3 wk of treatment, plasma levels of 20-HETE, epoxyeicosatrienoic acids (EETs)/dihydroxyeicosatrienoic acids (DHETs), and other HETEs in tumor mice given different treatments were determined by LC/MS/MS. The levels of different eicosanoids were quantified by comparing the ratio of eicosanoids vs. 15(S)-HETE-d8. The plasma 20-HETE, 5-HETE, 8-HETE, 11-HETE, and 15-HETE levels were 2.84 ± 0.5, 10 ± 1.4, 4 ± 0.9, 5.23 ± 0.8, and 1.34 ± 0.5 ng/ml, respectively, in the vehicle-treated group. The EETs-to-DHETs ratio in the vehicle-treated group was 0.9 ± 0.2. Values are means ± SE; n = 4. *P < 0.05 vs. vehicle. #P < 0.05 vs. rofecoxib group. B: MC38 colon tumor-bearing mice were treated with HET0016 (5 mg·kg−1·day−1 ip) or vehicle for 3 wk. At the end of treatment, plasma PGs levels were determined by LC/MS/MS. Values are means ± SE. C: effects of 3-wk treatment of rofecoxib on Cyp4a expression in the liver and kidney of MC38 tumor mice. C, control; Rof, rofecoxib treatment.
Expression of COX-1, COX-2, and Cyp4a in HCMECL, primary RBMEC, and MBM.
It is possible that increased circulating 20-HETE levels by rofecoxib are a result of a decrease in metabolism by COX-2 because 20-HETE can be metabolized by COX enzymes (6, 28, 37). To investigate whether the cerebrovasculature has the capacity to synthesize and metabolize 20-HETE, we determined the expression of COX-1, COX-2, and Cyp4a in HCMECL, primary RBMEC, and MBM. We found that COX-1 (∼70 kDa), COX-2 (∼72 kDa), and Cyp4a (∼52 kDa) were expressed in HCMECL (Fig. 8A), primary RBMEC (Fig. 8B), and MBM (Fig. 8C). These results provide evidence that the cerebrovasculature is involved in the synthesis and metabolism of 20-HETE across species.
Fig. 8.
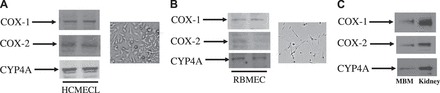
Expression of COX-1, COX-2, and Cyp4a in human cerebral microvascular endothelial cell line (HCMECL; A), rat brain microvascular endothelial cells (RBMEC; B), and mouse brain microvessels (MBM; C). Microscopic images shown on the right of A and B are views of cultured HCMECL and RBMEC, respectively. We used 30–50 μg of protein from different samples for Western blot analysis. Based on protein standards (Bio-Rad), the respective sizes of COX-1, COX-2, and Cyp4a are ∼70, ∼72, and ∼52 kDa, respectively.
Combined treatment with rofecoxib and HET0016 reduces the adverse effects of rofecoxib on ischemic stroke.
Since chronic treatment with rofecoxib significantly increased plasma 20-HETE levels (Fig. 7A), we investigated whether cotreatment with 20-HETE inhibitor would alter the adverse effects of rofecoxib on ischemic stroke during anti-tumor therapy. MC38 colon tumor-bearing mice treated with vehicle, rofecoxib, or rofecoxib plus HET0016, as well as control mice (nontumor), were subjected to ischemic stroke using a modified thromboembolic model that closely mimics ischemia-reperfusion injury in patients with acute ischemic stroke. We found that cerebrovascular damage was significantly increased after ischemic stroke in the rofecoxib-treated group, as shown by an 80% occurrence rate of bleeding as opposed to no visible bleeding in the other groups (Fig. 9A). In addition, mortality was ∼30% in the rofecoxib group compared with 15% in other groups, indicating that rofecoxib had deleterious effects on the brain during anti-tumor therapy.
Fig. 9.
