

Abstract
The phosphatidylinositiol 3-kinase (PI3K), AKT, mammalian target of rapamycin (mTOR) signaling pathway (PI3K/AKT/mTOR) is frequently dysregulated in disorders of cell growth and survival, including a number of pediatric hematologic malignancies. The pathway can be abnormally activated in childhood acute lymphoblastic leukemia (ALL), acute myelogenous leukemia (AML), and chronic myelogenous leukemia (CML), as well as in some pediatric lymphomas and lymphoproliferative disorders. Most commonly, this abnormal activation occurs as a consequence of constitutive activation of AKT, providing a compelling rationale to target this pathway in many of these conditions.
A variety of agents, beginning with the rapamycin analogue (rapalog) sirolimus, have been used successfully to target this pathway in a number of pediatric hematologic malignancies. Rapalogs demonstrate significant preclinical activity against ALL, which has led to a number of clinical trials. Moreover, rapalogs can synergize with a number of conventional cytotoxic agents and overcome pathways of chemotherapeutic resistance for drugs commonly used in ALL treatment, including methotrexate and corticosteroids. Based on preclinical data, rapalogs are also being studied in AML, CML, and non-Hodgkin’s lymphoma. Recently, significant progress has been made using rapalogs to treat pre-malignant lymphoproliferative disorders, including the autoimmune lymphoproliferative syndrome (ALPS); complete remissions in children with otherwise therapy-resistant disease have been seen.
Rapalogs only block one component of the pathway (mTORC1), and newer agents are under preclinical and clinical development that can target different and often multiple protein kinases in the PI3K/AKT/mTOR pathway. Most of these agents have been tolerated in early-phase clinical trials. A number of PI3K inhibitors are under investigation. Of note, most of these also target other protein kinases. Newer agents are under development that target both mTORC1 and mTORC2, mTORC1 and PI3K, and the triad of PI3K, mTORC1, and mTORC2. Preclinical data suggest these dual- and multi-kinase inhibitors are more potent than rapalogs against many of the aforementioned hematologic malignancies.
Two classes of AKT inhibitors are under development, the alkyl-lysophospholipids (APLs) and small molecule AKT inhibitors. Both classes have agents currently in clinical trials. A number of drugs are in development that target other components of the pathway, including eukaryotic translation initiation factor (eIF) 4E (eIF4E) and phosphoinositide-dependent protein kinase 1 (PDK1). Finally, a number of other key signaling pathways interact with PI3K/AKT/mTOR, including Notch, MNK, Syk, MAPK, and aurora kinase. These alternative pathways are being targeted alone and in combination with PI3K/AKT/mTOR inhibitors with promising preclinical results in pediatric hematologic malignancies. This review provides a comprehensive overview of the abnormalities in the PI3K/AKT/mTOR signaling pathway in pediatric hematologic malignancies, the agents that are used to target this pathway, and the results of preclinical and clinical trials, using those agents in childhood hematologic cancers.
The investigation and use of drugs that target signaling pathways in malignancies has grown exponentially since the discovery of imatinib, a BCR-ABL tyrosine kinase inhibitor that has revolutionized the treatment of chronic myelogenous leukemia (CML) and Philadelphia chromosome positive (Ph+) acute lymphoblastc leukemia (ALL) in children.[1,2]
One pathway that has been studied extensively in a large number of conditions is the phosphatidylinositiol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway. This evolutionarily conserved signaling pathway has key roles in cell growth, survival, and metabolism. It is aberrantly activated in a number of malignant and non-malignant diseases, which has led to preclinical studies and clinical trials investigating compounds that target the various components of the pathway. Drugs that target mTOR were the first to be studied, showing remarkable efficacy in a number of conditions. Subsequently, drugs were developed that can target PI3K and AKT as well as a number of intermediates in the PI3K/AKT/mTOR signaling pathway, including agents that target individual protein kinases and drugs that target multiple kinases in the pathway.[3,4]
Clinical trials investigating a number of agents are ongoing in pediatric ALL, lymphoblastic lymphoma, fibromatosis, and neuroblastoma, as well as a variety of childhood sarcomas, brain tumors, and lymphoproliferative disorders. In addition, there are promising preclinical data demonstrating activity of different agents against acute myelogenous leukemia (AML), CML, and a number of lymphomas. For a number of these malignancies the real promise of these pathway inhibitors is their ability to overcome chemotherapy resistance and synergize with existing cytotoxic therapies.
The aim of this review is to describe the efficacy and toxicity of agents that target the PI3K/AKT/mTOR signaling pathway in childhood hematologic cancer. PubMed was the main search engine used; keywords employed were ‘children’, ‘mTOR’, ‘PI3K’, ‘AKT’, ‘cancer’, ‘leukemia’, ‘lymphoma’, ‘hematologic’, and ‘lymphoproliferative’. In addition, each therapeutic agent described in the text was searched in combination with the keywords ‘children’ and ‘cancer’. Clinicaltrials.gov was also searched using the same search terms. Finally, the 2010 American Society of Hematology and 2011 American Society of Clinical Oncology annual meeting abstract search engine websites (www.hematology.org and www.asco.org, respectively) were searched using the same terms. All searches were limited to English-language articles. Abstract references were only included if they provided important information on recent and ongoing clinical trials. References were chosen based on their relevance to pediatric hematologic cancer. Adult data are presented where there are insufficient pediatric data.
1. Phosphatidylinositiol 3-Kinase (PI3K)/AKT/Mammalian Target of Rapamycin (mTOR) Signaling Pathway
The PI3K/AKT/mTOR signaling pathway is involved in a diverse number of cellular functions, including transcription, translation, cell-cycle progression, apoptosis, and metabolism (figure 1). PI3Ks are a family of related enzymes first described in the 1980s by Lewis Cantley and colleagues in a number of seminal papers that demonstrated viral oncoproteins needed an association with a lipid kinase (PI3K) for transformation.[5,6] PI3Ks are important for cell growth, metabolism, and survival. Their primary biochemical function is to phosphorylate the 3-hydroxyl group of phosphoinositides.[7] The PI3K family is divided into three classes (I, II, III) based on structure and function. Class I PBKs are the most studied, as they are the class involved in malignant transformation and in cellular growth and metabolism.[8] The roles of classes II and III PI3Ks are less clear; however, data suggest class II PI3Ks are involved in membrane trafficking, and class III PI3Ks may be involved in autophagy.[9]
Fig. 1.
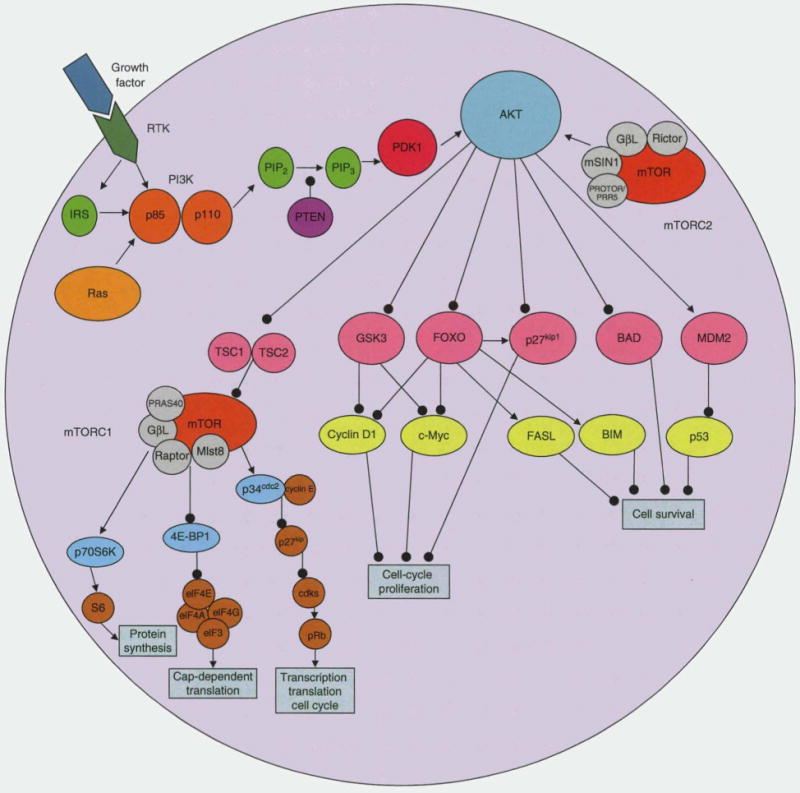
PI3K/AKT/mT0R signaling pathway. Growth factor or ligand binding to a receptor tyrosine kinase, including IGF-1R, PDGFR, or EGFR, leads to activation of IRS-1 which upregulates and activates PI3K by removing the inhibition of the regulatory (p85) subunit on the catalytic subunit (p110). PI3K can also be directly activated by RTK or RAS. After activation, PI3K phosphorylates PIP2 to make PIP3. PIP3 recruits PDK1 and AKT to the cell membrane. PDK1 and mTORC2 phosphorylate and activate AKT. PTEN negatively regulates AKT activation by converting PIP3 to PIP2. Activated AKT can regulate cell growth and protein synthesis by activating mTORC1 through TSC1/2. AKT can stimulate cell-cycle progression by modulating cell-cycle inhibitors (p27kip1 through FOXO and GSK3) and cell-cycle stimulators (cyclin D1 and c-Myc). AKT can also regulate programmed cell death by inhibition of FasL, BIM, or BAD, and by degradation of p53 through MDM2. mTOR can form two distinct complexes, mTORCI and mTORC2. mTORCI is composed of mTOR, GβL, Mlst8, PRAS40, and raptor. mTORC2 is composed of mTOR, GβL, mSIN1, and Rictor. mTORCI can regulate protein synthesis and cell-cycle progression through phosphorylating p70S6 kinase (S6K1) and 4E-BP1. mTORCI also facilitates the elimination of the p27kip1 through interactions with p34cdc2, allowing cell-cycle progression by cdks which can phosphorylate Rb. Arrows represent activation. Lines with circles represent inhibition. 4E-BP1 = elF4E binding protein; BAD = BCL-2-associated death promoter; BIM = BCL-2-interacting mediator of cell death; cdks = cyclin-dependent kinases; EGFR = epidermal growth factor receptor; elF = eukaryotic initiation factor; FasL = fas ligand; FOXO = Forkhead box O proteins; GSK3 = glycogen synthase kinase 3; GβL = G protein β subunit-like; IGF-1R = insulin-like growth factor-1 receptor; IRS-1 = insulin receptor substrate-1 ; KIDM2 = mouse double minute 2; Mlst8 = mammalian lethal with sec-13; mSIN1 = mammalian stress-activated protein kinase-interaction protein 1; mTOR = mammalian target of rapamycin; p27kip1 = cyclin-dependent kinase inhibitor kip1; p34cdc2 = cyclin-dependent controlling kinase p34; PDGFR = platelet-derived growth factor receptor; PDK1 = phosphoinositide-dependent protein kinase 1; PI3K = phosphatidylinositiol 3-kinase; PIP2 = phosphatidylinositol-4,5-biphosphate; PIP3 = phosphatidylinositol-3,4, 5-triphosphate; PRAS40 = proline-rich AKT substrate of 40 kDa; PROTOR/PRR5 = protein observed with Rictor-1/Proline-rich protein 5; PTEN = phosphatase and tensin homologue deleted on chromosome 10; RAS = a GTPase; Rb = retinoblastoma protein; RTK = receptor tyrosine kinase; TSC1/2 = tuberous sclerosis 1/2 complex.
Class I PI3Ks are subdivided into subtype A and B based on their activation. Class IA PI3Ks are activated by receptor tyrosine kinases (RTKs), G-protein coupled receptors, and RAS (a GTPase). Class IB PI3Ks are activated by G-protein-coupled receptors only.[7,8] Class IA PI3Ks are the family member subtype involved in the PI3K/AKT/mTOR signaling cascade, whereas class IB PI3Ks are primarily involved in immune function and inflammation.[7]
Class IA PI3Ks are heterodimers consisting of a catalytic subunit (p110) and a regulatory subunit (p85).[10,11] Each subunit can be encoded by three different genes: (i) regulatory, PIK3R1 (encodes p85α isoform), PIK3R2 (p85β), PIK3R3 (p85γ); and (ii) catalytic, PIK3CA (p110α), PIK3CB (p100β), and PIK3CC (p100δ).[9,12,13] A primary method of activation for Class IA PI3Ks is growth factor or ligand binding to a RTK, including insulin-like growth factor (IGF)-1 receptor, platelet-derived growth factor receptor, and epidermal growth factor receptor. The ligand-engaged RTK binds PI3K, removing the inhibition of the regulatory subunit on the catalytic subunit.[14,15] After activation, PI3K phosphorylates phosphatidylinositol-4,5-biphosphate (PIP2) to make phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 recruits phosphoinositide-dependent protein kinase 1 (PDK1) and AKT to the cell membrane and is an important mediator of early signaling events.[16] PDK1 and mTORC2 (discussed later in this section) phosphorylate AKT, thereby activating it.[7,8,17] PI3K is negatively regulated by S6K1, as well as phosphate and tensin homologue deleted on chromosome 10 (PTEN), which converts PIP3 to PIP2.
