

This paper describes a two-stage microfluidic system to enrich CD34-positive (CD34+) stem cells from mouse skin with high purity. Microfluidic devices minimize the processing time of stem cells in the starting population, are easily accessible, and can be multiplexed to separate large amounts of cell mixture. The released CD34+ stem cells were transplanted into full-thickness skin defects in nude mice. Follicles and hairs were regenerated, and sebaceous glands were formed in the graft sites.
Keywords: Microfluidic devices, CD34-positive stem cells, Skin graft site, Nude mice
Abstract
Skin stem cells resident in the bulge area of hair follicles and at the basal layer of the epidermis are multipotent and able to self-renew when transplanted into full-thickness defects in nude mice. Based on cell surface markers such as CD34 and the α6-integrin, skin stem cells can be extracted from tissue-derived cell suspensions for engraftment using the gold standard cell separation technique of fluorescence-activated cell sorting (FACS). This paper describes an alternative separation method using microfluidic devices coated with degradable antibody-functionalized hydrogels. The microfluidic method allows direct injection of tissue digestate (no preprocessing tagging of cells is needed), is fast (45 minutes from injected sample to purified cells), and scalable. This method is used in this study to isolate CD34-positive (CD34+) cells from murine skin tissue digestate, and the functional capability of these cells is demonstrated by transplantation into nude mice using protocols developed by other groups for FACS-sorted cells. Specifically, the transplantation of microfluidic isolated CD34+ cells along with dermal and epidermal cells was observed to generate significant levels of hair follicles and sebaceous glands consistent with those observed previously with FACS-sorted cells.
Introduction
The regenerative capability of stem cells and progenitor cells derived from various tissue types is well established and harnessed in early clinical trials in some areas, including repair of myocardium using cardiac progenitor cells and chronic wound treatment using bone marrow-derived mesenchymal stem cells [1–3]. Skin is one of few organs in mammals that constantly undergoes self-renewal and repair [4]. It is composed of two layers: an outer epidermal layer and an inner dermal layer. The epidermis is a multilayered structure consisting almost entirely of keratinocytes, whereas the dermis is primarily made up of fibroblasts [5].
The adnexal structures of skin include hair follicles, sebaceous glands, and sweat glands [5]. The hair follicle is a complex structure extending from deep within the dermis to the surface of the epidermis [6]. Each hair follicle goes through three major cyclic phases for self-renewal and hair growth, termed anagen, catagen, and telogen [4]. This continuous regenerative activity is mediated by stem cells that reside in the bulge area of hair follicles and at the basal layer of the epidermis [4]. The markers associated with these cells in both human and murine skin include CD34, the α6-integrin, K14, K15, LGR5, Lgr6, Sca1, and Lrig1 [4, 5, 7–11]. It is well known that tissue-derived hair follicles can be implanted into full-thickness dorsal skin defects in nude mice as a means to recapitulate and observe hair follicle development in vivo [6]. Recent work by several groups has built on this model to demonstrate that the transplantation of CD34-positive (CD34+) stem cells into such defects can result in the generation of interfollicular epidermis, hair follicles, and sebaceous glands [12–15]. Importantly, such regenerative activity required the transplantation of fresh dermal cells together with the CD34+ stem cells [4, 15], but the former alone could not achieve notable levels of structure generation [4, 15]. Consequently skin-derived CD34+ stem cells are associated with multipotency and self-renewal ability [4, 15].
The standard method of isolating CD34+ skin stem cells is fluorescence-activated cell sorting (FACS). Although magnet-activated cell sorting (MACS) has also been used to obtain these cells, FACS is more common because of its high sensitivity [4, 5, 16]. Although both FACS and MACS are reliable and well established, both techniques require prelabeling of cells with fluorescent or magnetic antibody tags and multiple steps of wash and centrifugation prior to selection of the CD34+ cells, all of which typically requires a minimum of 1 hour followed by an additional 30 minutes to 1 hour for sorting [4, 5]. These preprocessing steps prior to cell sorting typically result in reduced overall cell viability. In contrast, the microfluidic approach eliminates cell prelabeling process entirely, and the total time for cell isolation, from sample injection to completed elution of purified cells, is approximately 45 minutes. The microfluidic approach requires preparation of devices; however, these devices have the additional advantage of being able to be produced in large numbers and then stored until they are needed so that no additional preparation time is needed prior to cell isolation.
In summary, the major advantages of microfluidic approach relative to FACS and MACS are shorter overall processing time (typically 45 minutes from sample to purified cells) to preserve cell viability and minimize cell loss; scalability to easily process large volumes of sample in parallel fashion, as opposed to serial operation (a significant limitation of FACS); and simplicity in terms of the device and its operation (the separation involves only three fluid-injection steps), facilitating its use outside of laboratory settings.
Our group recently developed a method for cell isolation that requires no preprocessing and no attachment of fluorescent or magnetic tags onto the cells and that reduced overall processing time. This method involves the use of microfluidic devices coated with a degradable, antibody-functionalized hydrogel. Cells expressing the target antigen are captured by optimizing sample flow rates to ensure optimal binding from a complex sample, such as whole blood. Following a wash step to remove unbound sample cells and other material, the target cells are isolated by dissolving the coating under mild conditions [17, 18]. The hydrogel coating is a copolymer of alginate and polyethylene glycol (PEG), both of which are well-known and well-characterized implantable biodegradable polymers [19, 20]. On dissolution of the coating, the purified cells retain the capture antibody and some fragments of alginate and PEG. However, the retention of these entities on the cell surface does not affect in vitro culturability or, as demonstrated in the present work and in another recent publication from our group [21], the in vivo functionality.