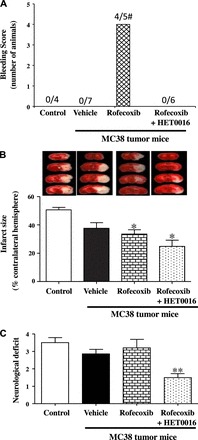
MC38 colon tumor-bearing mice treated with vehicle, rofecoxib (50 mg/l), or rofecoxib + HET0016 (5 mg·kg−1·day−1 ip), as well as control mice (nontumor), were subjected to ischemic stroke using a modified thromboembolic model. A: presence of bleeding was evaluated as a measure of vascular injury. Four out of five animals in the rofecoxib group developed macroscopic bleeding into the brain, while there was no visible bleeding in any other groups. #P = 0.0008 (Fisher's exact test). B: infarct size was significantly smaller in rofecoxib-treated groups. *P < 0.05 vs. vehicle. A representative image from each group is shown on top. C: functional outcome was greatly improved in the rofecoxib + HET0016 group, as indicated by a lower neurological deficit score. **P < 0.01 vs. other groups. Values are means ± SE; n = 4 in control and n = 5–7 in other groups.
We also found that rofecoxib reduced the size of infarcts compared with those in control mice. There was a trend toward further decrease in infarction size with HET0016 treatment (Fig. 9B). Although there appeared to be a decrease in infarct size in the vehicle-treated tumor mice, this did not reach statistical significance (Fig. 9B). Despite reduced infarct size, neurological deficit scores were higher in the rofecoxib group. However, treatment with rofecoxib + HET0016 significantly improved functional outcome (Fig. 9C). Thus combination treatment with HET0016 and rofecoxib significantly reduced the rofecoxib-induced adverse stroke events during anti-tumor therapy.
DISCUSSION
Although substantial evidence from APC, APPROVe, and PreSAP clinical trials demonstrates that coxibs reduce the incidence of CRC or even prevent it (3–5), the increased risk of side effects, including stroke and cardiovascular events, stopped the use of coxibs as cancer preventive agents. The prevailing theory to explain the adverse cardiovascular effects of coxibs is that they reduce the production of PGI2, a potent inhibitor of platelet aggregation, but do not affect the production of TxA2, a potent platelet-aggregating agent (9). Thus stroke and the cardiovascular complications caused by coxibs might be a consequence of a shifting balance between the levels of PGI2 and TxA2 (10). Although this theory is attractive, it cannot fully explain why other nonselective COX inhibitors, including diclofenac, ibuprofen, naproxen, and indomethacin, also significantly increase the risk of side effects (21). Hence, more complex mechanisms may be responsible for the coxib-induced side effects.
Interestingly, Liu et al. (21) recently reported that chronic administration of rofecoxib greatly increases 20-HETE levels without affecting other eicosanoid pathways. 20-HETE has well-characterized detrimental effects on cerebral circulation (33), and it promotes the growth of tumors in the brain (13), kidney (1), and lung (45). Since 20-HETE inhibitor effectively blocks tumor growth and reduces detrimental effects in cerebral circulation, we hypothesized that cotreatment with COX-2 and 20-HETE inhibitor could be a novel strategy, not only enhancing the anti-tumor efficacy of coxibs, but also attenuating the adverse effects of stroke induced by prolonged treatment with coxibs.
To determine the role of 20-HETE in colon cancer, we addressed the following questions. Is 20-HETE produced in colon cancer cells? If so, what is the action of 20-HETE in these cells? In addressing these questions, we found that the expression of Cyp4a is absence in normal colon tissue, whereas its expression is upregulated in colon carcinoma cells (Fig. 1A). These results suggest that increasing 20-HETE may play important role in CRC development because previous reports have shown that 20-HETE not only promotes the proliferation of renal adenocarcinoma cells (1) and human glioma cells (12), but also stimulates angiogenesis in non-small lung cancer cells (45). Further investigation is needed to determine whether Cyp4a is also upregulated in human colon cell lines and whether 20-HETE inhibitor is effective to block human colon tumor growth. Interestingly, a previous report (15) has demonstrated that the expression of Cyp epoxygenase 2J2 is undetectable in normal tissues, whereas its expression is upregulated in human carcinoma tissues. The exact mechanisms whereby the upregulation of Cyp4a and Cyp2J2 occurs in cancer tissues are still not clear, and it is required for further investigation.