AKT is a serine/threonine kinase that functions as an important regulator of cell growth, survival, and glucose metabolism. For full activation, AKT is phosphorylated at threonine-308 and serine-473 by PDK1 and mTORC2, respectively. The AKT family consists of three isoforms (AKT1, 2, and 3). AKT1 is the isoform most involved in regulating cellular survival and protein synthesis.[18] AKT2 is primarily involved in glucose transport through the insulin signaling pathway.[19] The function of AKT3 is poorly defined and this isoform is mostly limited to nervous system tissue.[18]
AKT can phosphorylate over 100 different substrates. AKT regulates cell growth and protein synthesis by activating mTOR through tuberous sclerosis 1/2 complex (TSC1/2). AKT stimulates cell-cycle progression by modulating cell-cycle inhibitors, including cyclin-dependent kinase inhibitor kip1 (p27kip1), through the FOXO family of Forkhead transcription factors, and glycogen synthase kinase 3 (GSK3), and cell-cycle stimulators, including cyclin D1 and c-Myc. AKT can regulate programmed cell death by inhibition of Fas ligand (FasL), BCL2-associated death promoter (BAD), BCL-2-interacting mediator of cell death (BIM), or BCL-2-associated X-protein (BAX), and degradation of p53.[20,21] Finally, either directly or indirectly, AKT regulates NF-kB (through IkB kinase), RAF, and c-JUN N-terminal kinase (JNK).[22]
mTOR is a serine/threonine kinase that regulates cell growth, survival, and metabolism. It does so by regulating ribosomal biogenesis, protein translation, expression of metabolism-related genes, and amino acid uptake, as well as increasing cell-cycle transit time, and inhibiting apoptosis and autophagy.[23–27] mTOR is activated by a number of signaling pathways, including PI3K/AKT, as well as RAS, BCR-ABL, and (T-cell leukemia/lymphoma 1 (TCL1).[28,29] mTOR can form two distinct complexes, mTORC1 and mTORC2.[30] The mTORC1 complex is composed of mTOR, G protein β subunit-like (GβL), mammalian lethal with sec-13 (Mlst8), proline-rich AKT substrate of 40 kDa (PRAS40), and regulatory-associated protein of mTOR (RAPTOR).[30] The mTORC2 complex includes mTOR, GβL, mammalian stress-activated protein kinase-interaction protein 1 (mSIN1), rapamycin-insensitive companion of mTOR (RICTOR), and protein observed with Rictor-1/Proline-rich protein 5 (PROTOR/PRR5).[30]
The main targets of activated mTORC1 are S6K1 and 4E-BP1.[23] Activated S6K1 induces 5′ terminal oligopyrimidine (TOP)-translation and ribosomal biosynthesis, and blocks apoptosis through BAD. S6K1 can also downregulate insulin receptor substrate-1 (IRS-1), resulting in feedback downregulation of the mTOR pathway.[31] Through phosphorylation of eukaryotic translation initiation factor (eIF) 4E binding protein (4E-BP1), mTORCl regulates cap-dependent protein translation.[32] When 4E-BP1 is phosphorylated by mTORC1, it is released from eIF4E, allowing eIF4E to associate with eIF4G and form the eIF4F translation initiation complex. The eIF4F complex is important for the cap-dependent translation of a number of proteins, including c-Myc and cyclin D1.[33] mTORCl also interacts with cyclin-dependent controlling kinase p34 (p34cdc2) to eliminate the cyclin-dependent kinase inhibitor p27kip1, resulting in cell-cycle progression under the regulation of cyclin-dependent kinases.[33,34] In contrast, mTORC2, in concert with PDK1, activates AKT by phosphorylation.
2. PI3K/AKT/mTOR Signaling in Cancer
Dysregulation of the PI3K/AKT/mTOR signaling pathway is a common event in a number of adult and pediatric malignancies. The dysregulation can be intrinsic to the signaling pathway or as a consequence of mutations in pathways that can activate or regulate the PI3K/AKT/mTOR pathway, including activating mutations in Fms-like tyrosine kinase receptor (FLT3), N- or KRAS, and c-Kit tyrosine kinase receptor.[35–37] Activating mutations in PI3K subunit genes PIK3CA and PIK3R1, and inactivating mutations in PTEN are common in a wide variety of malignancies, including colon, breast, liver, ovarian, leukemia, brain tumors, melanoma, prostate, thyroid, and lymphoma.[38–41] Gain of function mutations in all three AKT genes have been identified in adult malignancies, including breast, colon, melanoma, and ovarian.[7,42–44] No mutations have been identified in any AKT isoform in childhood cancer; however, chromosomal gains amplifying the AKT1 gene have been described recently in rare cases of childhood AML, T-cell ALL, and gliosarcoma.[45–47] Amplification of eIF4E, S6K1, and cyclin D have been reported in adult cancers, including breast and mantle cell lymphomas but not in pediatric tumors.[48–51] mTOR-specific mutations in human cancer are extremely rare, with only two reported cases in adult carcinomas.[52]
3. Agents that Target PI3K/AKT/mTOR
Table I summarizes agents that target the PI3K/AKT/mTOR pathway. The majority of these will be discussed in the text with focus on agents undergoing investigation in hematologic malignancies.
Table I.
Inhibitors of the phosphatidylinositiol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway
| Class | Agent | Target | Administration | Development |
|---|---|---|---|---|
| Rapalogs | Sirolimus | mTORC1 | Oral | FDA approveda |
| Everolimus | mTORC1 | Oral | FDA approveda | |
| Temsirolimus | mTORC1 | Parenteral | FDA approveda | |
| Ridaforolimus | mTORC1 | Oral and parenteral | Clinical trials | |
| mTOR kinase inhibitors | PP242 | mTORC1/2 | Parenteral | Pre-clinical |
| INK128 | mTORC1/2 | Oral | Clinical trials | |
| AZD8055 | mTORC1/2 | Oral | Clinical trials | |
| OSI-027 | mTORC1/2 | Oral | Clinical trials | |
| PI3K and PI3K/mTOR inhibitors | BKM120 | PI3K | Oral | Clinical trials |
| SAR245408 | PI3K | Oral | Clinical trials | |
| GS-1101 | PI3K (p110δ) | Oral | Clinical trials | |
| Wortmannin | PI3K, mTORC1, PI4K | Not for clinical use | Pre-clinical | |
| LY294002 | PI3K, mTORC1, PI4K, PIM1, PLK1, ATM, CK2, DNA-PK | Not for clinical use | Pre-clinical | |
| PX-866 | PI3K, mTORC1 | Oral | Clinical trials | |
| SF1126 | PI3K, PIM1, mTORC1/2, PLK1, DNA-PK, CK2, ATM | Oral | Clinical trials | |
| SAR245409, | PI3K, mTORC1 | Oral | Clinical trials | |
| Pictrelisib | PI3K, mTORC1, DNA-PK | Oral | Clinical trials | |
| GDC-0980 | PI3K, mTORC1 | Oral | Clinical trials | |
| PI-103 | PI3K, mTORC1 | Parenteral | Pre-clinical | |
| PF-4691502, | PI3K, mTORC1 | Oral | Clinical trials | |
| BGT226 | PI3K, mTORC1 | Oral | Clinical trials | |
| GSK2126458 | PI3K, mTORC1 | Oral | Clinical trials | |
| ZSTK474 | PI3K, mTORC1 | Oral | Clinical trials | |
| XL-499 | PI3K (p110δ), mTORC1 | Not published | Pre-clinical | |
| BEZ235 | PI3K, mTORC1/2, DNA-PK | Oral | Clinical trials | |
| AKT inhibitors | Perifosine | AKT | Oral | Clinical trialsb |
| Triciribine | AKT | Parenteral | Clinical trials | |
| MK-2206 | AKT | Oral | Clinical trials | |
| GSK690693 | AKT, PAK6, PKC, PrkX | Parenteral | Pre-clinical | |
| GSK2141795 | AKT | Oral | Clinical trials | |
| GSK2110183 | AKT | Oral | Clinical trials | |
| GDC-0068 | AKT | Oral | Clinical trials | |
| LY2780301 | AKT, p70S6K | Oral | Clinical trials | |
| PDK-1 inhibitors | UCN-01 | PDK-1 | Parenteral | Clinical trials |
| GSK2334470 | PDK-1 | Not published | Pre-clinical | |
| AR12 | PDK-1 | Oral | Pre-clinical | |
| Other | 4EGI-1 | elF4F | Not published | Pre-clinical |
| Ribavirin | elF4E | Oral, parenteral | FDA approveda |
Also approved in the EU.
Granted orphan drug status in the EU.
elF4F = eukaryotic translation initiation factor 4F; ATM = ataxia-telangiectasia mutated; CK2 = casein kinas II; DNA-PK = DNA-dependent protein kinase; mTOR = mammalian target of rapamycin; p70S6K = p70 ribosomal S6 kinase; PAK6 = p21-activated kinase 6; PDK-1 = phosphoinositide-dependent protein kinase 1; PI3K = phosphatidylinositiol 3-kinase; PIM1 = protocol-oncogene serine/threonine protein kinase PIM-1; PKC = protein kinase C; PLK1 = protein-like kinase 1; PrkX = protein kinase, X-linked.
3.1 Rapalogs
Rapalogs are the first class of agents targeting the PI3K/AKT/mTOR signaling pathway that were used to treat human malignancies. The first rapalog investigated in the clinical setting was sirolimus (rapamycin).[53] Sirolimus is a macrocyclic lactone isolated from Streptomyces hydroscopicus. It was originally isolated on Easter Island, also known as Rapa Nui, hence its trade name. Sirolimus is most commonly used to suppress the immune system and prevent rejection after kidney transplant; however, it is also used as an immune suppressant after other organ (liver, heart, and lung) and bone marrow transplants.[54–59]
Sirolimus and other rapalogs associate with binding protein FKBP12, primarily blocking the mTORC1 complex with little to no activity against the mTORC2 complex.[60] Newer second-generation small molecule inhibitors of mTOR kinase that target both mTORCl and mTORC2 (mTORC1/2 inhibitors) are under development (see section 3.2). It was not until 2002 that it was realized that mTOR could associate in two functionally distinct complexes and that rapalogs could not normally directly disrupt mTORC2.[61] Thus, older literature broadly uses the term mTOR inhibitors (MTIs) to describe rapalogs, and the term rapalogs was coined to distinguish this class of agents from other mTOR kinase inhibitors. In newer nomenclature, MTIs include all agents that can target mTOR regardless of mechanism or class. Confounding the issue further, while rapalogs were originally, and often, reported to have no activity against mTORC2, in certain cell types with prolonged treatment rapalogs can inhibit the assembly of the mTORC2 complex.[62,63]
Sirolimus has variable bioavailability and poor aqueous solubility, requiring therapeutic drug monitoring. To overcome these issues, a number of second-generation rapalogs were developed, including everolimus, temsirolimus, and ridaforolimus (formerly known as deforolimus). Sirolimus and everolimus are given enterally, and temsirolimus is given intravenously. Ridaforolimus is available in an oral and intravenous preparation.[64,65] The rapalogs have been studied in a large variety of malignant and non-malignant diseases. It is beyond the scope of this manuscript to give even a cursory overview of all the clinical trials; however, a detailed description of preclinical and clinical data supporting their use in pediatric hematologic malignancies are discussed in the disease-specific sections (see section 4).