In terms of microfluidic device design, we recently described the use of a two-stage version of the above system to enrich CD34+ stem cells from mouse epidermis [22]. The first stage was used to deplete CD71+ nonstem cells from murine epidermal cell suspensions. In the second stage, the device was coated with the degradable hydrogen functionalized with an antibody against CD34 to positively select CD34+ stem cells in the released cell population (Fig. 1A, 1B). In this initial demonstration, the purity of released CD34+ cells was around 55%, and recovered cells remained viable and formed colonies with characteristic morphologies in vitro [22]. In the present study, we describe enhancements to our prior approach that result in significantly higher purity of the CD34+ population and demonstrate the functional ability of these enriched cells in terms of hair follicle and sebaceous gland formation in vivo.
Figure 1.

Experimental setup of microfluidic cell separation and in vivo surgery. (A): The epidermal cell population was loaded in 1-ml syringes and flowed to devices via tubing. Flow rate was controlled using a pump, and released cells were collected in a 1.5-ml centrifuge tube. (B): A zoom-in view of the microfluidic device, which consists of two stages. The first stage of the device is to deplete CD71-positive nonstem cells from the epidermal cell population, and the second stage of the device is to enrich CD34-positive (CD34+) stem cells. The enriched stem cell population was collected in a 1.5-ml centrifuge tube. (C): The 6-mm-diameter full-thickness skin defects were created on the dorsal side of a Nu/Nu mouse. A silicone grafting chamber was inserted under the skin defect and secured by two Autoclips. (D): A mixture of microfluidic-enriched CD34+ stem cells, unenriched epidermal cells, and newborn dermal cells based on six different groups (Table 1) were injected into the graft sites of skin defect via the grafting chamber. Green food dye represents cell suspension and is only for the purpose of illustration.
Materials and Methods
Preparation of Epidermal Cell Populations From Adult Mice
Wild-typeC57BL/6 mice (male; Charles River Laboratories International, Inc., Wilmington, MA, http://www.criver.com) aged 6–8 weeks were used to isolate epidermal cell populations. All animals were housed following institutional animal care and use committee (IACUC) regulations at Northeastern University. Epidermal cell harvest and tissue digestion were carried out according to the protocol described by Nowak and Fuchs [4]. Next, the epidermal skin digestate was passed through a series of filters at sizes of 40 μm (twice) (Fisher Scientific International, Hampton, NH, http://www.fishersci.com) and 20 μm (once) (Partec North America, Swedesboro, NJ, http://www.partec.com). Cells were collected after they were flowed through each filter, and centrifuged at 500g for 8 minutes. Supernatant was discarded, and the resulting cell pellet was resuspended in serum-free medium (Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F-12 [DMEM:F12] at a 1:3 ratio without calcium [customized product]; Invitrogen-Life Technologies, Grand Island, NY, http://www.lifetechnologies.com) prior to cell separation experiments or cell transplantation experiments.
Preparation of Dermal Cell Populations From Postnatal Mice
BALB/C postnatal day 1 pups were used to acquire dermal cell populations for in vivo transplantation. All animals were housed following IACUC regulations at Northeastern University. The BALB/C strain was chosen as the source for dermal cells based on our intent to follow a well-established protocol [15] for comparison of in vivo functionality between our microfluidic cell separation technique with FACS-based studies.
Isolation of dermal cells was performed following the protocol described by Jensen and coworkers [5]. Briefly, skin of five pups was floated in dispase-trypsin solution to separate the dermis from the epidermis [5]. The dermis was further digested in 0.25% collagenase solution for 1 hour, and the resulting tissue digestate was filtered through a 70-μm filter (Fisher Scientific). The cell suspension obtained was centrifuged at 500g for 8 minutes to collect cell pellets, and the pellets was resuspended in serum-free medium (DMEM:F12 at 1:3 ratio without calcium; Invitrogen; customized product) on ice until the time for in vivo cell transplantation.
Microfluidic Device Design
A two-stage microfluidic device design was applied to this study, as described in our previous work [22]. The first stage was a device to deplete CD71+ cell populations in epidermal cell suspensions, and the second stage was designed to capture CD34+ stem cells in the cell mixture (Fig. 1A, 1B). In the first-stage device, silane chemistry was used to covalently bind CD71 antibody (catalog no. 14-0711; eBioscience Inc., San Diego, CA, http://www.ebioscience.com) onto the channel surface, and the second-stage device used a degradable antibody-functionalized hydrogel coating [22].
Microfluidic Device Fabrication: Soft Lithography
Microfluidic devices were fabricated via standard polydimethylsiloxane-based soft lithography [23], as described in prior work [17, 18].
Improvement of Microfluidic Surface Functionalization
In order to increase the specificity of alginate-antibody coating for stem cell capture, the following improvements were made when antibody was immobilized in alginic acid for the second-stage devices. First, the pH of the 4-morpholineethanesulfonic acid (MES) buffer (Thermo Scientific Pierce, Rockford, IL, http://www.piercenet.com;) was adjusted to 6.0 using NaOH particles (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) for better preservation of functional CD34 antibodies in all steps. The mixing procedure occurred at room temperature: 22.5 mg of 4-arm PEG amine (molecular weight: 10 kDa; Laysan Bio, Arab, AL, http://www.laysanbio.com), 4.8 mg of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), 13.2 mg of N-hydroxysulfosuccinimide (Sulfo-NHS; Thermo Scientific Pierce), and 45 mg of alginic acid (Sigma-Aldrich) were stirred in 2 ml MES buffer in an Ultra-Turrax tube drive (IKA Works, Inc., Wilmington, NC, http://www.ika.com) at maximal speed for 30 minutes. After an incubation period of 1 hour, the PEG-alginic acid solution was stored at 4°C.