To the best of our knowledge, the present study is the first to show that 20-HETE is produced in MC38 cells (Fig. 1). It is well known that 20-HETE synthesis is primarily catalyzed by the Cyp4a gene family (34). In the mouse, four isoforms have been identified: Cyp4a10, Cyp4a12a, Cyp4a12b, and Cyp4a14. Using baculovirus and Sf9 insect cells, Muller et al. (26), in a study of the hydroxylation of AA by mouse Cyp4a isoforms, demonstrated that AA ω-hydroxylation is catalyzed by Cyp4a10, Cyp4a12a, and Cyp4a12b. Cyp4a12a and Cyp4a12b have similar catalytic activity for 20-HETE production, with a Vmax value ∼10 min−1 and a Km value ∼20–40 μM. Furthermore, the ω-hydroxylase activity of AA for Cyp4a10 is ∼25- to 75-fold lower than that of Cyp4a12 isoforms. Therefore, it is possible that one of the mouse Cyp4a isoforms is responsible for 20-HETE synthesis in MC38 cells.
To determine the action of 20-HETE in colon cancer cells, we demonstrated that HET0016 dose-dependently inhibited the growth and proliferation of MC38 cells (Figs. 3A and 4A). Interestingly, a previous study (12) showed that overexpression of CYP4A1, a potent 20-HETE synthase, while having negligible ability to form EETs (12, 27), induces hyperproliferative phenotype in human glioma cells (U251). They also demonstrated that elevated 20-HETE levels not only increases oxidative stress and promote angiogenesis, but also induce the phosphorylation of extracellular signal-regulated kinase-1/2, cyclin D-1/2, and vascular endothelial growth factor in U251 cells. Therefore, it is possible that 20-HETE stimulates proliferation of MC38 cells through similar molecular mechanisms. Importantly, we found that 20-HETE blockade did not affect the proliferation of NCM460 cells, a normal human colon mucosal epithelial cell line (Fig. 5). These data demonstrate that the action of 20-HETE is mainly mediated in the colon cancer cells rather than in normal colon cells.
To determine whether 20-HETE inhibition enhances the anti-tumor effects of COX-2 inhibitor in colon cancer cells, we addressed the question of whether the combined therapy with rofecoxib and HET0016 would have an increased inhibitory effect on the proliferation of colon cancer cells in vitro and tumor growth in vivo. Our data showed that, although monotherapy with either HET0016 or rofecoxib reduced the proliferation of MC38 cells and tumor growth, greater anti-proliferative and anti-tumor effects were obtained when rofecoxib was administered in combination with HET0016 (Figs. 4 and 6).
Although the present study provided new information about the additional inhibitory effect of rofecoxib + HET0016 on the growth of colon tumor, the exact mechanisms whereby this combination blocked tumor growth were still not known. It is possible that PGE2, the major COX-2-derived PG, was mainly present in the medium of MC38 cells (Fig. 2B), which is consistent with the notions that PGE2 is secreted by cancer cells, and that multidrug resistance protein-4 is involved in this process (32). Thus the action of PGE2 to promote MC38 tumor growth may have been mediated through its paracrine and autocrine action. It is well known that PGE2 promotes the growth of CRC by stimulating angiogenesis and cell proliferation, as well as inhibiting apoptosis (42). Therefore, it is possible that the enhanced inhibitory effect of rofecoxib by HET0016 (Fig. 6B) on tumor growth was because this combination blocked both the paracrine/autocrine action of PGE2 and mitogenic and angiogenic action (1, 12, 45) of 20-HETE in colon cancer cells. Moreover, because CRC is a heterogeneous disease, involving multiple dysregulated pathways (40, 42), the cotreatment therapy with rofecoxib and HET0016 may increase therapeutic efficacy.