The rapalogs are generally well tolerated. The most common side effects include stomatitis and hyperlipidemia. Often the stomatitis is the most severe during the initial months of therapy. It is usually low grade, not requiring the discontinuation of the agent. When rapalogs are used in combination with cytotoxic chemotherapy, the stomatitis can be more severe and dose limiting. Hyperlipidemia may require treatment with a statin or other agent; hyperlipidemia occurs more commonly, and more often requires treatment, in adults. Other more common side effects include skin reactions, diarrhea, fatigue, and thrombocytopenia. Even though sirolimus can cause mild thrombocytopenia, it is active against immune-mediated thrombocytopenia and is under clinical investigation for this indication.[66,67]
Rarer side effects include peripheral edema, interstitial pneumonitis, and renal insufficiency. Edema and pneumonitis are more common in adults and with combination regimens. With single agent therapy, the risk of opportunistic infection is extremely low; however, there is a higher risk when used in combination with other immunosuppressant drugs.[68,69] Long-term use of any immune suppressant carries the theoretical risk of developing secondary malignancies from decreased cancer immune surveillance. Despite this concern, clinical studies have shown a reduced incidence of new cancers in patients given rapalogs after organ transplant compared with patients treated with other agents, supporting the antineoplastic properties of this compound.[70]
Most of the postmarketing side effect data from rapalog use has come from patients treated with sirolimus after solid organ or hematopoietic stem cell transplant (HSCT). In these settings, rapalogs are typically given with calcineurin inhibitors. In combination with calcineurin inhibitors, rapalogs significantly increase the risk of microangiopathy and veno-occlusive disease; however, these side effects have not been reported when rapalogs are used without calcineurin inhibitors. All rapalogs can delay wound and vascular healing. Accordingly, these effects may occur because the calcineurin inhibitors cause vascular damage and the rapalogs prevent healing, exacerbating the problem.[71,72]
3.2 mTOR Kinase Inhibitors (mTORC1/2 Inhibitors)
A large number of small molecule mTOR kinase inhibitors are under development, including PP242, INK128, AZD8055, and OSI-027.[72,73] Unlike rapalogs, the mTOR kinase inhibitors are active against both mTORCl and 2. These agents are ATP-competitive inhibitors of the kinase active sites of both mTORC1 and 2. Long-term exposure to mTORC1-specific inhibitors can lead to a compensatory increase of AKT that can, in theory, promote drug resistance and/or activate other AKT-regulated pathways.[74] PI3K and mTORC2 are both upstream of AKT. Targeting multiple intermediates along the PI3K/AKT/mTOR axis may overcome this potential problem.[75–77] Most of the small molecule inhibitors that target mTORC1/2 also target other proteins in the PI3K/AKT/mTOR signaling pathway, including PI3K, and are discussed in section 4.3. As with many kinase inhibitors, these agents can also affect other kinase pathways at higher concentrations, but the half maximal inhibitory concentration (IC50) values for mTOR kinase for many of these drugs, including PP242, are nearly 100-fold lower than their affinity for other kinases.[78] PP242 is the prototypical mTORC1/2 kinase inhibitor and is the most studied in preclinical models, demonstrating activity against a number of malignancies, including multiple myeloma and lymphoblastic leukemia.[79,80] PP242 has only been investigated in preclinical models; however, INK128, AZD8055, and OSI-027 are currently being evaluated in early-phase clinical trials.[81]
3.3 PI3K Inhibitors
A number of agents that target PI3K are under development although space does not permit discussing all of the agents in detail (table I).[72,73] The majority of these agents also target mTOR.[73] There are three agents that specifically target PI3K and have entered clinical trials: BKM120, SAR245408, and GS-1101. BKM120 is a PI3K catalytic subunit inhibitor.[82] It has been investigated in early-phase trials in adults with advanced solid tumors, demonstrating efficacy in breast tumors with KRAS or PIK3CA mutations.[83] The agent was well tolerated and toxicities included hyperglycemia, mucositis, rash, and mood alterations. SAR245408 is an ATP-competitive reversible PI3K inhibitor.[84,85] It has been studied as a single agent in adults with advanced malignancies, in combination with erolitinib in adults with advanced solid tumors, and in combination with paclitaxel and carboplatin in adults with advanced solid tumors.[86–88] Side effects included rash, nausea, vomiting, allergic reactions, and diarrhea. All the PI3K inhibitors in clinical trials, except GS-1101, target p110α. GS-1101 targets p110δ, and as p110δ is restricted to leukocytes, the focus of this agent is hematologic malignancies.[89,90] GS-1101 was well tolerated and demonstrated activity in chronic lymphocytic leukemia and non-Hodgkin’s lymphoma in phase I trials.[91,92]
The most studied PI3K inhibitors in preclinical models are wortmannin and LY294002. Both agents are potent PI3K inhibitors, targeting PI3K, PI4K, and mTOR. LY294002 also targets the serine/threonine protein kinase (PIM1), protein-like kinase 1 (PLK1), casein kinas II (CK2), and ataxia-telangiectasia mutated (ATM).[73] Neither are under clinical development as both have extremely poor bioavailability.[73] To overcome this issue, similar compounds have been developed. PX-866 is a potent wortmannin derivative that is currently being studied in early-phase trials in adults with solid tumors, with promising results and limited toxicity.[93] SF1126 is a prodrug of LY294002 that has been investigated in a phase I trial in adults with solid tumors and B-cell malignancies.[94] The drug was well tolerated and demonstrated activity in patients with chronic lymphocytic leukemia, gastrointestinal stromal tumor, and renal cell carcinoma. The only grade 3 or higher toxicity was diarrhea.
A large number of additional dual PI3k-mTOR inhibitors, including SAR245409, pictrelisib, GDC-0980, PF-4691502, BGT226, GSK2126458, and ZSTK474 are in early-phase clinical trials as single agents or in combination with cytotoxic drugs.[64] One agent that warrants special mention is BEZ235 as it is being investigated extensively in preclinical models of pediatric leukemias.[95–97] It is currently in early-phase trials in adults with solid tumors. BEZ235 inhibits PI3K, mTORC1, and mTORC2.[98] Early data demonstrate it is well tolerated. Nausea, diarrhea, and vomiting are the only common toxicities, and the dose-limiting toxicities are fatigue and thrombocytopenia.[99]
3.4 AKT Inhibitors
A number of AKT inhibitors are in preclinical development and early-phase clinic trials (table I).[72,73] AKT inhibitors primarily fall into two groups – alkyl-lysophospholipids (APLs) and small molecule inhibitors. APLs are a class of structurally related compounds that act on cellular membranes in actively dividing cells. They induce apoptosis by accumulating in lipid rafts, recruiting Fas family members, and blocking AKT translocation to the cell membrane, which is required for activation.[100,101] The first APL to undergo investigation was miltefosine, an agent used to treat visceral leishmaniasis.[102] Unfortunately, the dose required to inactivate AKT was too toxic. Perifosine is an APL that is currently being investigated in early-phase clinical trials in advanced solid tumors, breast cancer, melanoma, head and neck carcinoma, Waldstrom’s macroglobulinemia, multiple myeloma, and a number of leukemias and lymphomas.[103–105] Unlike miltefosine, perifosine can block AKT activation at a tolerable dose. Perifosine is generally well tolerated with toxicities, including cytopenias, arthritis, fatigue, and diarrhea.[103]
A number of AKT small molecule inhibitors are in preclinical development and a few are in clinical trials, including triciribine (tricyclic nucleoside phosphate), MK-2206, and GSK2141795. Triciribine has been tested in clinical trials for the longest period of time as the first phase I published trial results were in 1983.[106] At that time it was not known that the drug inhibited AKT, and it was hypothesized to be a nonspecific DNA synthesis inhibitor.[107] In early clinical trials, triciribine was found to have a number of significant side effects, including electrolyte abnormalities (hyperglycemia and hypercalcemia) and hepatotoxicity, limiting its use.[106] Recently, early-phase trials have resumed investigating the agent, specifically in tumors with activated AKT.[108] MK-2206 is an allosteric AKT inhibitor with activity against all three AKT isoforms. Unlike triciribine, it is an oral medication. MK-2206 is currently being investigated in a large number of clinical trials as a single agent and in combination.[73] Early study findings suggest it is tolerable with side effects including rash, fatigue, and hyperglycemia.[109] GSK2141795 is a potent oral AKT inhibitor with activity against all three isoforms. The agent appears to be tolerable with side effects including hyper- and hypoglycemia, rash, fatigue, diarrhea, and nausea.[110]
3.5 Other Agents that Target the P13K/AKT/mTOR Pathway
A number of additional compounds are in development that can target other proteins in the PI3K/AKT/mTOR pathway. These include a number of compounds in preclinical development, such as 4EGI-1 (eIF4F inhibitor) and UCN-01, GSK2334470, and AR12 (PDK-1 inhibitors).[111–114] Ribavirin is a small molecule inhibitor that has been used primarily for its anti-viral properties in immunocompromised individuals. It can suppress eIF4E by inhibiting its binding to 5′methyl-7 GTP-capped messenger RNA.[115,116] Based on its activity against eIF4E, it has been studied in a number of hematologic malignancies (discussed in section 4). Finally, as a number of extrinsic pathways can regulate PI3K/AKT/mTOR, drugs targeting alternative pathways are being used in part to down-regulate the PI3K/AKT/mTOR pathway. These include drugs that target Notch, MAP kinase interacting kinase (MNK), the tyrosine kinase Syk, and aurora kinase.[117–120]
4. Targeting PI3K/AKT/mTOR in Pediatric Hematologic Malignancies
4.1 Acute Lymphoblastic Leukemia
ALL is the most common pediatric malignancy. The development and use of novel therapeutics to treat ALL is one of the great success stories of modern medicine as pediatric ALL was the first malignancy to be treated successfully and eventually cured with chemotherapy. Currently, patients are treated with 2–3 years of rotating blocks of therapy, utilizing ten or more different drugs. The key to improving outcome in these populations will most likely require the inclusion of novel targeted therapies.
ALL is a lymphoid malignancy resulting from transforming events in early B- and T-cell precursors. Recently, using modern genomic techniques, a number of potentially targetable genetic alterations have been identified in a wide variety of signaling pathways, including Jak/Stat, Ras/Raf/Mek/Erk, TP53/RB, and the PI3K/AKT/mTOR pathway.[121–124] While novel agents targeting these pathways are unlikely to cure ALL individually, the addition of targeted agents to multi-agent cytotoxic backbones has the potential to significantly improve cure rates, as evidenced by markedly improved survival in Ph+ ALL patients when the BCR-ABL kinase inhibitor imatinib was added to a multi-agent chemotherapy backbone.[1]
The PI3K/AKT/mTOR pathway can be dysregulated in ALL patients by a number of mechanisms. Activation of the PI3K/AKT/mTOR pathway is a common event in precursor T-ALL. Most commonly, this is through inactivation of PTEN, resulting in activation of AKT.[125,126] PTEN inactivation can occur as a consequence of mutations or deletions in the PTEN gene, or as a consequence of defects in other signaling pathways that can lead to decreased transcription or post-translational modification of PTEN.[127] The PI3K/AKT/mTOR pathway can also be activated directly in T-ALL by mutations in AKT1, PIK3CA, and PIK3R1, and indirectly through mutations in RAS and Notch family members.[126,128] In Ph+ pre-B-cell ALL (B-ALL), the PI3K/AKT/mTOR pathway is activated directly by BCR-ABL.[129] Recently, genetic rearrangements in cytokine receptor-like factor 2 (CRLF2) have been identified in high-risk subsets of pre-B-ALL, affecting up to 10% of patients, and genetic rearrangements in interleukin (IL)-7 receptor α (IL7R) have been identified in pre-B- and pre-T-ALL.[123,130] IL7R heterodimerizes with CRLF2 to form the receptor for thymic stromal-derived lymphoietin (TSLP), a protein that our group had previously demonstrated can stimulate proliferation of pre-B-ALL cells and activate signaling through PI3K/AKT/mTOR.[131]
Our group hypothesized that targeting mTOR may be effective in ALL before it was established that the pathway was dysregulated in ALL.[132] Our hypothesis was based on the activity of mTOR inhibitors against normal lymphocytes and the knowledge that PI3K/AKT/mTOR signaling was activated in a number of other malignancies. We demonstrated that sirolimus could inhibit growth and induce cell death in ALL cell lines and improve survival in an Eu-RET[133] transgenic mouse model of leukemia/lymphoma.[132] Since that initial report, the activity of a number of rapalogs, including everolimus, sirolimus, and temsirolimus have been extensively studied in ALL in preclinical models by our group and others, establishing activity not only in cell lines and transgenic mouse models, but also against primary human ALL samples supported in vitro with stromal cells and in vivo with immunodeficient mouse strains.[131,132,134–140] Our work and the work of others suggest that rapalogs may have the highest degree of activity against ALL types with the worst prognosis, including pre-T, adult, BCR-ABL and Ikaros mutant.[29,135,141,142] Supporting this are recent data demonstrating that early engraftment of pre-B-ALL in immune-deficient mice is associated with the activation of the mTOR pathway and a very high risk of relapse.[143]
Experience with chemotherapy for decades, as well as more recent experience with targeted therapies demonstrates that no single agent is likely to be curative. Thus, it is important to study new agents in combination with conventional drugs used to treat ALL. The goal should be to select rationally designed combinations based on the mechanism(s) of action of the new agent and the standard agents. Rapalogs have been studied extensively with conventional agents in preclinical models of ALL. Our group, and others, have demonstrated that rapalogs have an additive or synergistic effect when combined with many agents in vitro, including methotrexate, corticosteroids, doxorubicin, etoposide, and asparaginase.[136,144–146] An added benefit was not found when rapalogs were combined with vincristine in vitro; however, the combination was found to be more effective than either drug alone in vivo.[136,139,146]
Rapalogs have been studied in vivo in combination with methotrexate and corticosteroids. The combination of the rapalog temsirolimus with methotrexate was found to have a profound effect against xenografted pre-B-ALL, curing some animals.[136] One mechanism of ALL blast resistance to methotrexate is over-expression of dihydrofolate reductase (DHFR). We demonstrated rapalogs can decrease DHFR in ALL as a consequence of downregulation of cyclin D1. Thus, rapalogs may be useful in overcoming methotrexate resistance in ALL. Similarly, rapalogs have been shown to reverse glucocorticoid resistance in ALL through downregulation of MCL-1.[145] As corticosteroid resistance is common in relapsed ALL, and corticosteroids are one of the most active and important classes of agents used to treat ALL, the ability to restore corticosteroid sensitivity is promising. Rapalogs are being investigated in combination with other novel therapeutics in ALL with the goal of inhibiting multiple activated signaling pathways. Encouraging results have been found in preclinical studies combining rapalogs with inhibitors of Notch signaling, Jak/stat and the proteasome.[141,147,148]
Based on the large amount of preclinical data supporting their use, rapalogs are being investigated in clinical trials in patients with ALL. Two adult patients with ALL have been treated in early-phase trials using single-agent rapalogs. Both of these patients were enrolled in studies open to patients with relapsed or refractory malignancies, and both patients tolerated therapy but did not have an objective response.[149,150] Recently, we completed a single-institution phase I trial of sirolimus in children with relapsed and/or refractory ALL. An interim report of the study demonstrated stable disease in three of seven patients.[151] Based on our preclinical work, we have opened a single institution study of sirolimus in combination with methotrexate in relapsed pediatric ALL (NCT01162551). Recently, a pilot trial of sirolimus and glucocorticoids given for 5 days as an investigational window prior to multi-agent therapy for relapsed ALL was completed at the Dana Farber Cancer Institute (Boston, MA, USA). The goals of the study were to determine if the combination could be given safely and whether sirolimus would downregulate MCL-1. A corticosteroid-only control arm window was used as a comparison. An early report of the trial suggested the combination is tolerated and that MCL-1 is downregulated in ALL blasts in the sirolimus arm in most patients. One patient had reduction in peripheral blasts during the 5-day window, with an increase in blasts after sirolimus was discontinued.[152] Based on these results, the Dana Farber Cancer Institute is opening a multiagent re-induction protocol, including a rapalog. Ongoing and opening clinical trials include a pilot study of sirolimus and pegaspargase at Emory Children’s Hospital (Atlanta, GA, USA) [NCT00957320] and a multi-institutional phase I trial through the Children’s Oncology Group (COG; ADVL1114), adding temsirolimus to an intensive re-induction backbone. Finally, as sirolimus can also be used for graft-versus-host disease (GVHD) prophylaxis, a large multi-institutional COG-initiated clinical trial (ASCT0431) was opened to test the hypothesis that post-HSCT sirolimus could be used to prevent GVHD and treat ALL, improving survival. This trial was recently suspended after an interim futility analysis suggested that the trial would not meet its event-free survival goal.