Prior to the surface modification described below, each device was filled with a 1 g/ml solution of calcium chloride in double-distilled water and incubated overnight. On the day of cell-isolation experiments, the calcium chloride solution was aspirated using a dry 1-ml syringe, and alginic acid solution was hand injected into each device and incubated for 45 minutes at room temperature. An antibody solution was prepared by adding 7.5 μg of anti-mouse CD34 purified antibody (catalog no. 14-0341; eBioscience), 0.6 mg of EDC, and 1.65 mg of Sulfo-NHS in 100 μl of MES buffer. All second-stage devices requiring surface functionalization were connected serially, then the antibody solution was injected inside at a flow rate of 10 μl/minute for 100 μl using a Harvard Apparatus PHD 2000 syringe pump (Harvard Apparatus, Holliston, MA, http://www.harvardapparatus.com). After another incubation of 45 minutes, MES buffer (pH 6.0) containing 10 mM CaCl2 was injected through the second-stage devices at the same flow rate using a syringe pump. The devices were then immediately used in cell-capture experiments.
Microfluidic Isolation of CD34+ Stem Cells
To ensure the sterility of cell-isolation process, all surfaces and instruments were wiped once with Wex-Cide (“Ready To Use” format; Wexford Laboratories, Kirkwood, MO, http://www.wexfordlabs.com), followed by another wipe with 2-propanol. Surfaces cleaned in this way included the bench top, the Harvard Apparatus PHD 2000 syringe pump, the pump rack, and the tube rack (Fig. 1A). All connecting tubing and tools contacting the microfluidic devices were autoclaved at 121°C and 15 psi beforehand. As soon as the cells were released from microfluidic devices and collected in 10-ml centrifuge tubes, the tubes were capped and transferred to a class II biological safety cabinet for subsequent steps.
Tissue-derived epidermal cells at 13.2 × 105 cells per milliliter were loaded into 1-ml syringes (Becton, Dickinson and Company, Franklin Lakes, NJ, http://www.bd.com) filled to a volume of 0.4 ml. Ten syringes of cell suspension were clamped onto a 10-port Harvard Apparatus PHD 2000 syringe pump. Cells were flowed into 10 two-stage devices at 3 μl/minute and a total injected volume of 50 μl, followed by a rinsing step using serum-free medium at 5 μl/minute and a total volume of 50 μl. A total of 6.6 × 104 cells passed through two-stage devices. The cells flowed through in sample effluent were discarded. After the rinsing step, the first- and second-stage devices were disconnected from each other. Cells captured in the second-stage devices were released by flowing 50 mM EDTA (in distilled water; Sigma-Aldrich) through devices at 15 μl/minute and 300-μl total volumes. Released cells were collected in a tube containing 3 ml culture medium supplemented with 15% chelated fetal bovine serum (FBS) to neutralize EDTA. The medium volume was set at a ratio of 10:1 relative to the EDTA volume to minimize the effect of EDTA on cells. Collected cell suspension was centrifuged at 500g for 8 minutes and resuspended in staining buffer (phosphate-buffered saline [PBS] with 2% calcium-free chelated FBS) either for flow cytometry analysis or directly applied to in vivo transplantation experiments. Details on preparation of chelated FBS can be found in Nowak and Fuchs’s protocol [4].
Flow Cytometry Analysis to Determine CD34+ Cell Population
Each cell specimen was collected from three two-stage devices, which yielded approximately 3,000 cells (1,000 cells per device). Cell specimens were incubated with FITC-conjugated anti-mouse CD34 antibody (catalog no. 11-0341; eBioscience) following the protocol described in our previous work [22]. Flow cytometry analysis was carried out using a Beckman Coulter Quanta SC bench-top flow cytometer (Beckman Coulter, Brea, CA, http://www.beckmancoulter.com). Cell viability was assessed using propidium iodide (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com) by adding 5 μl of dye into each cell specimen 1 minute prior to flow cytometry.
In Vivo Cell Transplantation
Male Nu/Nu mice aged 7 weeks were used as recipients in cell transplantation experiments. All animals were housed following IACUC regulations at Northeastern University. Mice were anesthetized using isoflurane inhalant (3%–5%) administered with 100% O2. The 6-mm-diameter full-thickness skin defects were created on the backs of Nu/Nu mice [24]. A silicone grafting chamber (Renner GMBH, Maulbronn-Schmie, Germany, http://www.renner-pumpen.de) was then inserted under the skin defect with its dome covering the wound, and the chamber was secured by two Autoclips (MikRonPrecision Inc., Gardena, CA) (Fig. 1C) [5]. Previously isolated cell populations were mixed and injected into the chamber on each mouse at 5 × 106 dermal cells, 3 × 106 unenriched epidermal cells, and 105 microfluidic-enriched CD34+ cells, according to six different study groups (Fig. 1D). Following survival of surgery, buprenorphine at 0.07 mg/kg was injected into mice subcutaneously twice daily for up to 3 days as needed, and silicone chambers were removed from the backs of Nu/Nu mice after 7 days. At the 40-day study endpoint, Nu/Nu mice were sacrificed using CO2, and the graft site of the full skin defect was harvested.
