

Abstract
Clustering of acetylcholine receptors (AChR) in muscle fibers prior to innervation by motor neurons is thought to be involved in neuromuscular junction formation. Jing et al. now report in Neuron that this prepatterning of AChRs, via a novel MuSK-dependent Wnt pathway, may guide motor axons to the central region of muscle fibers for synapse formation in zebrafish.
Chemical synapses are specialized contacts between two neurons or a neuron and a nonneuronal cell. Synapses enable neurons to build complex circuits in the brain that underlie mental perception, thought processes, and the control of other systems in the body. Recent years have witnessed significant advances in understanding of anatomic structures and protein components of synapses. Less is known about the mechanism of synapse assembly. One example of a well-studied synapse is the neuromuscular junction (NMJ), a peripheral synapse that uses acetylcholine (ACh) as a neurotransmitter. NMJ formation depends on interactions between motor neurons and skeletal muscle fibers. Prior to the arrival of nerve terminals, aneural ACh receptor (AChR) clusters form in the central region of muscle fibers, forming a band around the fibers. This phenomenon is called prepatterning (Kummer et al., 2006). The current hypothesis of mammalian NMJ formation is that innervation disperses aneural AChR clusters in nonsynaptic areas via muscle activity elicited by ACh; however, in synaptic regions, this negative activity is overpowered by positive factors delivered by motor nerve terminals to induce AChR clusters. Subsequently, AChR becomes highly concentrated only in the membrane facing the terminal where the NMJ forms.
One positive factor is agrin, a protein synthesized in motor neurons that stimulates AChR clustering in vitro. Agrin binds LRP4, a member of the LDL receptor (LDLR) family, and LRP4 binds and activates the receptor tyrosine kinase MuSK ([Kim et al., 2008] and [Zhang et al., 2008]). Mutation of any of the three genes prevents NMJ formation in mice ([Kummer et al., 2006] and [Weatherbee et al., 2006]). However, the phenotypes of LRP4 and MuSK mutants are more severe than those of the agrin mutant. In particular, prepatterning of muscle fibers disappears in LRP4 or MuSK mutants, but not in agrin mutants, suggesting the existence of a pathway that requires MuSK and LRP4, but not agrin. An open question then is how MuSK or LRP4 is regulated before neuronal agrin is delivered. One possibility is that MuSK is activated by dimerization or interaction with other proteins ([Kim et al., 2008] and [Zhang et al., 2008]). Intriguingly, however, MuSK has a cysteine-rich domain (CRD) that shows homology to the Wnt receptor Frizzled. Like Frizzled, it interacts on the membrane with an LDLR family member (LRP4) ([Kim et al., 2008] and [Zhang et al., 2008]) and in the cytosol with Dishevelled (Dvl, or Dsh in Drosophila), an adaptor protein that interacts with Frizzled to initiate Wnt signaling (Luo et al., 2002) (Figure 1). In addition to Dvl, adenomatous polyposis coli (APC) and β-catenin, two other key proteins in the Wnt canonical pathway, have been implicated in AChR clustering in vitro and/or postsynaptic and presynaptic differentiation in NMJ formation in vivo ([Li et al., 2008], [Luo et al., 2002] and [Wang et al., 2003]). It seems possible, then, that MuSK activity is regulated by a Wnt ligand.
Figure 1. Working Models for Wnt Regulation of NMJ Formation.
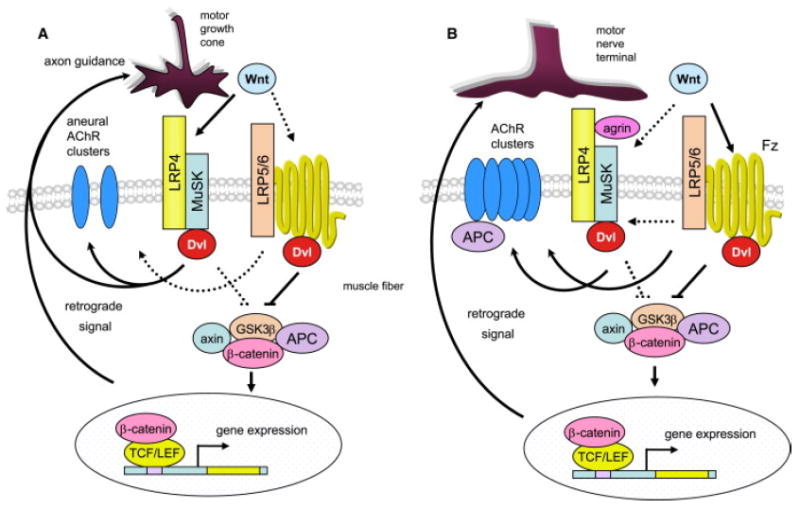
(A) In zebrafish, prior to innervation, Wnt11r interacts with MuSK to activate Dvl-dependent pathways necessary for forming aneural clusters and guiding motor growth cones to the middle region of muscle fibers.
(B) In vitro studies suggest that Wnts regulate agrin-induced AChR clustering by binding to the receptor Frizzled (Fz). They may also act by interacting with MuSK. Differentiation of motor nerve terminals is regulated by a β-catenin-dependent pathway. Dashed lines indicate pathways to be determined.