To determine whether 20-HETE inhibition reduces the adverse effects of rofecoxib on stroke during anti-tumor therapy, we addressed the following questions. Does prolonged use of rofecoxib elevate 20-HETE levels? If so, does 20-HETE inhibitor attenuate the adverse effects on stroke during anti-tumor therapy? We found that 3 wk of anti-tumor therapy with rofecoxib significantly increased circulating 20-HETE levels (Fig. 7). This increase was highly selective because rofecoxib treatment did not affect levels of EETs/DHETs, 5-HETE, 8-HETE, 11-HETE, or 15-HETE (Fig. 7). These results are consistent with the finding in a previous report (21) that, in C57BL/6J mice, the administration of rofecoxib causes greatly induction of 20-HETE levels without affecting circulating levels of LOX- and COX-derived eicosanoids.
The exact mechanisms whereby rofecoxib induces 20-HETE are still unknown. Since rofecoxib treatment does not affect the expression of Cyp4a in the liver and kidney of MC38 tumor mice (Fig. 7C), the reason for the induction of circulating 20-HETE levels by rofecoxib (Fig. 7A) may not be a consequence of the increases in endogenous 20-HETE synthesis. It is well established that 20-HETE can be metabolized by COXs into PGs, such as 20-OH PGE2 and 20-OH PGF2-α (6, 21). Therefore, it is possible that the increase in 20-HETE levels induced by prolonged use of rofecoxib in vivo is caused by inhibition of the metabolizing pathways of 20-HETE by COX enzymes in brain microvessels. To test this possibility, we determined whether the cerebrovasculature has the capacity to carry out 20-HETE synthesis and metabolism. We found that COX-1, COX-2, and Cyp4a are expressed in HCMECL, RBMECs, and MBM (Fig. 8), providing evidence that the cerebrovasculature is involved in 20-HETE synthesis and metabolism.
Although the clinical trials have established that regular use of coxibs over 10–15 yr reduces the risk of CRC by 40–50% (42), prolonged use of coxibs was associated with higher risk of stroke and cardiovascular events. Thus a great deal of effort has focused on reducing the side effects of coxibs. One approach to achieve this goal is to use lower doses of coxibs in combination with other complementary agents. Accumulating evidence has demonstrated that combination treatment provides better efficacy in anti-cancer therapy with coxibs. For example, combining celecoxib with sorafenib, a multikinase inhibitor, enhanced inhibition of the growth of hepatocellular carcinoma cells (24). Combined therapy of rofecoxib with S-1, an oral fluoropyryzine drug, produced more potent efficacy to restrain liver metastasis of LM-H3 cells than each agent alone (39); and celecoxib combined with AEE788, a tyrosine kinase inhibitor, potentiated celecoxib-mediated inhibition of proliferation and angiogenesis in HCT 15 colon cancer cells (40). In the present study, we showed that rofecoxib + HET0016 provides better blockage of MC38 tumor growth than does rofecoxib alone (Fig. 6). Therefore, combined treatment with rofecoxib and HET0016 could be clinically important for the treatment of CRC because it would allow the use of a lower dose of rofecoxib, which could reduce complications induced by its prolonged use.
To determine whether chronic treatment with rofecoxib causes the adverse effects associated with stroke, nontumor mice and MC38 tumor mice treated with vehicle, rofecoxib, or rofecoxib + HET0016 were subjected to embolic stroke using a humanized clot model. HT, which is secondary bleeding into the brain after an ischemic event, is an important complication of ischemic stroke. Studies have shown that disability and death occur more frequently in ischemic stroke patients who develop HT (18). We found that HT was significantly increased in rofecoxib-treated mice. Four out of the five animals in that group developed HT, whereas there was no visible bleeding in the other groups (Fig. 9). Thus chronic administration of rofecoxib was associated with increased complications of ischemic stroke. These results are in agreement with clinical data showing long-term use of rofecoxib is associated with a risk of stroke (5, 36). It is well known that 20-HETE is a potent vasoconstrictor of cerebral arteries (11); it also increases platelet aggregation and shortens bleeding time (21); it contributes to the development of cerebral vasospasm (35); it also contributes to neuronal cell death after ischemia-reperfusion injury (33); and it is involved in neuronal excitotoxicity in vivo (43). It is possible that elevated levels of 20-HETE induced by rofecoxib during anti-tumor therapy (Fig. 7) trigger the detrimental action of 20-HETE in cerebral circulation, thereby causing the adverse effects associated with ischemic stroke. Notably, combined treatment with rofecoxib + HET0016 attenuated the adverse effects associated with ischemic stroke induced by rofecoxib (Fig. 9). These results demonstrate that this combination therapy can enhance anti-tumor effects, but reduce deleterious cerebrovascular events. Since the relative degree of selectivity for COX-2 inhibition is rofecoxib >> celecoxib (2), further investigation is needed to determine whether combination therapy with 20-HETE inhibition and celecoxib is also effective to prevent celecoxib-induced side effects.