Tissue Preservation
Pieces of full skin grafts were fixed in 4% paraformaldehyde in PBS (Thermo Scientific Pierce) overnight at 4°C and then were transferred to 20% sucrose in PBS for 24 hours. Next they were transferred to 30% sucrose in PBS for 2 days until the tissue was submerged. Skin grafts were then embedded in Tissue-Tek OCT compound (Electron Microscopy Sciences, Hatfield, PA, http://www.emsdiasum.com/microscopy), being sectioned to 30-μm thickness using a Leica cryostat (Leica Microsystems Inc., Buffalo Grove, IL, http://www.leicabiosystems.com), and were mounted onto Superfrost Plus Gold slides (Electron Microscopy Sciences).
Histology
Tissue sections were stained with hematoxylin and eosin using Gill III hematoxylin and Oil Red O staining according to standard protocols for frozen sections. In Oil Red O, Harris-modified hematoxylin solution (Sigma-Aldrich) was used to stain cell nuclei. Air-dried slides were mounted in mounting medium (Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com) and covered with coverslips to be observed under a light microscope. Length and width of hair follicles, shafts, and sebaceous gland coverage were measured using ImageJ software (National Institutes of Health, Bethesda, MD, http://imagej.nih.gov/ij/).
Statistical Analysis
For quantitative measurements of hair shafts and sebaceous gland coverage, at least four samples were used for each group (Table 1). Data were expressed as mean ± SD. A one-way analysis of variance (ANOVA) was performed between various groups. If statistical difference was determined by one-way ANOVA, the Student-Newman-Keuls test was performed to determine the statistical significance (p < .05, p < .01) between two individual groups.
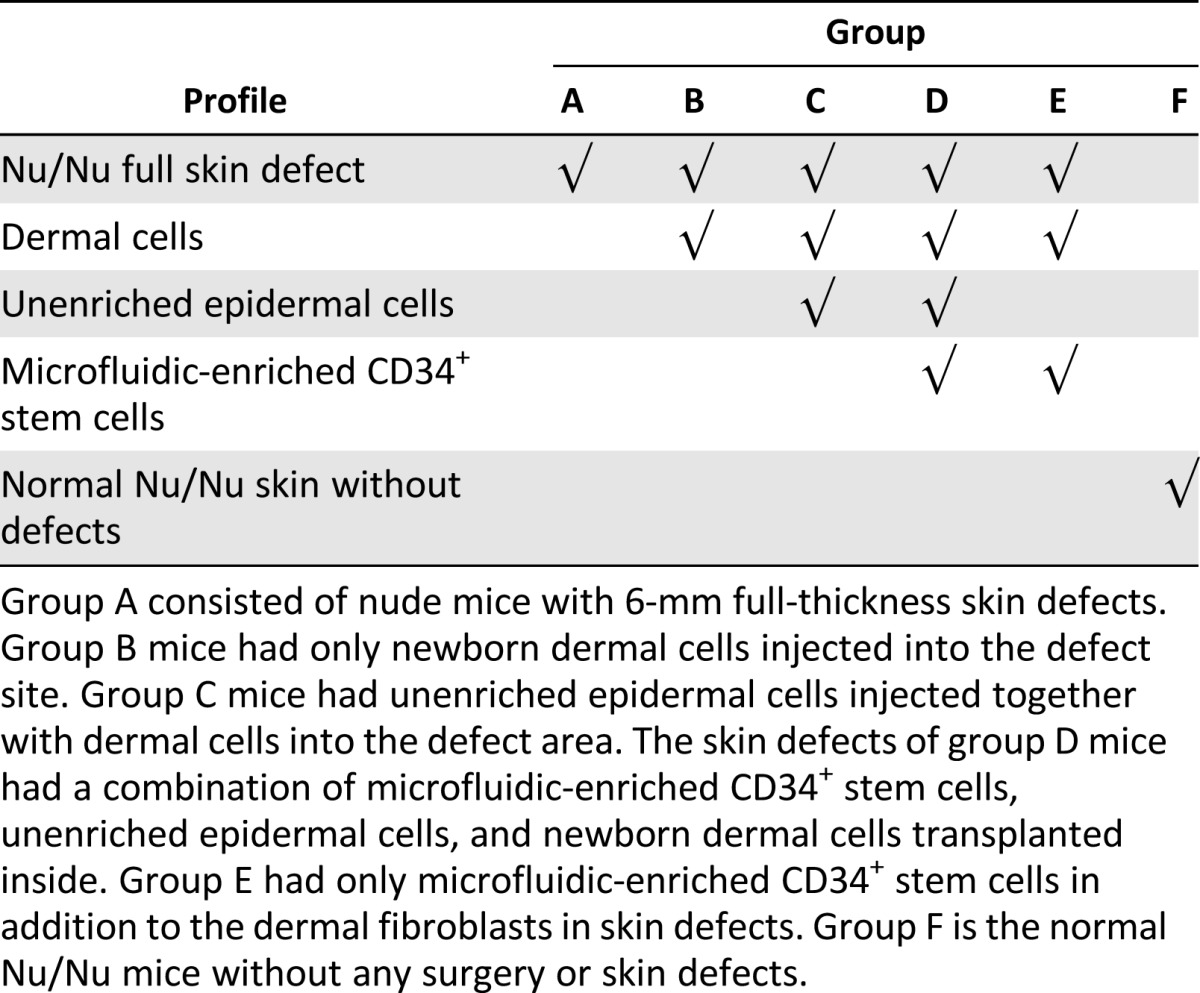
Table 1.