While the majority of studies thus far in pediatric ALL have focused on rapalogs, more recent preclinical studies have investigated other drugs that target the PI3K/AKT/mTOR pathway. Preliminary data by our group suggest targeting eIF4E with ribavirin may be active against ALL.[153] As mentioned, the most common means of abnormally activating the pathway is through AKT. mTORC1 is downstream of AKT, and as rapalogs are the agents furthest in clinical development, the initial focus on rapalogs to treat ALL was logical and prudent. Nevertheless, AKT activates pathways other than mTORCl and long-term treatment with a rapalog may lead to feedback activation of AKT. Thus, in theory, for some ALL subsets (especially pre-T-ALL), a more ideal approach would be either to target AKT directly, or to target proteins up- and downstream of AKT. This could be accomplished with either an mTORC1/2 inhibitor or an agent that targets PI3K and mTORC1.
The PI3K inhibitors wortmannin and LY294002 have pre-clinical activity against both pre-T- and pre-B-ALL comparable to rapalogs.[29,154,155] Dual inhibition of PI3K and mTORC1, however, using either a PI3K inhibitor with a rapalog or an agent that targets both (e.g. PI-103) is superior to PI3K inhibitors or rapalogs alone in preclinical models.[29,154] Furthermore, the dual mTORC1/2 inhibitors PP242 and OSI-027 are very active in vitro against pre-clinical models of pre-T-ALL (cell lines and primary cells) and are superior to rapalogs as these agents successfully inactivate AKT and mTOR.[156] BEZ235, an inhibitor of mTORC1/2 and PI3K, was also shown to be very active against pre-T-ALL in vitro, inactivating both AKT and mTOR.[95] Preliminary studies demonstrate similar results with BEZ235 in pre-B-ALL in vitro and in vivo.[96,97] PP242, OSI-027, and BEZ235 were combined with conventional cytotoxics in these studies, demonstrating in vitro synergy with a number of agents, including vincristine, doxorubicin, dexamethasone, cytarabine, and cyclophosphamide. The AKT inhibitor GSK690693 was found to have significant activity against both pre-B- and pre-T-ALL in vitro; however, it had limited activity when tested in vivo, emphasizing the importance of murine studies for all of these agents.[157,158]
4.2 Acute and Chronic Myelogenous Leukemia
AML is a clonal disorder of early myeloid lineages. Very intensive multi-agent chemotherapy strategies and HSCT have improved survival in childhood AML; however, approximately 40% of patients are incurable and children with very high-risk features, including monosomy 7 and high FLT3-ITD (internal tandem duplicated), have very poor prognosis.
The PI3K/AKT/mTOR pathway is abnormally activated in 50–80% of AML cases, most commonly as constitutive activation of AKT.[159,160] The upregulation of the PI3K/AKT/mTOR pathway can occur as a consequence of activating mutations in the FLT3 receptor, c-KIT receptor, NRAS, or KRASP.[35–37] In addition, upregulation can occur through abnormal autocrine/paracrine secretion of IGF-1 or vascular endothelial growth factor (VEGF), overexpression of PBKp100β, PI3Kp100δ or PDK1, or underexpression of protein phosphatase 2 (PP2A).[161–168] Unlike T-ALL, PTEN inactivation in AML is a very rare mechanism of AKT activation.[159,160]
As with ALL, rapalogs were the first agents to be studied in AML. Rapalogs have demonstrated modest activity in preclinical studies against AML; however, single–agent, early-phase trials in adults did not find activity.[149,150,169,170] Preclinical studies combining rapalogs with cytotoxics and targeted agents, including etoposide, doxorubicin, cytarabine, histone deacetylase inhibitors, and glycolytic inhibitors, are more promising, often showing pronounced combined effects.[171–176] Based on these results, combination therapy trials are ongoing in adults, and it is anticipated that studies will begin in children if the adult data are promising. Recently, a phase I study of sirolimus and mitoxantrone, etoposide, cytarabine (MEC) chemotherapy was completed at the University of Pennsylvania School of Medicine.[177] Twenty-nine subjects were treated, and the combination was tolerable and somewhat active, with a complete response (CR) plus partial response (PR) rate of 22%. Of note, five subjects had blasts evaluated to determine if sirolimus was downregulating mTOR, and only one subject’s blasts showed definitive inhibition. Preliminary results from a phase Ib trial combining everolimus and low-dose cytarabine in previously untreated elderly patients were promising, demonstrating improved median overall survival in poor risk subjects treated with the combination compared with low-dose cytarabine alone (175 vs 44 days).[178] Similarly, early results of a phase Ib/II trial combining everolimus with azacitidine in relapsed or refractory AML demonstrated considerable activity, with an overall response rate (ORR) of 36% in subjects with refractory or relapsed AML.[179]
The focus in AML is shifting to investigating other agents that target the PI3K/AKT/mTOR pathway. Direct targeting of PI3K with wortmannin and LY294002 have shown strong cytotoxic effects in preclinical AML models.[37,180,181] Because eIF4E is upregulated in 30% of AML patients with the French-American-British (FAB) classification subtype M4/M5, ribavirin, an eIF4E inhibitor, was studied in preclinical models and in a pilot study.[116,182] It was found to be active against M4/M5 AML in vitro and in patients as 5 of 11 subjects had objective responses (one CR, two PRs, and two blast reductions). Because constitutive activation of AKT is the most common pathway abnormality in AML, dual PI3K/mTOR inhibitors (PI-103, BEZ235), mTORC1/2 inhibitors (OSI-027, PP242), and AKT inhibitors (perifosine) have been investigated in preclinical models and, at least in vitro, appear to be more effective than rapalogs.[183–187] Based on these results, early-phase clinical trials are ongoing in adults.
CML is a myeloproliferative disorder characterized by clonal myeloid cells with an abnormal fusion protein, BCR-ABL, that has tyrosine kinase activity. Until recently, the only curative therapy was HSCT; however, the disease is now treated and often potentially cured with BCR-ABL-targeting tyrosine kinase inhibitors (TKIs), including imatinib, nilotinib, and dasatinib. The successful treatment of CML with these TKIs presents the single strongest clinical example of the power of targeted therapy, but it may also represent a unique response in a unique disease. CML can present in chronic phase, accelerated phase, or blast crisis. Accelerated phase and blast crisis have significantly poorer prognosis, are very rare in childhood, and often need more aggressive therapy. The majority of children in chronic phase will respond to first- (imatinib) or second-line (nilotinib or dasatinib) TKIs; however, some patients have innate resistance and others develop TKI resistance with time. New agents are needed for patients with resistant disease and for those who present in accelerated phase and/or blast crisis. The PI3K/AKT/mTOR pathway is upregulated in CML (and Ph+ ALL) because BCR-ABL activates S6 and 4E-BP1 through mTOR.[188] Rapalogs are active in preclinical models against both TKI-naïve and -resistant CML, including blasts with the very resistant T315I mutation.[189–192] In addition, rapalogs are synergistic against CML blasts when combined with TKIs.[189–191] Based on the preclinical results, clinical trials investigating the activity of rapalogs are underway in both resistant CML and Ph+ ALL. Preliminary results from early-stage trials are encouraging, demonstrating measurable responses in most subjects.[191]
4.3 Lymphomas
A large number of groups have investigated the PI3K/AKT/mTOR pathway in lymphomas and lymphoproliferative disorders. Most of the focus has been dedicated toward two adult lymphoma types, mantle cell lymphoma and follicular lymphoma. As neither of these lymphomas present in childhood, except for a few case reports of follicular lymphoma, the role of PI3K/AKT/mTOR signaling and the activity of inhibitors in these diseases will not be discussed; however, it can be found in other reviews.[60,65,193] The PI3K/AKT/mTOR pathway may be dysregulated in three lymphoma types that present in childhood: Hodgkin lymphoma, diffuse large B-cell lymphoma (DLBCL), and ALK-positive anaplastic large-cell lymphoma (ALCL).
DLBCL is the most common lymphoma in adults and one of the more common types found in children. Unlike adults where prognosis is mixed, the majority of children have disease that is curable with aggressive chemotherapy. Nevertheless, in approximately 20–30% of children, disease is not curable with current treatment regimens and novel therapies are needed. A recent study identified genetic amplification of RBS6KB1, resulting in abnormal activation of S6K1 in over 90% of DLBCL tumors.[194,195] These tumors were all from adults and future studies will need to determine if over-expression of this intermediate is also present in pediatric DLBCL. Based on this work, rapalogs have been studied in preclinical models of DLBCL, finding considerable activity as a single agent and synergy with histone deacetylase inhibitors and rituximab.[194–196] These results have been validated in early-stage clinical trials in adults, finding single-agent ORR of 28–30%, using everolimus or temsirolimus.[197,198]
Hodgkin’s disease is a highly curable tumor in children; however, more aggressive or relapsed Hodgkin’s disease can be difficult to treat. Preclinical studies have demonstrated that AKT is often constitutively activated in Hodgkin’s disease cell lines; however, no studies have confirmed or refuted this finding in primary tumors.[199] Preclinical studies of inhibitors of the PI3K/AKT/mTOR signaling pathway have been mixed, as neither sirolimus nor LY294002 demonstrated in vitro activity; however, everolimus demonstrated significant activity in a Hodgkin’s disease xenograft model.[199,200] Clinical trials using rapalogs are ongoing in adults and early results are encouraging. Everolimus was found to have modest single-agent activity in one series of 19 adult patients with refractory Hodgkin’s disease.[201] The ORR was 47%, with eight PRs and one CR.
ALK-positive ALCL is a rare lymphoma that can occasionally present in childhood. The PI3K/AKT/mTOR signaling pathway is often dysregulated in ALCL, most frequently as a consequence of constitutive activation of AKT. Targeting mTOR with rapalogs was found to be effective in preclinical models.[200,202] A complete response in an adult with refractory cutaneous ALCL was recently reported.[203] This patient was treated outside the context of a clinical trial; however, early-phase clinical trials are currently ongoing.