Previous studies documented that 20-HETE analogs, including WIT0013 and N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine (5,14–20-HEDGE), and 20-HETE antagonist (WIT002) are important tools to study the function of 20-HETE pathway in vivo. For example, Regner et al. (31) found that 5,14,20-HEDGE protects kidney from ischemia-reperfusion injury. Another report (44) demonstrated that WIT0013 increases cerebral vascular tone, whereas WIT002 attenuates 20-HETE-induced vasoconstriction response in vascular smooth muscle cells. Thus it will be interesting to determine the roles of 20-HETE analogs and antagonists in coxib-induced stroke event.
Perspectives and Significance
We have made the novel findings that 20-HETE is produced in colon cancer cells, where it promotes the proliferation of cancer cells and tumor growth. In vitro and in vivo, combined treatment with rofecoxib + HET0016 displayed greater anti-proliferative and anti-tumor effects than did rofecoxib alone. Our LC/MS/MS analysis supported the notion that prolonged use of rofecoxib increases circulating 20-HETE levels, which could trigger cerebrovascular events, during anti-tumor therapy. It appears that combination with rofecoxib and HET0016 significantly reduces rofecoxib-induced cerebrovascular damage and stroke outcomes. Therefore, this study provides a novel intervention strategy that concurrent inhibition of 20-HETE and COX-2 can not only enhance anti-tumor efficacy of coxibs, but also prevent coxib-induced adverse stroke events.
GRANTS
This study was supported by the following grants: American Heart Association Grant-in-Aid grants (AHASE00054; AHASE00090) to M.-H. Wang; National Institute of Neurological Disorders and Stroke Grant NS-054688 to A. Ergul; National Natural Science Foundation of China (NSFCN81000341) to P. Luo; and Division of Research Resources Grant S10-RR-027926 to K. R. Maddipati.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.Z., M.N.H., X.Z., W.L., K.R.M., T.S., and M.-H.W. performed experiments; Y.Z., M.N.H., X.Z., K.R.M., A.E., and M.-H.W. analyzed data; Y.Z. and M.-H.W. prepared figures; P.L. and M.-H.W. conception and design of research; P.L., T.S., A.E., and M.-H.W. edited and revised manuscript; T.S., A.E., and M.-H.W. drafted manuscript; M.-H.W. interpreted results of experiments; M.-H.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Jeanne D. Cole for editorial assistance.