Study groups used in the in vivo experiments
Results
Microfluidic Isolation of CD34+ Stem Cells
The primary component of our microfluidic separation system is the degradable hydrogel coating used to capture the CD34+ cells. This hydrogel is a copolymer of alginic acid and PEG, which, like homopolymer alginate hydrogels, is a viscous liquid that transforms into a semisolid gel on contact with calcium ions. Exposure of the gel to a calcium chelating molecule such as EDTA can reverse this transformation. Our cell-isolation method involves adsorbing the gel precursor within the microfluidic devices and then forming the gel by flowing a solution of calcium chloride. Following capture of a target cell type and a wash step, captured cells are recovered by flowing an EDTA solution through the device.
In order to increase the purity of the CD34+ cells isolated using our two-stage approach, we improved the surface chemistry of the antibody-hydrogel complex in the second-stage devices by incorporating the following considerations (details provided in Materials and Methods). First, the MES buffer used to store components of the hydrogel (i.e., alginic acid and PEG) and cause them to react using carbodiimide chemistry was modified using sodium hydroxide to have a pH of 6.0. Second, rather than cause the alginic acid, PEG, and capture antibody to react prior to adsorption of the pregel solution within the microfluidic devices, as we did in our prior work, the antibody solution was flowed after the pregel material was adsorbed within the microfluidic channels.
The original tissue digestate contained 8.0% ± 0.1% CD34+ cells. In our prior work, we described enrichment of the CD34+ fraction to 56% ± 5% [22]. On making the improvements summarized above and detailed in Materials and Methods, our two-stage configuration provided a purity level of 82% ± 2%, significantly higher (p < .01) than in our prior work (Fig. 2). The original tissue digestate had an overall cell viability of 63% ± 1%, and the viability of the microfluidic isolated CD34+ cells was identical, namely, 63% ± 1%.
Figure 2.

A two-stage microfluidic device with improved antibody density on the device surface enriched CD34+ stem cells in high purity. When antibody was rearranged and concentrated on the top surface of hydrogel inside the second-stage microfluidic device, the purity of collected CD34+ stem cells reached 82 ± 2% (mean ± SD). This purity level is significantly higher than our previous two-stage device design [22] (n = 4 per group, p < .01). Abbreviation: CD34+, CD34-positive.
In Vivo Transplantation of CD34+ Stem Cells Isolated via Microfluidic Method
In order to evaluate in vivo function of CD34+ stem cells isolated using our microfluidic method, these cells were mixed with other cell types, and several control groups were added to the study. A total of six study groups were included in this project (Table 1). As described above, dermal cells have been identified as an essential component for the survival of transplanted CD34+ cells [4, 15]. To compare the results of our study with prior work by other groups, our experimental design mirrored that of transplantation studies described by other groups in which mixed suspensions of dermal cells, unenriched epidermal cells, and FACS-sorted enriched CD34+ stem cells were transplanted into nude mice [4, 5]. Groups A and B are two negative control groups with either no cells transplanted (group A) or only neonatal dermal cells transplanted (group B). Nu/Nu mice typically do not have visible hair on the back, but a small amount of short hair can be seen in a small number of these mice. We include examples of Nu/Nu mice free of surgery in group F as another negative control group that shows the degree of hair nude mice can have naturally. Large amounts (∼106 cells) of dermal cells and unenriched epidermal cells are two key components for the successful transplantation of skin grafts, and these are represented in group C. The inclusion of CD34+ cells isolated using our microfluidic method with the cells of group C is defined as group D. A large number of unenriched epidermal cells (106 cells) usually accompanied CD34+ stem cells (105 cells) in skin grafts for successful transplantation unless ∼106 stem cells can be collected to substitute the mass population of the former unenriched epidermal cells [5, 15]. An additional group, group E, was defined as having all of the component cells of group D except the unenriched epidermal cells, to examine and compare the role of these cells.
Figure 3 shows side-view dorsal skin photographs of recipient nude mice taken 40 days after transplantation surgeries. Full-thickness skin defects were healed in all six groups. Groups A and B did not show any hair at the transplantation sites. Group F animals showed short hair grown on the back of nude mice naturally. Longer hair was observed on animals from groups C, D, and E, but group D animals had the longest and densest hair growth at the transplantation site.
Figure 3.

Groups C, D, and E had visible hair outgrowth from the skin defect sites. Group D, with the injection of both microfluidic-enriched CD34-positive stem cells and unenriched epidermal cells, had the most extensive hair growth among all groups. Without the epidermal cell population, the wound healed in groups A and B, but no hair was formed. Group F shows the extreme case of natural hair in Nu/Nu without surgery. A thin layer of hair can be seen on the dorsal skin of nude mice.