4.4 Lymphoproliferative Disorders
In addition to lymphomas, we, and others, have studied the PI3K/AKT/mTOR signaling pathway in pediatric lymphoproliferative disorders. Two of these disorders, viral-associated lymphoproliferative disorders (viral-LPDs) and autoimmune lymphoproliferative syndrome (ALPS) warrant mention. Viral-LPDs are characterized by abnormal B-cell proliferation, because of a poor cytotoxic T-cell response to viral insult in the setting of immune compromise. The vast majority of these LPDs occur as a consequence of Epstein-Barr virus (EBV) infection after solid organ transplant or HSCT, and are appropriately named post-transplant lymphoproliferative disorders (PTLDs).[204,205] Children can also develop viral-LPDs with inherited immune deficiencies, including, but not limited to, ataxia telangiectasia, Wiscott-Aldrich syndrome, severe combined immune deficiency, and common variable immunodeficiency.[206,207] PTLDs and other viral-LPDs have the potential for malignant transformation and can be life threatening. Treatment of PTLD often includes the reduction, withdrawal, or alteration of immunosuppressant therapy. This approach may not be feasible in some cases because of potential allograft rejection after solid organ transplant or risk for development or worsening of GVHD after HSCT. In addition, antiviral agents, anti-B-cell monoclonal antibodies (rituximab), adoptive immunotherapy with EBV-specific cytotoxic T cells, and systemic chemotherapy may be used.[204,205] Constitutive activation of mTOR signaling is common in PTLD and preclinical studies have demonstrated rapalogs are active against PTLD.[208,209] Based on these data, a number of investigators have proposed transitioning immune suppression to a rapalog. This approach has been recently shown to improve PTLD in many cases, occasionally inducing a complete remission.[210–213]
ALPS is a disorder of abnormal lymphocyte survival caused by defective Fas-mediated apoptosis.[135,214,215] Children with ALPS often present with lymphoproliferation (massive lymphadenopathy and/or splenomegaly) and autoimmune disease (most commonly autoimmune cytopenias). ALPS is considered a pre-malignant condition as approximately 10% of patients develop cancer. ALPS is diagnosed by a combination of clinical findings and laboratory abnormalities, including elevation of an atypical T-cell population in peripheral blood termed double negative T-cells (DNTs; cell phenotype CD3+, CD4−, CD8−, T-cell receptor [TCR] α/β+), biomarkers (elevated plasma IL-10, IL-18, vitamin B12, or soluble-FasL, as well as polyclonal hypergammaglobulinemia), mutations in ALPS causative genes (FAS, FASL, CASPIO), and in vitro evidence of defective Fas-mediated apoptosis.[216]
Many patients with ALPS require treatment, most commonly because of autoimmune cytopenias. Some patients only require corticosteroid bursts with disease flares; however, other patients require daily treatment. ALPS patients are often treated with non-specific long term immune suppression. The most studied agent is mycophenolate mofetil,[217] which improves autoimmune disease in many patients and is often used as first-line treatment; however, some patients have partial responses, some relapse, and mycophenolate mofetil has little to no activity against lymphoproliferation.
We hypothesized that the PI3K/AKT/mTOR signaling pathway may be dysregulated and that targeting the pathway would be effective in ALPS. We studied the activity of sirolimus in a mouse model of ALPS, finding marked disease improvement, superior to other therapies including mycophenolate mofetil.[218] We subsequently opened a clinical trial that continues to enroll (NCT00392951).[66,219] Our initial experience is that sirolimus is uniquely active in these patients, and we have published early results of the trial, finding complete responses in the majority of treated patients.[66] Disease often rapidly resolved as many patients had a complete response within the first month. The majority of patients had failed other treatments, including mycophenolate mofetil, and were often previously treated with multiple agents to maintain stable disease. Patients had resolution of both autoimmune disease and lymphoproliferation, as well as elimination of the abnormal DNTs with sirolimus. These results have been confirmed in other patients by other groups.[220,221] Based on the promising results from this trial we broadened the inclusion criteria to include children with other autoimmune cytopenia syndromes and are currently broadening it to include other lymphoproliferative disorders.
5. Conclusions
Considerable data suggest the PI3K/AKT/mTOR signaling pathway is often dysregulated in a number of hematologic malignancies and lymphoproliferative disorders. Promising preclinical data and early clinical trial results demonstrate targeting this pathway may be effective in many of these diseases. A number of drugs that target individual and multiple protein kinases in the pathway are in preclinical and clinical development, and we believe that targeting the pathway at multiple nodes may present the most effective approach. Most of these agents are well tolerated. An important future step is to improve the understanding of the biology underlying the pathway dysregulation in these conditions in order to select the best agent(s) to use in a given disease. Also, as most hematologic malignancies require a multi-agent approach to treatment, developing the most effective combinations of PI3K/AKT/mTOR inhibitors and conventional cytotoxic agents is very important. As children are not little adults, it is important that preclinical studies test pathway inhibitors in models relevant to childhood malignancies and that well designed clinical trials are performed in children.
Acknowledgments
The work described in this review was supported by grants from the United States Immunodeficiency Network (USIDNET) through the National Institute of Allergy and Infectious Diseases (NIAID) grant numbers N01-A1-30070 and R56A1091791, a Foerderer-Murray Award, the Goldman Philanthropic Partnerships and the Rockefeller Brothers Fund, the Partnership for Cures Patient Impact Initiative, a Larry and Helen Hoag Foundation Clinical Translational Research Career Development Award, and the Leukemia and Lymphoma Society (David Teachey); National Institutes of Health (NIH) 1 K08 CA104882-01A1, grant number IRG-78-002-30 from the American Cancer Society, the Children’s Cancer Fund, the Kimmel Foundation, the Florence R.C. Murray Program at the Children’s Hospital of Philadelphia and W.W. Smith Charitable Trust (Valerie Brown); and NIH CA102646, CA1116660, ACS RSG0507101, and the Weinberg Fund of the Children’s Hospital of Philadelphia (Stephan Grupp). Stephan Grupp and Valerie Brown have a patent covering methods of treatment for ALL, including treatment with rapamycin or a derivative thereof (patent number 7026330). David Barrett has no conflicts of interest to declare that are directly relevant to the content of this review.
References
- 1.Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol. 2009 Nov 1;27(31):5175–81. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001 Apr 5;344(14):1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 3.Yap TA, Garrett MD, Walton MI, et al. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8(4):393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 4.LoPiccolo J, Blumenthal GM, Bernstein WB, et al. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updaat. 2008;11(1–2):32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sugimoto Y, Whitman M, Cantley LC, et al. Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglycerol. Proc Natl Acad Sci USA. 1984 Apr;81(7):2117–21. doi: 10.1073/pnas.81.7.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitman M, Kaplan DR, Schaffhausen B, et al. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985 May 16–22;315(6016):239–42. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- 7.Markman B, Atzori F, Perez-Garcia J, et al. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010 Apr;21(4):683–91. doi: 10.1093/annonc/mdp347. [DOI] [PubMed] [Google Scholar]
- 8.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010 Feb 20;28(6):1075–83. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006 Aug;7(8):606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 10.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 11.Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005 Dec;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005 Feb 18;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 13.Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002 Sep;4(9):648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 14.Kang S, Denley A, Vanhaesebroeck B, et al. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci USA. 2006 Jan 31;103(5):1289–94. doi: 10.1073/pnas.0510772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008 Sep;27(41):5486–96. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grupp SA, Harmony JA. Increased phosphatidylinositol metabolism is an important but not an obligatory early event in B lymphocyte activation. J Immunol. 1985 Jun;134(6):4087–94. [PubMed] [Google Scholar]
- 17.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997 Apr 1;7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 18.Yang ZZ, Tschopp O, Baudry A, et al. Physiological functions of protein kinase B/Akt. Biochem Soc Trans. 2004 Apr;32(Pt 2):350–4. doi: 10.1042/bst0320350. [DOI] [PubMed] [Google Scholar]
- 19.Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003 Jul;112(2):197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castaneda CA, Cortes-Funes H, Gomez HL, et al. The phosphatidyl inositol 3-kinase/AKT signaling pathway in breast cancer. Cancer Metastasis Rev. 2010 Dec;29(4):751–9. doi: 10.1007/s10555-010-9261-0. [DOI] [PubMed] [Google Scholar]
- 21.Hirsch E, Ciraolo E, Ghigo A, et al. Taming the PI3K team to hold inflammation and cancer at bay. Pharmacol Ther. 2008 May;118(2):192–205. doi: 10.1016/j.pharmthera.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Martelli AM, Tazzari PL, Evangelisti C, et al. Targeting the phosphatidyl-inositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: from bench to bedside. Curr Med Chem. 2007;14(19):2009–23. doi: 10.2174/092986707781368423. [DOI] [PubMed] [Google Scholar]
- 23.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006 Feb 10;124(3):471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Gera JF, Mellinghoff IK, Shi Y, et al. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004 Jan 23;279(4):2737–46. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 25.Majumder PK, Febbo PG, Bikoff R, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004 Jun;10(6):594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 26.Asnaghi L, Calastretti A, Bevilacqua A, et al. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene. 2004 Jun 21;23(34):5781–91. doi: 10.1038/sj.onc.1207698. [DOI] [PubMed] [Google Scholar]
- 27.Zeng X, Kinsella TJ. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008 Apr 1;68(7):2384–90. doi: 10.1158/0008-5472.CAN-07-6163. [DOI] [PubMed] [Google Scholar]
- 28.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006 May 25;441(7092):424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 29.Kharas MG, Janes MR, Scarfone VM, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008 Sep;118(9):3038–50. doi: 10.1172/JCI33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007 Apr;12(4):487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 31.Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005 Jan;30(1):35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003 May-Jun;2(3):222–32. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 33.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006 Aug;5(8):671–88. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 34.Ewen ME, Sluss HK, Sherr CJ, et al. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993 May 7;73(3):487–97. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- 35.Muranyi AL, Dedhar S, Hogge DE. Combined inhibition of integrin linked kinase and FMS-like tyrosine kinase 3 is cytotoxic to acute myeloid leukemia progenitor cells. Exp Hematol. 2009 Apr;37(4):450–60. doi: 10.1016/j.exphem.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Faderl S, Pal A, Bornmann W, et al. Kit inhibitor APcK110 induces apoptosis and inhibits proliferation of acute myeloid leukemia cells. Cancer Res. 2009 May 1;69(9):3910–7. doi: 10.1158/0008-5472.CAN-08-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birkenkamp KU, Geugien M, Schepers H, et al. Constitutive NF-kappaB DNA-binding activity in AML is frequently mediated by a Ras/PI3-K/PKB-dependent pathway. Leukemia. 2004 Jan;18(1):103–12. doi: 10.1038/sj.leu.2403145. [DOI] [PubMed] [Google Scholar]
- 38.Bader AG, Kang S, Zhao L, et al. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005 Dec;5(12):921–9. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 39.Philp AJ, Campbell IG, Leet C, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001 Oct 15;61(20):7426–9. [PubMed] [Google Scholar]