REFERENCES
- 1.Alexanian A, Rufanova VA, Miller B, Flasch A, Roman RJ, Sorokin A. Down-regulation of 20-HETE synthesis and signaling inhibits renal adenocarcinoma cell proliferation and tumor growth. Anticancer Res 29: 3819–3824, 2009 [PMC free article] [PubMed] [Google Scholar]
- 2.Antman EM, DeMets D, Loscalzo J. Cyclooxygenase inhibition and cardiovascular risk. Circulation 112: 759–770, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Arber N, Spicak J, Racz I, Zavoral M, Breazna A, Gerletti P, Lechuga MJ, Collins N, Rosenstein RB, Eagle CJ, Levin B. Five-year analysis of the prevention of colorectal sporadic adenomatous polyps trial. Am J Gastroenterol 106: 1135–1146, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 355: 873–884, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 352: 1092–1102, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Carroll MA, Pilar Gracia M, Falck JR, McGiff JC. Cyclooxygenase dependency of the renovascular actions of cytochrome P450-derived arachidonate metabolites. J Pharmacol Exp Ther 260: 104–109, 1992 [PubMed] [Google Scholar]
- 7.Chen L, Fan C, Zhang Y, Bakri M, Dong H, Morisseau C, Maddipati KR, Luo p Wang CY, Hammock BD, Wang MH. Beneficial effects of inhibition of soluble epoxide hydrolase on glucose homeostasis and islet damage in a streptozotocin-induced diabetic mouse model. Prostaglandins Other Lipid Mediat 104–105: 42–48, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crago EA, Thampatty BP, Sherwood PR, Kuo CW, Bender C, Balzer J, Horowitz M, Poloyac SM. Cerebrospinal fluid 20-HETE is associated with delayed cerebral ischemia and poor outcomes after aneurysmal subarachnoid hemorrhage. Stroke 42: 1872–1877, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.FitzGerald GA. Coxibs and cardiovascular disease. N Engl J Med 351: 1709–1711, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol 50: 470–479, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER, Harder DR. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J Physiol 507: 771–781, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo AM, Sheng J, Scicli GM, Arbab AS, Lehman NL, Edwards PA, Falck JR, Roman RJ, Scicli AG. Expression of CYP4A1 in U251 human glioma cell induces hyperproliferative phenotype in vitro and rapidly growing tumors in vivo. J Pharmacol Exp Ther 327: 10–19, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo M, Roman RJ, Fenstermacher JD, Brown SL, Falck JR, Arbab AS, Edwards PA, Scicli AG. 9L gliosarcoma cell proliferation and tumor growth in rats are suppressed by N-hydroxy-N′-(4-butyl-2-methylphenol) formamidine (HET0016), a selective inhibitor of CYP4A. J Pharmacol Exp Ther 317: 97–108, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Hoda MN, Li W, Ahmad A, Ogbi S, Zemskova MA, Johnson MH, Ergul A, Hill WD, Hess DC, Sazonova IY. Sex-independent neuroprotection with minocycline after experimental thromboembolic stroke. Exp Transl Stroke Med 3: 16, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, Wang DW. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res 65: 4707–4715, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Konson A, Ben-Kasus T, Mahajna JA, Danon A, Rimon G, Agbaria R. Herpes simplex virus thymidine kinase gene transduction enhances tumor growth rate and cyclooxygenase-2 expression in murine colon cancer cells. Cancer Gene Ther 11: 830–840, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Konson A, Mahajna JA, Danon A, Rimon G, Agbaria R. The involvement of nuclear factor-kappa B in cyclooxygenase-2 overexpression in murine colon cancer cells transduced with herpes simplex virus thymidine kinase gene. Cancer Gene Ther 13: 1093–1104, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Kunte H, Busch MA, Trostdorf K, Vollnberg B, Harms L, Mehta RI, Castellani RJ, Mandava P, Kent TA, Simard JM. Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas. Ann Neurol 72: 799–806, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li JX, Zhang Y. Emerging drug targets for pain treatment. Eur J Pharmacol 681: 1–5, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Lieberman DA. Screening for colorectal cancer. Clin Cornerstone 4: 1–10, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Liu JY, Li N, Yang J, Li N, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc Natl Acad Sci U S A 107: 17017–17022, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maddipati KR, Zhou SL. Stability and analysis of eicosanoids and docosanoids in tissue culture media. Prostaglandins Other Lipid Mediat 94: 59–72, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Marumo T, Eto K, Wake H, Omura T, Nabekura J. The inhibitor of 20-HETE synthesis, TS-011, improves cerebral microcirculatory autoregulation impaired by middle cerebral artery occlusion in mice. Br J Pharmacol 161: 1391–1402, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morisaki T, Umebayashi M, Kiyota A, Koya N, Tanaka H, Onishi H, Katano M. Combining celecoxib with sorafenib synergistically inhibits hepatocellular carcinoma cells in vitro. Anticancer Res 33: 1387–1395, 2013 [PubMed] [Google Scholar]