Analysis of Hair Follicles and Hair Shafts at Transplantation Site
Hair follicles and hair shafts are the key features of a successful skin graft, and these structures were examined by histological staining, as shown in Figure 4. Skin tissue from group A and B animals did not contain any observable hair follicles or hair shafts. In contrast, tissue samples from groups C, D, E, and F contained varying levels of hair follicle and hair shaft structures (Fig. 4A). In terms of the average width of hair shafts, group C had a width of 51 ± 5 μm, which is narrower than that of group D (73 ± 13 μm; p < .05) (Fig. 4B). Group D also had wider hair shafts on average relative to group E (45 ± 8 μm; p < .05) and group F (43 ± 3 μm; p < .01). The width of hair follicles was measured at the bulb area located at the bottom of individual hair follicles. Group F had an average follicle width of 63 ± 11 μm, which was the narrowest width across all of the study groups (Fig. 4C). By comparison with this control group, the average follicle widths for groups E, D, and C were wider, at 80 ± 11 μm (p < .01), 79 ± 16 μm (p < .05), and 77 ± 6 μm (p < .01), respectively. When analyzing the length of hair follicles, no single group had dominantly longer follicles than the other groups (Fig. 4D). Group D had an average follicle length of 1,004 ± 66 μm, longer than that of group C (862 ± 42 μm; p < .01) and group F (839 ± 123 μm; p < .01). Group E had a follicle length of 1,065 ± 86 μm, which is longer than those of group C (p < .01) and group F (p < .01). In summary, with both the participation of CD34+ microfluidic-enriched stem cells and unenriched epidermal cells, group D had the widest hair shaft in skin grafts and longer hair follicles than groups C and F. With the effects of microfluidic-enriched CD34+ stem cells and dermal fibroblasts, group E had a longer length of follicles than groups C and F. The natural skin of nude mice had the narrowest width of hair follicles among all study groups.
Figure 4.

Follicles and outgrown hair were observed in Nu/Nu mouse skin defect areas in groups C, D, and E. (A): Hematoxylin and eosin staining showed healing of skin defects in groups A and B. Groups C, D, and E had various degrees of follicle and hair formation in defect areas. Group F had follicles and hair shafts on the dorsal skin of the natural nude mice. (B): With the transplantation of both microfluidic-enriched CD34-positive (CD34+) cells and unenriched epidermal cells, group D had the widest hair shafts grow in defect areas (n = 6, mean ± SD; p < .05 versus group C; p < .05 versus group E; p < .01 versus group F). (C): Groups C, D, and E had similar width of hair follicles in skin grafts, but all of their hair follicles were wider than the natural skin of nude mice in group F (n = 8, mean ± SD; p < .01 groups C and E versus group F, respectively; p < .05 group D versus group F). (D): With both effects of microfluidic-enriched CD34+ cells and unenriched epidermal cell population, group D had longer hair follicles than groups C and F, respectively (n = 8, mean ± SD; p < .01). With microfluidic-enriched CD34+ cells and dermal fibroblasts, group E still had longer hair follicles than group C (with injection of only unenriched epidermal cells and dermal fibroblasts) (p < .01) and group F (the natural hair of nude mice) (n = 8, mean ± SD, p < .01, respectively). Scale bar = 250 μm (groups A–F).
Sebaceous Glands at Transplantation Sites
Sebaceous glands are connected to hair follicles and secrete an oily substance called sebum. Sebum also travels to skin surfaces to protect the skin barrier. Oil Red O staining was used to stain sebum and characterize sebaceous gland formation in each study group (Fig. 5). Groups C, D, and E showed sebaceous glands surrounding hair follicles (Fig. 5A), and sebum was observed on the surface of the skin of animals from these groups. Tissue samples from nude mice (group F) contained sebaceous glands, whereas groups A and B did not show any staining for sebaceous glands. In terms of sebaceous gland coverage, as measured by Oil Red O stain coverage in each image, group D had average sebaceous gland coverage of 46% ± 4%, significantly higher than that of Group E (32% ± 8%) and group F (34% ± 9%; p < .05 for both) (Fig. 5B). Sebaceous gland coverage in group C was not significantly different from that of other groups.
Figure 5.

Groups C, D, and E regenerated sebaceous glands, an important component of skin. (A): Oil Red O staining showed that groups A and B did not form sebaceous glands in the healed area of skin defects. Groups C, D, and E regenerated sebaceous glands, and sebum secretion was observed on the skin surface. The skin samples from nude mice (group F) also showed sebaceous glands. (B): When both microfluidic-enriched CD34-positive (CD34+) stem cells and unenriched epidermal cells were transplanted into skin defects together with dermal fibroblasts, group D had higher sebaceous gland coverage relative to group E (microfluidic-enriched CD34+ stem cells and dermal cells only) and group F (the natural skin of nude mice) (n = 10 per group, mean ± SD; p < .01, respectively). Scale bar = 250 μm (groups A–F).
Discussion
The objective of this study was to demonstrate the in vivo functional efficacy of CD34+ skin stem cells purified using our affinity-based microfluidic method. We used well-known protocols for transplantation along with an improved version of our previously described separation system. Our previous two-stage system device enriched CD34+ cells contained in murine skin tissue digestate from a starting level of approximately 8% to approximately 56% [22]. To increase the purity of CD34+, we modified the method of fabricating the antibody-alginate complex in the second-stage device. In the previous method, CD34 antibodies were mixed within the PEG-alginate solution prior to application of this gel-coating precursor onto the microfluidic device surfaces. With this approach, a significant quantity of antibody is contained within the coating and not exposed to the surface and thus is not available to capture target cells. With the modified technique used in the present study, the antibody was added following adsorption of the gel precursor on the surfaces of the second-stage devices at higher concentration (0.075 μg/μl). This change resulted in a greater level of target cell capture and significantly higher purity (82%). Although this purity level is below that reported for FACS-based purification of these cells (∼94% [5]), further improvements in the design of the microfluidic system, such as increased density and controlled orientation of the capture antibodies, are expected to allow higher purity levels to be attained. In terms of recovery metrics, approximately 6.6 × 104 cells from the tissue digestate were injected into each first-stage device, of which 8%, or 5.3 × 103 cells, were CD34+; of the ∼1,000 cells collected from each second-stage device at the end of the separation process, approximately 82% (around 820 cells) were CD34+. This translates to a recovery rate of around 16%, reflecting the compromise made in optimizing the microfluidic system for purity and overall simplicity.