- 40.Feilotter HE, Coulon V, McVeigh JL, et al. Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. Br J Cancer. 1999 Feb;79(5–6):718–23. doi: 10.1038/sj.bjc.6690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ligresti G, Militello L, Steelman LS, et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009 May 1;8(9):1352–8. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007 Jul 26;448(7152):439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 43.Bilbao C, Rodriguez G, Ramirez R, et al. The relationship between micro-satellite instability and PTEN gene mutations in endometrial cancer. Int J Cancer. 2006 Aug 1;119(3):563–70. doi: 10.1002/ijc.21862. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997 Mar 28;275(5308):1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 45.Armengol G, Canellas A, Alvarez Y, et al. Genetic changes including gene copy number alterations and their relation to prognosis in childhood acute myeloid leukemia. Leuk Lymphoma. 2010 Jan;51(1):114–24. doi: 10.3109/10428190903350397. [DOI] [PubMed] [Google Scholar]
- 46.Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003 Oct;13(4):507–18. doi: 10.1111/j.1750-3639.2003.tb00481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Remke M, Pfister S, Kox C, et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-beta and PI3K-AKT pathways and deletions at 6q 15–16.1 as a genomic marker for unfavorable early treatment response. Blood. 2009 Jul 30;114(5):1053–62. doi: 10.1182/blood-2008-10-186536. [DOI] [PubMed] [Google Scholar]
- 48.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. Apr;7(4):209–19. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 49.Sorrells DL, Black DR, Meschonat C, et al. Detection of eIF4E gene amplification in breast cancer by competitive PCR. Ann Surg Oncol. 1998 Apr-May;5(3):232–7. doi: 10.1007/BF02303778. [DOI] [PubMed] [Google Scholar]
- 50.Perez-Tenorio G, Karlsson E, Waltersson MA, et al. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res Treat. 2011 Oct 16;128(3):713–23. doi: 10.1007/s10549-010-1058-x. [DOI] [PubMed] [Google Scholar]
- 51.Uchimaru K, Taniguchi T, Yoshikawa M, et al. Detection of cyclin D1 (bel-1, PRAD1) overexpression by a simple competitive reverse transcription-polymerase chain reaction assay in t(11;14)(q13;q32)-bearing B-cell malignancies and/or mantle cell lymphoma. Blood. 1997 Feb 1;89(3):965–74. [PubMed] [Google Scholar]
- 52.Sato T, Nakashima A, Guo L, et al. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010 May 6;29(18):2746–52. doi: 10.1038/onc.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmelzte T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 54.Baur B, Oroszlan M, Hess O, et al. Efficacy and safety of sirolimus and everolimus in heart transplant patients: a retrospective analysis. Transplant Proc. 2011 Jun;43(5):1853–61. doi: 10.1016/j.transproceed.2011.01.174. [DOI] [PubMed] [Google Scholar]
- 55.Cutler C, Antin JH. Sirolimus for GVHD prophylaxis in allogeneic stem cell transplantation. Bone Marrow Transplant. 2004 Sep;34(6):471–6. doi: 10.1038/sj.bmt.1704604. [DOI] [PubMed] [Google Scholar]
- 56.Harper SJ, Gelson W, Harper IG, et al. Switching to sirolimus-based immune suppression after liver transplantation is safe and effective: a single-center experience. Transplantation. 2011 Jan 15;91(1):128–32. doi: 10.1097/tp.0b013e3181fe131b. [DOI] [PubMed] [Google Scholar]
- 57.Kahan B. Toxicity spectrum of inhibitors of mammalian target of rapamycin in organ transplantation: etiology, pathogenesis and treatment. Expert Opin Drug Saf. 2011 Sep;10(5):727–49. doi: 10.1517/14740338.2011.579898. [DOI] [PubMed] [Google Scholar]
- 58.McMahon G, Weir MR, Li XC, et al. The evolving role of mTOR inhibition in transplantation tolerance. J Am Soc Nephrol. 2011 Mar;22(3):408–15. doi: 10.1681/ASN.2010040351. [DOI] [PubMed] [Google Scholar]
- 59.Shitrit D, Yussim A, Kramer MR. Role of siroliumus, a novel immunosuppressive drug in heart and lung transplantation. Respir Med. 2004 Sep;98(9):892–7. doi: 10.1016/j.rmed.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Teachey DT, Grupp SA, Brown VI. Mammalian target of rapamycin inhibitors and their potential role in therapy in leukaemia and other haematological malignancies. Br J Haematol. 2009 Jun;145(5):569–80. doi: 10.1111/j.1365-2141.2009.07657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002 Sep;10(3):457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 62.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006 Apr 21;22(2):159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 63.Rosner M, Hengstschlager M. Cytoplasmic and nuclear distribution of the protein complexes mTORCl and mTORC2: rapamycin triggers dephosphorylation and derealization of the mTORC2 components rictor and sinl. Hum Mol Genet. 2008 Oct 1;17(19):2934–48. doi: 10.1093/hmg/ddn192. [DOI] [PubMed] [Google Scholar]
- 64.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010 Apr;7(4):209–19. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 65.Younes A, Samad N. Utility of mTOR inhibition in hematologic malignancies. Oncologist. 2011 May 31;16(6):730–41. doi: 10.1634/theoncologist.2010-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009 Apr;145(1):101–6. doi: 10.1111/j.1365-2141.2009.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teachey DT, Jubelirer T, Baluarte HJ, et al. Treatment with sirolimus ameliorates tacrolimus-induced autoimmune cytopenias after solid organ transplant. Pediatr Blood Cancer. 2009 Dec;53(6):1114–6. doi: 10.1002/pbc.22183. [DOI] [PubMed] [Google Scholar]
- 68.Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008 Aug 9;372(9637):449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 69.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007 May 31;356(22):2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 70.Kapoor A. Malignancy in kidney transplant recipients. Drugs. 2008;68(Suppl 1):11–9. doi: 10.2165/00003495-200868001-00003. [DOI] [PubMed] [Google Scholar]
- 71.Rosenthal J, Pawlowska A, Bolotin E, et al. Transplant-associated thrombotic microangiopathy in pediatric patients treated with sirolimus and tacrolimus. Pediatr Blood Cancer. 2011 Jul 15;57(1):142–6. doi: 10.1002/pbc.22861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dienstmann R, Rodon J, Markman B, et al. Recent developments in anticancer agents targeting PI3K, Akt and mTORC1/2. Recent Pat Anticancer Drug Discov. 2011 May 1;6(2):210–36. doi: 10.2174/157489211795328503. [DOI] [PubMed] [Google Scholar]
- 73.Ogita S, Lorusso P. Targeting phosphatidylinositol 3 kinase (PI3K)-Akt beyond rapalogs. Target Oncol. 2011 May 6;6(2):103–17. doi: 10.1007/s11523-011-0176-7. [DOI] [PubMed] [Google Scholar]
- 74.Sun SY, Rosenberg LM, Wang X, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005 Aug 15;65(16):7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 75.Roccaro AM, Sacco A, Husu EN, et al. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood. 2010 Jan 21;115(3):559–69. doi: 10.1182/blood-2009-07-235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bhatt AP, Bhende PM, Sin SH, et al. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood. 2010 Jun 3;115(22):4455–63. doi: 10.1182/blood-2009-10-251082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cirstea D, Hideshima T, Rodig S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther. 2010 Apr;9(4):963–75. doi: 10.1158/1535-7163.MCT-09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009 Feb 10;7(2):e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Janes MR, Limon JJ, So L, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010 Feb;16(2):205–13. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoang B, Frost P, Shi Y, et al. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood. 2010 Nov 25;116(22):4560–8. doi: 10.1182/blood-2010-05-285726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schenone S, Brullo C, Musumeci F, et al. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18(20):2995–3014. doi: 10.2174/092986711796391651. [DOI] [PubMed] [Google Scholar]
- 82.Sanchez CG, Ma CX, Crowder RJ, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011 Mar 1;13(2):R21. doi: 10.1186/bcr2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baselga J, De Jonge M, Rodon J, et al. A first-in-human phase 1 study of BKMI20, an oral panclass I PI3K inhibitor, in patients with advanced solid tumors. ASCO Meet Abstracts. 2010;28(15 Suppl):3003. [Google Scholar]
- 84.Chakrabarty A, Sanchez V, Kuba MG, et al. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci USA. 2012;109(8):2718–23. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grana B, Burris H, Rodon J, et al. Oral PI3K kinase inhibitor BKM120 monotherapy in patients with advanced solid tumors: an update on safety and efficacy. ASCO Meet Abstracts. 2011;29(15 Suppl):3043. [Google Scholar]
- 86.Edelman G, Bedell C, Shapiro G, et al. A phase 1 dose escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients with advanced maligancies. ASCO Meet Abstracts. 2010;28(15 Suppl):3004. [Google Scholar]
- 87.Moldovan C, Soria J, LoRusso P, et al. A phase 1 safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with erlotinib in patients with advanced solid tumors. ASCO Meet Abstracts. 2010;28(15 Suppl):3070. [Google Scholar]
- 88.Traynor A, Kurzrock R, Bailey H, et al. A phase 1 safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with paclitaxel and carboplatin in patients with advanced solid tumors. ASCO Meet Abstracts. 2010;28(15 Suppl):3078. [Google Scholar]
- 89.Vanhaesebroeck B, Welham MJ, Kotani K, et al. PI10delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997 Apr 29;94(9):4330–5. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Herman SE, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010 Sep 23;116(12):2078–88. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Furman R, Byrd J, Brown JR, et al. An isoform-selective inhibitor of phosphatidylinositol 3-Kinase P110delta, demonstrates clinical activity and pharmacodynamic effects in patients with relapsed or refractory chronic lymphocytic leukemia. ASH Ann Meet Abstr. 2010;116(21):66. [Google Scholar]
- 92.Kahl B, Byrd JC, Flinn I, et al. Clinical safety and activity in a phase 1 study of CAL-101, an isoform-selective inhibitor of PBKdelta, in patients with relapsed or refractory non-hodgkin lymphoma. ASH Ann Meet Abstr. 2010;116(21):1777. [Google Scholar]
- 93.Jimeno A, Herbst R, Falchook G, et al. Final results from a phase 1, dose-escalation study of PX–866, an irreversible, pan-isoform inhibitor of PI3 kinase. ASCO Meet Abstracts. 2010;28(15 Suppl):3005. [Google Scholar]
- 94.Ogita S, Lorusso P. Targeting phosphatidylinositol 3 kinase (PI3K)-Akt beyond rapalogs. Target Oncol. 2011;6(2):103–17. doi: 10.1007/s11523-011-0176-7. [DOI] [PubMed] [Google Scholar]
- 95.Chiarini F, Grimaldi C, Ricci F, et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010 Oct 15;70(20):8097–107. doi: 10.1158/0008-5472.CAN-10-1814. [DOI] [PubMed] [Google Scholar]
- 96.Wong J, Welschinger R, Baraz R, et al. Comparison of dual PI3K7mTOR inhibitors with mTOR inhibitors using a pre-clinical model of acute lymphoblastic leukemia. ASH Ann Meet Abstr. 2010;116(21):3258. [Google Scholar]
- 97.Schultz C, Dahlhaus M, Sekora A, et al. The dual PI3K and mTOR kinase inhibitor NVP-BEZ235 induces cell cycle arrest in acute lymphoblastic leukemia cells and potentiates the cytotoxicity of dexamethasone, cytarabine, and doxorubicin. ASH Ann Meet Abstr. 2010;116(21):4115. [Google Scholar]
- 98.Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K7mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008 Oct 1;68(19):8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 99.Peyton J, Rodon J, Burris H, et al. A dose-escalation study with the novel formulation of the oral pan-class I PI3K inhibitor BEZ235, solid dipersion system (SDS) sachet, in patients with advanced or solid tumors. ASCO Meet Abstracts. 2011;29(15 Suppl):3066. [Google Scholar]
- 100.Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood. 2007 Jan 15;109(2):711–9. doi: 10.1182/blood-2006-04-016824. [DOI] [PubMed] [Google Scholar]
- 101.van Blitterswijk WJ, Verheij M. Anticancer alkylphospholipids: mechanisms of action, cellular sensitivity and resistance, and clinical prospects. Curr Pharm Des. 2008;14(21):2061–74. doi: 10.2174/138161208785294636. [DOI] [PubMed] [Google Scholar]
- 102.Sundar S, Jha TK, Thakur CP, et al. Oral miltefosine for Indian visceral leishmaniasis. N Engl J Med. 2002 Nov 28;347(22):1739–46. doi: 10.1056/NEJMoa021556. [DOI] [PubMed] [Google Scholar]