In a recent issue of Neuron, Granato and colleagues (Jing et al., 2009) provide evidence that a MuSK isoform indeed binds a Wnt ligand, at least in zebrafish. As in mice, zebrafish myotubes adjacent to the notocord (adaxial myotubes) form aneural AChR clusters in advance of motor axon innervation. These myotubes are soon replaced by lateral fast muscle fibers. Subsequently, motor axons innervate fast muscle fibers at sites of aneural AChR clusters. Earlier work of the same group showed that unplugged (the zebrafish ortholog of MuSK) null mutants lack AChR prepatterning and show NMJ and axonal guidance defects. They now show that the MuSK isoform SV1, which lacks the Ig domains but contains the CRD, is the one required for prepatterning. Granato and colleagues then searched for a MuSK ligand and noticed Wnt11r, which is expressed near the dorsal adaxial myotubes. Remarkably, knockdown of Wnt11r reduces adaxial AChR prepatterning and causes axonal stalling and branching, phenotypes reminiscent of MuSK/unplugged mutants, suggesting that Wnt11r acts through MuSK/unplugged. In support of this idea, the two genes interact both genetically and biochemically. Moreover, coexpression of Wnt11r with wild-type, but not a CRD-deletion mutant that is unable to bind Wnt11r, is sufficient to induce ectopic AChR prepatterning. These observations provide compelling evidence that Wnt11r, via an interaction with the CRD of MuSK, regulates prepatterning of muscle fibers in zebrafish (Figure 1). Recent in vitro studies show that some Wnt may regulate agrin-induced AChR clusters ([Henriquez et al., 2008] and [Wang et al., 2008]), suggesting a role of Wnt signaling in NMJ formation at a later stage. It is unclear whether these effects were mediated by Wnt binding to the CRD in MuSK or Frizzled.
An equally significant finding of this paper regards the role of prepatterning in NMJ formation. The existence of prepatterning raises the possibility that muscle fibers do not passively wait to be innervated, but rather play an instructive role. This idea was surprising when prepatterning was first discovered. However, the role of prepatterning in NMJ formation has not been vigorously investigated because of the lack of mutants that specifically disrupt aneural AChR clusters without altering neural AChR clusters or vice versa. The authors of this paper cleverly developed ways to tease apart these two events (Jing et al., 2009). They show that expression of a MuSK/unplugged SV1-myc transgene completely restores the AChR prepattern but fails to induce neural AChR clusters in MuSK/unplugged null mutants. Remarkably, knockdown of Wnt11r reduces adaxial AChR prepattern, but has no apparent effect on neural AChR clusters, suggesting that prepatterning may not be important for later NMJ formation. To test this idea further, the authors generated transgenic zebrafish whose expression of MuSK/unplugged was inducible by heat shock. They then crossed these fish into MuSK/unplugged null mutants to see whether they were able to rescue neural AChR clusters when induced after prepatterning. Heat-shock-treated transgenic embryos are fully motile, suggesting functional NMJs. Morphological characterization indicates that in vivo neural synapses can form in the absence of prepatterned AChRs.
What is the function of prepatterning, then? Results from this paper seem to suggest a connection to confinement or guidance of motor nerve terminals to the center of muscle fibers, identifying a novel function of prepatterning. This connection could bring ingrowing motor nerve terminals into spatial register with future sites of postsynaptic specialization and thus make NMJ formation more efficient. Earlier work by Granato and colleagues showed that axon pathway finding is normal in rapsyn zebrafish mutants that lack prepatterned AChRs; therefore, they may not be involved in directing growth cones of motor nerves. Thus, in zebrafish at least, aneural AChR clusters are not necessary for axon guidance and subsequent formation of nerve-induced AChR clusters. Their role appears to be to outline a region in the middle of muscle fibers that is important for guiding motor nerve growth cones. Further experiments are needed to determine whether this applies to mammalian NMJs that are larger and take longer to form.
References
- Henriquez JP, Webb A, Bence M, Bildsoe H, Sahores M, Hughes SM, Salinas PC. Proc Natl Acad Sci USA. 2008;105:18812–18817. doi: 10.1073/pnas.0806300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Lefebvre JL, Gordan LR, Granato M. Neuron. 2009;61:721–733. doi: 10.1016/j.neuron.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer TT, Misgeld T, Sanes JR. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Li XM, Dong XP, Luo SW, Zhang B, Lee DH, Ting AK, Neiswender H, Kim CH, Carpenter-Hyland E, Gao TM, et al. Nat Neurosci. 2008;11:262–268. doi: 10.1038/nn2053. [DOI] [PubMed] [Google Scholar]
- Luo Z, Wang Q, Zhou J, Wang J, Liu M, He X, Wynshaw-Boris A, Xiong W, Lu B, Mei L. Neuron. 2002;35:489–505. doi: 10.1016/s0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- Wang J, Jing Z, Zhang L, Zhou G, Braun J, Yao Y, Wang ZZ. Nat Neurosci. 2003;6:1017–1018. doi: 10.1038/nn1128. [DOI] [PubMed] [Google Scholar]
- Wang J, Ruan NJ, Qian L, Lei WL, Chen F, Luo ZG. J Biol Chem. 2008;283:21668–21675. doi: 10.1074/jbc.M709939200. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Anderson KV, Niswander LA. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]