In the present study, 10 two-stage devices were used to obtain approximately 10,000 purified CD34+ cells from each run. Although these runs were performed with manually loaded syringes, our group and others have developed automated systems for fluid pumping into banks for 20–40 microfluidic devices. The advantage of such systems is the consolidation of the sample and all reagent reservoirs into single locations and an automated operation (enabled by various automation systems) that does not require any user actions beyond initiation of the process. Such systems are not intrinsically limited in the number of devices for which they are designed, and depending on needs and frequency of use, systems capable of operating more than 40 devices can easily be designed. For the cell types examined in this study, an automated and scaled-up system with 40 devices could produce four times as many cells in one run as in the present study, specifically ∼40,000 CD34+ cells.
For in vivo transplantation, CD34+ epidermal stem cells were isolated from wild-type C5BL6 mice. In similar work by other groups, green fluorescent protein-labeled CD34+ cells from transgenic mice [5, 15] were typically used along with FACS to isolate these cells. Although the use of such transgenic models greatly simplifies sorting and imaging of these cells, our objective was to recapitulate the functional abilities of these cells using CD34+ cells from a wild-type source to validate our cell-separation technique.
The formation of key structures such as hair follicles and hair shafts is a key indicator of successful skin stem cell transplantation. In this study, the full-thickness skin defects in groups A and B healed, but no hair growth was observed (Fig. 4A). The addition of epidermal cells and dermal fibroblasts resulted in some hair growth, but when combined with CD34+ cells isolated using our microfluidic method (group D), the observed hair growth and hair shaft width was significantly greater (Fig. 4B). Although CD34+ cells isolated via microfluidics were present in both groups D and E, the hair growth observed in the latter was similar to groups without these cells. This distinction highlights the important role of the unenriched epidermal cells that are present in group D but not group E, consistent with the observations of others made using FACS-sorted cells [5].
The hair follicles in groups C, D, and E were wider than their natural counterparts in nude mice, as shown in the control group F. In addition, the average width of hair follicles in our study groups was between 60 μm and 80 μm, close to the average width (90 μm) of natural hair in nude mice measured by Asakawa and coworkers [25]. With the addition of microfluidic-enriched CD34+ stem cells, groups D and E had longer hair follicles than groups C and F individually. The average length of hair follicles across all four groups was approximately 900 μm (Fig. 4D), shorter than the 1,600-μm average length reported by Asakawa and coworkers [25]. In that study, the hair follicles were divided into three types based on morphology, and only the “awl” type of hair follicles were measured by length. In our study, all hair follicles, regardless of their appearance, were included in the length measurement.
Sebaceous glands are an important supportive component of skin. The engraftment of the dermal cell population alone (group B) did not regrow this gland in skin graft sites, whereas the addition of epidermal cells regenerated sebaceous glands in groups C, D, and E (Fig. 5A). Our Oil Red O staining data are consistent with those reported by Blanpain et al., in whose study FACS-sorted CD34+ stem cells grew sebaceous glands in skin grafts of nude mice [15]. Furthermore, the locations of sebaceous glands in our study were similar to those reported by Sato et al. [26] and Cui et al. [27]. These glands were observed adjacent to hair follicles, and sebum traveled to the skin surface via hair shafts. Few studies show the fraction of sebaceous gland coverage in different groups of skin grafts. In this study, our semiquantitative measurements (Fig. 5B) indicated that sebum coverage in groups C, D, and E was similar to that in the natural skin of nude mice (group F), a desirable result from the standpoint of transplantation.
Taken together, the hair follicle and sebaceous gland data in our study suggest that microfluidic-enriched CD34+ stem cells were able to regenerate hair similar to the natural hair of nude mice. When the enriched CD34+ stem cells were combined with unenriched epidermal cells, this group had the widest hair shafts, long hair follicles, and highest sebaceous gland coverage in skin grafts. These data indicate that the microfluidic-enriched CD34+ stem cells contributed significantly to hair regeneration in full-thickness skin defects on nude mice. Our results further demonstrate that the functional in vivo capabilities of microfluidic-enriched cells are equivalent to those of FACS-sorted cells used by other research teams, a finding that represents an important validation of the microfluidic cell-isolation method. Considering the relative speed of the microfluidic technique (45 minutes from sample injection to CD34+ cell elution), the absence of preprocessing in the form of cell tagging, and the ability to automate and scale this technique, this approach to cell isolation may enable broader translational use of stem cells resident in tissue.
Conclusion
This paper describes a two-stage microfluidic system to enrich CD34+ stem cells from mouse skin, with high purity. Compared with the standard FACS and MACS technologies, microfluidic devices minimize the processing time of stem cells in the starting population, are easily accessible, and can be multiplexed to separate large amounts of cell mixture. The released CD34+ stem cells were transplanted into full-thickness skin defects in nude mice. Follicles and hairs were regenerated, and sebaceous glands were formed in the graft sites.
Acknowledgments
We thank Adam Hatch for assistance in synthesizing antibody-alginate solutions. This work was supported by the U.S. National Institutes of Health under Grant R01-EB009327 and by the U.S.-Israel Binational Science Foundation under Grant 2013002.