- 103.Ghobrial IM, Roccaro A, Hong F, et al. Clinical and translational studies of a phase II trial of the novel oral Akt inhibitor perifosine in relapsed or relapsed/refractory Waldenstrom’s macroglobulinemia. Clin Cancer Res. 2010 Feb 1;16(3):1033–41. doi: 10.1158/1078-0432.CCR-09-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009 Mar;11(2):102–10. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Unger C, Berdel W, Hanauske AR, et al. First-time-in-man and pharmacokinetic study of weekly oral perifosine in patients with solid tumours. Eur J Cancer. 2010 Mar;46(5):920–5. doi: 10.1016/j.ejca.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 106.Mittelman A, Casper ES, Godwin TA, et al. Phase I study of tricyclic nucleoside phosphate. Cancer Treat Rep. 1983 Feb;67(2):159–62. [PubMed] [Google Scholar]
- 107.Feun LG, Savaraj N, Bodey GP, et al. Phase I study of tricyclic nucleoside phosphate using a five-day continuous infusion schedule. Cancer Res. 1984 Aug;44(8):3608–12. [PubMed] [Google Scholar]
- 108.Garrett CR, Coppola D, Wenham RM, et al. Phase I pharmacokinetic and pharmacodynamic study of triciribine phosphate monohydrate, a small-molecule inhibitor of AKT phosphorylation, in adult subjects with solid tumors containing activated AKT. Invest New Drugs. 2011;29(6):1381–9. doi: 10.1007/s10637-010-9479-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yap T, Yan L, Patnaik A, et al. Final results of a translational phase 1 study assessing a QOD schedule of the potent AKT inhibitor MK-2206 in-corporatin predictive, pharmacodynamic (PD), and functional imaging biomarkers. ASCO Meet Abstracts. 2011;29(15 Suppl):3001. [Google Scholar]
- 110.Burris H, Siu LL, Infante J, et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical activityi of the oral AKT inhibitor GSK2141795 (GSK795) in a phase 1 first-in-human study. ASCO Meet Abstracts. 2011;29(15 Suppl):3003. [Google Scholar]
- 111.Moerke NJ, Aktas H, Chen H, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007 Jan 26;128(2):257–67. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 112.Zhu J, Huang JW, Tseng PH, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004 Jun 15;64(12):4309–18. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 113.Gopalsamy A, Shi M, Boschelli DH, et al. Discovery of dibenzo[c,f][2,7] naphthyridines as potent and selective 3-phosphoinositide-dependent kinase-1 inhibitors. J Med Chem. 2007 Nov 15;50(23):5547–9. doi: 10.1021/jm070851i. [DOI] [PubMed] [Google Scholar]
- 114.Sato S, Fujita N, Tsuruo T. Interference with PDKl-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine) Oncogene. 2002 Mar 7;21(11):1727–38. doi: 10.1038/sj.onc.1205225. [DOI] [PubMed] [Google Scholar]
- 115.Hagner PR, Schneider A, Gartenhaus RB. Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood. 2010 Mar 18;115(11):2127–35. doi: 10.1182/blood-2009-09-220020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kentsis A, Topisirovic I, Culjkovic B, et al. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc Natl Acad Sci USA. 2004 Dec 28;101(52):18105–10. doi: 10.1073/pnas.0406927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo D, Teng Q, Ji C. NOTCH and phosphatidylinositide 3-kinase/phosphatase and tensin homolog deleted on chromosome ten/AKT/mammalian target of rapamycin (mTOR) signaling in T-cell development and T-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2011 Apr 4;52(7):1200–10. doi: 10.3109/10428194.2011.564696. [DOI] [PubMed] [Google Scholar]
- 118.Konicek BW, Stephens JR, McNulty AM, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011 Mar 1;71(5):1849–57. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 119.Leseux L, Hamdi SM, Al Saati T, et al. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006 Dec 15;108(13):4156–62. doi: 10.1182/blood-2006-05-026203. [DOI] [PubMed] [Google Scholar]
- 120.Taga M, Hirooka E, Ouchi T. Essential roles of mTOR/Akt pathway in Aurora-A cell transformation. Int J Biol Sci. 2009;5(5):444–50. doi: 10.7150/ijbs.5.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007 Apr 12;446(7137):758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 122.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008 Nov 28;322(5906):1377–80. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009 Nov;41(11):1243–6. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118(11):3080–7. doi: 10.1182/blood-2011-03-341412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guo W, Lasky JL, Chang CJ, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008 May 22;453(7194):529–33. doi: 10.1038/nature06933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009 Jul 16;114(3):647–50. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008 Nov;118(11):3762–74. doi: 10.1172/JCI34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guo D, Ye J, Dai J, et al. Notch-1 regulates Akt signaling pathway and the expression of cell cycle regulatory proteins cyclin D1, CDK2 and p21 in T-ALL cell lines. Leuk Res. 2009 May;33(5):678–85. doi: 10.1016/j.leukres.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 129.Kharas MG, Deane JA, Wong S, et al. Phosphoinositide 3-kinase signaling is essential for ABL oncogene-mediated transformation of B-lineage cells. Blood. 2004 Jun 1;103(11):4268–75. doi: 10.1182/blood-2003-07-2193. [DOI] [PubMed] [Google Scholar]
- 130.Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukomas. J Exp Med. 2011 May 9;208(5):901–8. doi: 10.1084/jem.20110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brown VI, Hulitt J, Fish J, et al. Thymic stromal-derived lymphopoietin induces proliferation of pre-B leukemia and antagonizes mTOR inhibitors, suggesting a role for interleukin-7Ralpha signaling. Cancer Res. 2007 Oct 15;67(20):9963–70. doi: 10.1158/0008-5472.CAN-06-4704. [DOI] [PubMed] [Google Scholar]
- 132.Brown VI, Fang J, Alcorn K, et al. Rapamycin is active against B-precursor leukemia in vitro and in vivo, an effect that is modulated by IL-7-mediated signaling. Proc Natl Acad Sci USA. 2003 Dec 9;100(25):15113–8. doi: 10.1073/pnas.2436348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wasserman R, Zeng XX, Hardy RR. The evolution of B precursor leukemia in the Emu-ret mouse. Blood. 1998;92(1):273–82. [PubMed] [Google Scholar]
- 134.Avellino R, Romano S, Parasole R, et al. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood. 2005 Aug 15;106(4):1400–6. doi: 10.1182/blood-2005-03-0929. [DOI] [PubMed] [Google Scholar]
- 135.Teachey DT, Obzut DA, Cooperman J, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood. 2006 Feb 1;107(3):1149–55. doi: 10.1182/blood-2005-05-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Teachey DT, Sheen C, Hall J, et al. mTOR inhibitors are synergistic with methotrexate: an effective combination to treat acute lymphoblastic leukemia. Blood. 2008 Sep 1;112(5):2020–3. doi: 10.1182/blood-2008-02-137141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hirase C, Maeda Y, Takai S, et al. Hypersensitivity of Ph-positive lymphoid cell lines to rapamycin: possible clinical application of mTOR inhibitor. Leuk Res. 2009 Mar;33(3):450–9. doi: 10.1016/j.leukres.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 138.Crazzolara R, Bradstock KF, Bendali LJ. RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy. 2009 Jul;5(5):727–8. doi: 10.4161/auto.5.5.8507. [DOI] [PubMed] [Google Scholar]
- 139.Crazzolara R, Cisterne A, Thien M, et al. Potentiating effects of RAD001 (everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood. 2009 Apr 2;113(14):3297–306. doi: 10.1182/blood-2008-02-137752. [DOI] [PubMed] [Google Scholar]
- 140.Houghton PJ, Morton CL, Kolb EA, et al. Initial testing (stage 1) of the mTOR inhibitor rapamycin by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008 Apr;50(4):799–805. doi: 10.1002/pbc.21296. [DOI] [PubMed] [Google Scholar]
- 141.Cullion K, Draheim KM, Hermance N, et al. Targeting the Notchl and mTOR pathways in a mouse T-ALL model. Blood. 2009 Jun 11;113(24):6172–81. doi: 10.1182/blood-2008-02-136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Teachey DT, Vincent T, Willman CL, et al. Targeting mTOR signaling is an effective strategy for IKAROS and JAK kinase mutated acute lymphoblastic leukemia (ALL) ASH Ann Meet Abstr. 2010;116(21):3251. [Google Scholar]
- 143.Meyer LH, Eckhoff SM, Queudeville M, et al. Early relapse in all is identified by time to leukemia in NOD/SCID mice and is characterized by a gene signature involving survival pathways. Cancer Cell. 2011 Feb 15;19(2):206–17. doi: 10.1016/j.ccr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 144.Saydam G, Celikkaya H, Cole P, et al. mTOR inhibition leads to increased sensitivity to methotrexate. [abstract no. 3303 online] Available from URL: http://www.aacrmeetingabstracts.Org/cgi/content/abstract/2005/l/777-c [Accessed 2012 May 28]
- 145.Wei G, Twomey D, Lamb J, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006 Oct;10(4):331–42. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 146.Houghton PJ, Morton CL, Gorlick R, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Mol Cancer Ther. 2010 Jan;9(1):101–12. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Batista A, Barata JT, Raderschall E, et al. Targeting of active mTOR inhibits primary leukemia T cells and synergizes with cytotoxic drugs and signaling inhibitors. Exp Hematol. 2011 Apr;39(4):457–72e3. doi: 10.1016/j.exphem.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 148.Saunders P, Cisterne A, Weiss J, et al. The mammalian target of rapamycin inhibitor RAD001 (everolimus) synergizes with chemotherapeutic agents, ionizing radiation and proteasome inhibitors in pre-B acute lymphocytic leukemia. Haematologica. 2011 Jan;96(1):69–77. doi: 10.3324/haematol.2010.026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yee KW, Zeng Z, Konopleva M, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2006 Sep 1;12(17):5165–73. doi: 10.1158/1078-0432.CCR-06-0764. [DOI] [PubMed] [Google Scholar]
- 150.Rizzieri DA, Feldman E, Dipersio JF, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2008 May 1;14(9):2756–62. doi: 10.1158/1078-0432.CCR-07-1372. [DOI] [PubMed] [Google Scholar]
- 151.Rheingold S, Sacks N, Chang YJ, et al. A phase I trial of sirolimus (rapamycin) in pediatric patients with relapsed/refractory leukemia. Blood (ASH Ann Meet Abstr) 2007 Nov;110:2834. [Google Scholar]
- 152.Schlis KD, Stubbs M, DeAngelo DJ, et al. A pilot of rapamycin with glucocorticoids in children adults with relapsed ALL. ASH Ann Meet Abstr. 2010;116(21):3244. [Google Scholar]
- 153.Teachey DT, Sheen C, Brown VI, et al. Targeting eIF4E with ribavirin demonstrates a potentially effective strategy against acute lymphoblastic leukemia (ALL) ASPHO Annual Meeting Abstr 2008 [online] Available from URL: http://onlinelibrary.wiley.com/store/10.1002/(ISSN)1545-5017/asset/homepages/ASPHO_2008_-final_paginated.pdf?v=l&s=fde2eal0743d8e3d9ebl8ad379856bb03b224de4 [Accessed 2012 Jul 20]
- 154.Chiarini F, Fala F, Tazzari PL, et al. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009 Apr 15;69(8):3520–8. doi: 10.1158/0008-5472.CAN-08-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brown VI, Seif AE, Reid GS, et al. Novel molecular and cellular therapeutic targets in acute lymphoblastic leukemia and lymphoproliferative disease. Immunol Res. 2008;42(1–3):84–105. doi: 10.1007/s12026-008-8038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Evangelisti C, Ricci F, Tazzari P, et al. Targeted inhibition of mTORCl and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia. 2011 May;25(5):781–91. doi: 10.1038/leu.2011.20. [DOI] [PubMed] [Google Scholar]
- 157.Carol H, Morton CL, Gorlick R, et al. Initial testing (stage 1) of the Akt inhibitor GSK690693 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2010 Dec 15;55(7):1329–37. doi: 10.1002/pbc.22710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Levy DS, Kahana JA, Kumar R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood. 2009 Feb 19;113(8):1723–9. doi: 10.1182/blood-2008-02-137737. [DOI] [PubMed] [Google Scholar]
- 159.Martelli AM, Evangelisti C, Chiarini F, et al. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010 Jun;1(2):89–103. doi: 10.18632/oncotarget.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Altman JK, Sassano A, Platanias LC. Targeting mTOR for the treatment of AML: new agents and new directions. Oncotarget. 2011 Jun;2(6):510–7. doi: 10.18632/oncotarget.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sujobert P, Bardet V, Cornillet-Lefebvre P, et al. Essential role for the p100delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005 Aug 1;106(3):1063–6. doi: 10.1182/blood-2004-08-3225. [DOI] [PubMed] [Google Scholar]
- 162.Billottet C, Grandage VL, Gale RE, et al. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006 Oct 26;25(50):6648–59. doi: 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]
- 163.Doepfner KT, Spertini O, Arcaro A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase/Akt pathway. Leukemia. 2007 Sep;21(9):1921–30. doi: 10.1038/sj.leu.2404813. [DOI] [PubMed] [Google Scholar]
- 164.Tazzari PL, Tabellini G, Bortul R, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007 May;21(5):886–96. doi: 10.1038/sj.leu.2404643. [DOI] [PubMed] [Google Scholar]