- 25.Mu Y, Klamerus MM, Miller TM, Rohan LC, Graham SH, Poloyac SM. Intravenous formulation of N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine (HET0016) for inhibition of rat brain 20-hydroxyeicosatetraenoic acid formation. Drug Metab Dispos 36: 2324–2330, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller DN, Schmidt C, Barbosa-Sicard E, Wellner M, Gross V, Hercule H, Markovic M, Honeck H, Luft FC, Schunck WH. Mouse Cyp4a isoforms: enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem J 403: 109–118, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen X, Wang MH, Reddy KM, Falck JR, Schwartzman ML. Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol Regul Integr Comp Physiol 276: R1691–R1700, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Oyekan AO. Differential effects of 20-hydroxyeicosatetraenoic acid on intrarenal blood flow in the rat. J Pharmacol Exp Ther 313: 1289–1295, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J Cereb Blood Flow Metab 26: 1551–1561, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Prakash R, Somanath PR, El-Remessy AB, Kelly-Cobbs A, Stern JE, Dore-Duffy P, Johnson M, Fagan SC, Ergul A. Enhanced cerebral but not peripheral angiogenesis in the Goto-Kakizaki model of type 2 diabetes involves VEGF and peroxynitrite signaling. Diabetes 61: 1533–1542, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ. Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int 75: 511–517, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M, Wijnholds J, Borst P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 100: 9244–9249, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, Koehler RC, Harder DR, Roman RJ. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 29: 629–639, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roman RJ. P450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Roman RJ, Renic M, Dunn KM, Takeuchi K, Hacein-Bey L. Evidence that 20-HETE contributes to the development of acute and delayed cerebral vasospasm. Neurol Res 28: 738–749, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Roumie CL, Mitchel EF, Jr, Kaltenbach L, Arbogast PG, Gideon P, Griffin MR. Nonaspirin NSAIDs, cyclooxygenase 2 inhibitors, and the risk for stroke. Stroke 39: 2037–2045, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Schwartzman ML, Falck JR, Yadagiri P, Escalante B. Metabolism of 20-hydroxyeicosatetraenoic acid by cyclooxygenase. Formation and identification of novel endothelium-dependent vasoconstrictor metabolites. J Biol Chem 264: 11658–11662, 1989 [PubMed] [Google Scholar]
- 38.Sheehan KM, Sheahan K, O'Donoghue DP, MacSweeney F, Conroy RM, Fitzgerald DJ, Murray FE. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA 282: 1254–1257, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Tachimori A, Yamada N, Amano R, Ohira M, Hirakawa K. Combination therapy of S-1 with selective cyclooxygenase-2 inhibitor for liver metastasis of colorectal carcinoma. Anticancer Res 28: 629–638, 2008 [PubMed] [Google Scholar]
- 40.Venkatesan P, Bhutia SK, Singh AK, Das SK, Dash R, Chaudhury K, Sarkar D, Fisher PB, Mandal M. AEE788 potentiates celecoxib-induced growth inhibition and apoptosis in human colon cancer cells. Life Sci 91: 789–799, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Wang D, DuBois RN. Eicosanoids and cancer. Nat Rev Cancer 10: 181–193, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D, DuBois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29: 781–788, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang ZJ, Carter EL, Kibler KK, Kwansa H, Crafa DA, Martin LJ, Roman RJ, Harder DR, Koehler RC. Attenuation of neonatal ischemic brain damage using a 20-HETE synthesis inhibitor. J Neurochem 121: 168–179, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu M, Cambj-Sapunar L, Kehl F, Maier KG, Takeuchi K, Miyata N, Ishimoto T, Reddy LM, Falck JR, Gebremedhin D, Harder DR, Roman RJ. Effects of a 20-HETE antagonist and agonists on cerebral vascular tone. Eur J Pharmacol 486: 297–306, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Yu W, Chen L, Yang YQ, Falck JR, Guo AM, Li Y, Yang J. Cytochrome P450 omega-hydroxylase promotes angiogenesis and metastasis by upregulation of VEGF and MMP-9 in non-small cell lung cancer. Cancer Chemother Pharmacol 68: 619–629, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]