Author Contributions
B.Z.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; Y.N.: conception and design, financial support, final approval of manuscript; M.L.Y.: provision of study material or patients, financial support, final approval of manuscript; S.K.M.: conception and design, data analysis and interpretation, financial support, administrative support, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Bolli R, Chugh AR, D’Amario D, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): Initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Chugh AR, Beache GM, Loughran JH, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: The SCIPIO trial: Surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126(suppl 1):S54–S64. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dabiri G, Heiner D, Falanga V. The emerging use of bone marrow-derived mesenchymal stem cells in the treatment of human chronic wounds. Expert Opin Emerg Drugs. 2013;18:405–419. doi: 10.1517/14728214.2013.833184. [DOI] [PubMed] [Google Scholar]
- 4.Nowak JA, Fuchs E. Isolation and culture of epithelial stem cells. Methods Mol Biol. 2009;482:215–232. doi: 10.1007/978-1-59745-060-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen KB, Driskell RR, Watt FM. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc. 2010;5:898–911. doi: 10.1038/nprot.2010.39. [DOI] [PubMed] [Google Scholar]
- 6.Weinberg WC, Goodman LV, George C, et al. Reconstitution of hair follicle development in vivo: Determination of follicle formation, hair growth, and hair quality by dermal cells. J Invest Dermatol. 1993;100:229–236. doi: 10.1111/1523-1747.ep12468971. [DOI] [PubMed] [Google Scholar]
- 7.Trempus CS, Morris RJ, Bortner CD, et al. Enrichment for living murine keratinocytes from the hair follicle bulge with the cell surface marker CD34. J Invest Dermatol. 2003;120:501–511. doi: 10.1046/j.1523-1747.2003.12088.x. [DOI] [PubMed] [Google Scholar]
- 8.Tumbar T, Guasch G, Greco V, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snippert HJ, Haegebarth A, Kasper M, et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–1389. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]
- 10.Roh C, Roche M, Guo Z, et al. Multi-potentiality of a new immortalized epithelial stem cell line derived from human hair follicles. In Vitro Cell Dev Biol Anim. 2008;44:236–244. doi: 10.1007/s11626-008-9084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin KK, Andersen B. Have hair follicle stem cells shed their tranquil image? Cell Stem Cell. 2008;3:581–582. doi: 10.1016/j.stem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Zouboulis CC, Adjaye J, Akamatsu H, et al. Human skin stem cells and the ageing process. Exp Gerontol. 2008;43:986–997. doi: 10.1016/j.exger.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Amici AW, Yamato M, Okano T, et al. The multipotency of adult vibrissa follicle stem cells. Differentiation. 2009;77:317–323. doi: 10.1016/j.diff.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Cotsarelis G. Epithelial stem cells: A folliculocentric view. J Invest Dermatol. 2006;126:1459–1468. doi: 10.1038/sj.jid.5700376. [DOI] [PubMed] [Google Scholar]
- 15.Blanpain C, Lowry WE, Geoghegan A, et al. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Huang E, Lian X, Chen W, et al. Characterization of rat hair follicle stem cells selected by vario magnetic activated cell sorting system. Acta Histochem Cytochem. 2009;42:129–136. doi: 10.1267/ahc.09016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatch A, Hansmann G, Murthy SK. Engineered alginate hydrogels for effective microfluidic capture and release of endothelial progenitor cells from whole blood. Langmuir. 2011;27:4257–4264. doi: 10.1021/la105016a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatch A, Pesko DM, Murthy SK. Tag-free microfluidic separation of cells against multiple markers. Anal Chem. 2012;84:4618–4621. doi: 10.1021/ac300496q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ungerleider JL, Christman KL. Concise review: Injectable biomaterials for the treatment of myocardial infarction and peripheral artery disease: Translational challenges and progress. Stem Cells Translational Medicine. 2014 doi: 10.5966/sctm.2014-0049. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Meel R, Vehmeijer LJ, Kok RJ, et al. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv Drug Deliv Rev. 2013;65:1284–1298. doi: 10.1016/j.addr.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Lin RZ, Hatch A, Antontsev VG, et al. Microfluidic capture of endothelial colony-forming cells from human adult peripheral blood: Phenotypic and functional validation in vivo. Tissue Eng Part C Methods. 2014 doi: 10.1089/ten.tec.2014.0323. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu B, Smith J, Yarmush ML, et al. Microfluidic enrichment of mouse epidermal stem cells and validation of stem cell proliferation in vitro. Tissue Eng Part C Methods. 2013;19:765–773. doi: 10.1089/ten.tec.2012.0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia Y, Whitesides GM. Soft lithography. Angew Chem Int Ed Engl. 1998;37:550–575. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 24.Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asakawa K, Toyoshima KE, Ishibashi N, et al. Hair organ regeneration via the bioengineered hair follicular unit transplantation. Sci Rep. 2012;2:424. doi: 10.1038/srep00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato T, Takahashi A, Kojima M, et al. A citrus polymethoxy flavonoid, nobiletin inhibits sebum production and sebocyte proliferation, and augments sebum excretion in hamsters. J Invest Dermatol. 2007;127:2740–2748. doi: 10.1038/sj.jid.5700927. [DOI] [PubMed] [Google Scholar]
- 27.Cui CY, Durmowicz M, Ottolenghi C, et al. Inducible mEDA-A1 transgene mediates sebaceous gland hyperplasia and differential formation of two types of mouse hair follicles. Hum Mol Genet. 2003;12:2931–2940. doi: 10.1093/hmg/ddg325. [DOI] [PubMed] [Google Scholar]