- 165.Wahner Hendrickson AE, Haluska P, Schneider PA, et al. Expression of insulin receptor isoform A and insulin-like growth factor-1 receptor in human acute myelogenous leukemia: effect of the dual-receptor inhibitor BMS-536924 in vitro. Cancer Res. 2009 Oct 1;69(19):7635–43. doi: 10.1158/0008-5472.CAN-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Imai N, Miwa H, Shikami M, et al. Growth inhibition of AML cells with specific chromosome abnormalities by monoclonal antibodies to receptors for vascular endothelial growth factor. Leuk Res. 2009 Dec;33(12):1650–7. doi: 10.1016/j.leukres.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 167.Bohm A, Aichberger KJ, Mayerhofer M, et al. Targeting of mTOR is associated with decreased growth and decreased VEGF expression in acute myeloid leukaemia cells. Eur J Clin Invest. 2009 May;39(5):395–405. doi: 10.1111/j.1365-2362.2009.02101.x. [DOI] [PubMed] [Google Scholar]
- 168.Pearn L, Fisher J, Burnett AK, et al. The role of PKC and PDK1 in monocyte lineage specification by Ras. Blood. 2007 May 15;109(10):4461–9. doi: 10.1182/blood-2006-09-047217. [DOI] [PubMed] [Google Scholar]
- 169.Recher C, Dos Santos C, Demur C, et al. mTOR, a new therapeutic target in acute myeloid leukemia. Cell Cycle. 2005 Nov;4(11):1540–9. doi: 10.4161/cc.4.11.2159. [DOI] [PubMed] [Google Scholar]
- 170.Perl AE, Carroll M. Exploiting signal transduction pathways in acute myelogenous leukemia. Curr Treat Options Oncol. 2007 Aug;8(4):265–76. doi: 10.1007/s11864-007-0043-z. [DOI] [PubMed] [Google Scholar]
- 171.Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005 Dec 15;106(13):4261–8. doi: 10.1182/blood-2004-11-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu RH, Pelicano H, Zhang H, et al. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia. 2005 Dec;19(12):2153–8. doi: 10.1038/sj.leu.2403968. [DOI] [PubMed] [Google Scholar]
- 173.Nishioka C, Ikezoe T, Yang J, et al. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia. 2008 Dec;22(12):2159–68. doi: 10.1038/leu.2008.243. [DOI] [PubMed] [Google Scholar]
- 174.Janus A, Linke A, Cebula B, et al. Rapamycin, the mTOR kinase inhibitor, sensitizes acute myeloid leukemia cells, HL-60 cells, to the cytotoxic effect of arabinozide cytarabine. Anticancer Drugs. 2009 Sep;20(8):693–701. doi: 10.1097/CAD.0b013e32832e89b4. [DOI] [PubMed] [Google Scholar]
- 175.Akers LJ, Fang W, Levy AG, et al. Targeting glycolysis in leukemia: a novel inhibitor 3-BrOP in combination with rapamycin. Leuk Res. 2011;35(6):814–20. doi: 10.1016/j.leukres.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Calabro A, Tai J, Allen SL, et al. In-vitro synergism of m-TOR inhibitors, statins, and classical chemotherapy: potential implications in acute leukemia. Anticancer Drugs. 2008 Aug;19(7):705–12. doi: 10.1097/CAD.0b013e328304ae19. [DOI] [PubMed] [Google Scholar]
- 177.Perl AE, Kasner MT, Tsai DE, et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res. 2009 Nov 1;15(21):6732–9. doi: 10.1158/1078-0432.CCR-09-0842. [DOI] [PubMed] [Google Scholar]
- 178.Wei A, Sadawarte S, Catalano J, et al. A phase lb study combining the mTOR inhibitor everolimus (RAD001) with low dose cytarabine in untreated elderly AML patients. ASH Ann Meet Abstr. 2010;116(21):3299. [Google Scholar]
- 179.Wei A, Sadawarte S, Catalano J, et al. Clinical activity of azacitidine in combination with the oral mTOR inhibitor everolimus (RAD001) in relapsed and refractory AML: interim analysis of a phase lb/II study. ASH Ann Meet Abstr. 2010;116(21):3301. [Google Scholar]
- 180.Xu Q, Simpson SE, Scialla TJ, et al. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003 Aug 1;102(3):972–80. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 181.Neri LM, Borgatti P, Tazzari PL, et al. The phosphoinositide 3-kinase/AKTl pathway involvement in drug and all-trans-retinoic acid resistance of leukemia cells. Mol Cancer Res. 2003 Jan;1(3):234–46. [PubMed] [Google Scholar]
- 182.Assouline S, Culjkovic B, Cocolakis E, et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood. 2009 Jul 9;114(2):257–60. doi: 10.1182/blood-2009-02-205153. [DOI] [PubMed] [Google Scholar]
- 183.Chapuis N, Tamburini J, Green AS, et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010 Nov 15;16(22):5424–35. doi: 10.1158/1078-0432.CCR-10-1102. [DOI] [PubMed] [Google Scholar]
- 184.Altman JK, Sassano A, Kaur S, et al. Dual mTORC2/mTORCl targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res. 2011 Jul 1;17(13):4378–88. doi: 10.1158/1078-0432.CCR-10-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Park S, Chapuis N, Bardet V, et al. PI-103, a dual inhibitor of class IA phosphatidylinositide 3-kinase and mTOR. has antileukemic activity in AML. Leukemia. 2008 Sep;22(9):1698–706. doi: 10.1038/leu.2008.144. [DOI] [PubMed] [Google Scholar]
- 186.Zeng Z, SHi Y, Tsao T, et al. Targeting mTORC1/2 by a mTOR kinase inhibitor (PP242) induces apoptosis in AML cells under conditions mimicking the bone marrow environment. ASH Ann Meet Abstr. 2010;116(21):778. [Google Scholar]
- 187.Papa V, Tazzari PL, Chiarini F, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008 Jan;22(1):147–60. doi: 10.1038/sj.leu.2404980. [DOI] [PubMed] [Google Scholar]
- 188.Ly C, Arechiga AF, Melo JV, et al. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. Cancer Res. 2003 Sep 15;63(18):5716–22. [PubMed] [Google Scholar]
- 189.Mohi MG, Boulton C, Gu TL, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci USA. 2004 Mar 2;101(9):3130–5. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mayerhofer M, Aichberger KJ, Florian S, et al. Identification of mTOR as a novel bifunctional target in chronic myeloid leukemia: dissection of growth-inhibitory and VEGF-suppressive effects of rapamycin in leukemic cells. FASEB J. 2005 Jun;19(8):960–2. doi: 10.1096/fj.04-1973fje. [DOI] [PubMed] [Google Scholar]
- 191.Sillaber C, Mayerhofer M, Bohm A, et al. Evaluation of antileukaemic effects of rapamycin in patients with imatinib-resistant chronic myeloid leukaemia. Eur J Clin Invest. 2008 Jan;38(1):43–52. doi: 10.1111/j.1365-2362.2007.01892.x. [DOI] [PubMed] [Google Scholar]
- 192.Mancini M, Corradi V, Petta S, et al. mTOR inhibitor RAD001 (everolimus) enhances the effects of imatinib in chronic myeloid leukemia by raising the nuclear expression of c-ABL protein. Leuk Res. 2010 May;34(5):641–8. doi: 10.1016/j.leukres.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 193.Witzig TE, Gupta M. Signal transduction inhibitor therapy for lymphoma. Hematology Am Soc Hematol Educ Program. 2010:265–70. doi: 10.1182/asheducation-2010.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhao XF, Gartenhaus RB. Phospho-p70S6K and cdc2/cdk1 as therapeutic targets for diffuse large B-cell lymphoma. Expert Opin Ther Targets. 2009 Sep;13(9):1085–93. doi: 10.1517/14728220903103833. [DOI] [PubMed] [Google Scholar]
- 195.Zhao MY, Auerbach A, D’Costa AM, et al. Phospho-p70S6K/p85S6K and cdc2/cdk1 are novel targets for diffuse large B-cell lymphoma combination therapy. Clin Cancer Res. 2009 Mar 1;15(5):1708–20. doi: 10.1158/1078-0432.CCR-08-1543. [DOI] [PubMed] [Google Scholar]
- 196.Gupta M, Ansell SM, Novak AT, et al. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009 Oct 1;114(14):2926–35. doi: 10.1182/blood-2009-05-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Witzig TE, Reeder CB, LaPlant BR, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011 Feb;25(2):341–7. doi: 10.1038/leu.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Smith SM, van Besien K, Karrison T, et al. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: the University of Chicago phase II consortium. J Clin Oncol. 2010 Nov 1;28(31):4740–6. doi: 10.1200/JCO.2010.29.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Dutton A, Reynolds GM, Dawson CW, et al. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin’s lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol. 2005 Mar;205(4):498–506. doi: 10.1002/path.1725. [DOI] [PubMed] [Google Scholar]
- 200.Jundt F, Raetzel N, Muller C, et al. A rapamycin derivative (everolimus) controls proliferation through down-regulation of truncated CCAAT enhancer binding protein {beta} and NF-{kappa}B activity in Hodgkin and anaplastic large cell lymphomas. Blood. 2005 Sep 1;106(5):1801–7. doi: 10.1182/blood-2004-11-4513. [DOI] [PubMed] [Google Scholar]
- 201.Johnston PB, Inwards DJ, Colgan JP, et al. A Phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol. 2010 May;85(5):320–4. doi: 10.1002/ajh.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vega F, Medeiros LJ, Leventaki V, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. 2006 Jul 1;66(13):6589–97. doi: 10.1158/0008-5472.CAN-05-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chumsri S, Zhao M, Garofalo M, et al. Inhibition of the mammalian target of rapamycin (mTOR) in a case of refractory primary cutaneous anaplastic large cell lymphoma. Leuk Lymphoma. 2008 Feb;49(2):359–61. doi: 10.1080/10428190701809214. [DOI] [PubMed] [Google Scholar]
- 204.Evens AM, Roy R, Sterrenberg D, et al. Post-transplantation lymphoproliferative disorders: diagnosis, prognosis, and current approaches to therapy. Curr Oncol Rep. 2010 Nov;12(6):383–94. doi: 10.1007/s11912-010-0132-1. [DOI] [PubMed] [Google Scholar]
- 205.Wudhikarn K, Holman CJ, Linan M, et al. Post-transplant lymphoproliferative disorders in lung transplant recipients: 20-yr experience at the University of Minnesota. Clin Transplant. 2011;25(5):705–13. doi: 10.1111/j.1399-0012.2010.01332.x. [DOI] [PubMed] [Google Scholar]
- 206.Filipovich AH, Mathur A, Kamat D, et al. Lymphoproliferative disorders and other tumors complicating immunodeficiencies. Immunodeficiency. 1994;5(2):91–112. [PubMed] [Google Scholar]
- 207.Tran H, Nourse J, Hall S, et al. Immunodeficiency-associated lymphomas. Blood Rev. 2008 Sep;22(5):261–81. doi: 10.1016/j.blre.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 208.El-Salem M, Raghunath PN, Marzec M, et al. Constitutive activation of mTOR signaling pathway in post-transplant lymphoproliferative disorders. Lab Invest. 2007 Jan;87(1):29–39. doi: 10.1038/labinvest.3700494. [DOI] [PubMed] [Google Scholar]
- 209.Majewski M, Korecka M, Joergensen J, et al. Immunosuppressive TOR kinase inhibitor everolimus (RAD) suppresses growth of cells derived from posttransplant lymphoproliferative disorder at allograft-protecting doses. Transplantation. 2003 May 27;75(10):1710–7. doi: 10.1097/01.TP.0000063934.89714.19. [DOI] [PubMed] [Google Scholar]
- 210.Andres AM, Lopez Santamaria M, Ramos E, et al. The use of sirolimus as a rescue therapy in pediatric intestinal transplant recipients. Pediatr Transplant. 2010 Nov;14(7):931–5. doi: 10.1111/j.1399-3046.2010.01363.x. [DOI] [PubMed] [Google Scholar]
- 211.Khalpey Z, Miller DV, Schmitto JD, et al. Long-term maintenance therapy for post-cardiac transplant monoclonal lymphoproliferative disorder: caveat Mammalian target of rapamycin. Transplant Proc. 2011 Jun;43(5):1893–9. doi: 10.1016/j.transproceed.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 212.Manuelli M, De Luca L, Iaria G, et al. Conversion to rapamycin immunosuppression for malignancy after kidney transplantation. Transplant Proc. 2011 May;42(4):1314–6. doi: 10.1016/j.transproceed.2010.03.051. [DOI] [PubMed] [Google Scholar]
- 213.Salassa JR, Ozgursoy OB, Weindling SM, et al. Computed tomographic angiography with three-dimensional reconstruction for transoral laser microsurgery. Otolaryngol Head Neck Surg. 2010 Mar;142(3):351–4. doi: 10.1016/j.otohns.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 214.Bleesing JJ, Straus SE, Fleisher TA. Autoimmune lymphoproliferative syndrome: a human disorder of abnormal lymphocyte survival. Pediatr Clin North Am. 2000 Dec;47(6):1291–310. doi: 10.1016/s0031-3955(05)70272-8. [DOI] [PubMed] [Google Scholar]
- 215.Lenardo MJ, Oliveira JB, Zheng L, et al. ALPS-ten lessons from an international workshop on a genetic disease of apoptosis. Immunity. 2010 Mar 26;32(3):291–5. doi: 10.1016/j.immuni.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116(14):e35–40. doi: 10.1182/blood-2010-04-280347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rao VK, Dugan F, Dale JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005 May;129(4):534–8. doi: 10.1111/j.1365-2141.2005.05496.x. [DOI] [PubMed] [Google Scholar]
- 218.Teachey DT, Obzut DA, Axsom K, et al. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS) Blood. 2006 Sep 15;108(6):1965–71. doi: 10.1182/blood-2006-01-010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Teachey DT. Autoimmune lymphoproliferative syndrome: new approaches to diagnosis and management. Clin Adv Hematol Oncol. 2011;9(3):233–5. [PubMed] [Google Scholar]
- 220.Janic MD, Brasanac CD, Jankovic JS, et al. Rapid regression of lymphadenopathy upon rapamycin treatment in a child with autoimmune lymphoproliferative syndrome. Pediatr Blood Cancer. 2009 Dec;53(6):1117–9. doi: 10.1002/pbc.22151. [DOI] [PubMed] [Google Scholar]
- 221.Tommasini A, Valencic E, Piscianz E, et al. Immunomodulatory drugs in autoimmune lymphoproliferative syndrome (ALPS) Pediatr Blood Cancer. 2012;58(2):310. doi: 10.1002/pbc.23205. reply 311. [DOI] [PubMed] [Google Scholar]