

Abstract
Mutations of spliceosome components are common in myeloid neoplasms. One of the affected genes, PRPF8, encodes the most evolutionarily conserved spliceosomal protein. We identified either recurrent somatic PRPF8 mutations or hemizygous deletions in 15/447 and 24/450 cases, respectively. 50% of PRPF8 mutant and del(17p) cases were found in AML and conveyed poor prognosis. PRPF8 defects correlated with increased myeloblasts and ring sideroblasts in cases without SF3B1 mutations. Knockdown of PRPF8 in K562 and CD34+ primary bone marrow cells increased proliferative capacity. Whole RNA deep sequencing of primary cells from patients with PRPF8 abnormalities demonstrated consistent missplicing defects. In yeast models, homologous mutations introduced into Prp8 abrogated a block experimentally produced in the second step of the RNA splicing process suggesting that the mutants have defects in proof-reading functions. In sum, the exploration of clinical and functional consequences suggests that PRPF8 is a novel leukemogenic gene in myeloid neoplasms with a distinct phenotype likely manifested through aberrant splicing.
Keywords: PRPF8, mutations, MDS, AML, alternative splicing
Introduction
The application of next generation sequencing (NGS) has enabled the discovery of novel somatic mutations and their genetic significance in myeloid neoplasms including myelodysplastic syndrome (MDS).1–6 Various cell functions are affected by these mutations including epigenetic regulation of gene expression at various levels (DNMT3A, TET2, EZH2),7–11 as well as signal transduction (SETBP1, RAS pathway).12–14 Furthermore, NGS projects have revealed a novel class of leukemogenic mutations affecting spliceosomal genes. Such mutations are present in more than 50% of MDS patients and can be associated with particular phenotypes.3,4,15–17
The spliceosome is an intricate multi-component RNA-protein complex composed of small nuclear ribonucleoprotein particles (snRNPs) that catalyzes the splicing reaction—the excision of introns and joining of the remaining exon sequences to form a functional messenger RNA. Splicing proceeds through 2 consecutive steps: i) the branch site initiates a nucleophilic attack on the 5′ splice site, producing a lariat intermediate and a cleaved 5′ exon; ii) the 5′ exon then attacks the 3′ splice site, yielding spliced mRNA and lariat intron products.18,19
The PRPF8 gene encodes the largest and most evolutionarily conserved protein of the spliceosome, with 61% amino-acid homology between yeast Prp8 and the human protein.20 As a key part of the catalytic core of the spliceosome, it forms interactions with all substrates, including the 5′ splice site, branch point, and 3′ splice site in the pre-mRNA, as well as with snRNAs and the excised intron.21,22 Research indicates that it is essential for vast majority of pre-mRNA splicing and is required in all tissues.23,24 In higher eukaryotes, PRPF8 is responsible for processing of majority of intron-containing transcripts, including alternatively spliced mRNAs.18”
Research indicates that it is essential for pre-mRNA splicing and is required in all tissues. Germline mutations clustered in the C-terminus of human PRPF8 lead to type 13 autosomal dominant retinitis pigmentosa (RP13).25,26
Recurrent somatic mutations of other components of the spliceosome, such as U2AF1, SRSF2, and SF3B1, are common in myeloid neoplasms and are associated with specific phenotypes. For example, roughly 65% of refractory anemia with ringed sideroblasts (RARS) cases have mutations in SF3B1, a component of the U2 snRNP.4 Ringed sideroblasts (RS) are characterized by aberrant accumulation of non-heme iron within mitochondria, but the precise mechanism of RS formation in MDS or SF3B1 mutant cells remains unknown.
During the investigation of spliceosomal mutations in myeloid neoplasms, we have identified novel, recurrent, somatic mutations in PRPF8.16 However, clinical correlations, prevalence, and the functional relevance of PRPF8 mutations in MDS and AML have not been systematically evaluated. Here, we show that PRPF8 mutations and deletions correlate with the RS phenotype and play a significant role in the second step of splicing. In addition, we demonstrate that PRPF8 mutations lead to widespread aberrant splicing of numerous gene transcripts, most notably those known to be involved in the hematopoietic pathway and iron metabolism in mitochondria.
Materials and methods
Patients
Informed consent was obtained according to protocols approved by the institutional review board of Cleveland Clinic. Diagnoses were assigned according to WHO 2008 criteria.27
Validation of mutations by Sanger and deep sequencing
All exons of PRPF8 were amplified and underwent direct genomic sequencing by the ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA) and deep sequencing by an IlluminaMiSeq sequencer as previously described.13,28,29 All mutations were detected by bidirectional sequencing and scored as somatic if not present in paired CD3 positive cell-derived DNA.
RNA deep sequencing
For detecting exon inclusion/exclusion ratios of targeted genes, we amplified the region of interest by reverse transcription and PCR using primers spanning the adjacent exons. Sequencing libraries were generated and subjected to deep sequencing as previously described.13
Single nucleotide polymorphism array (SNP-A)
SNP-A analysis was performed using Affymetrix 250K and 6.0 Kit (Affymetrix, Santa Clara, CA) according to the standard protocols, followed by copy number analysis using CNAG (v3.0) (http://www.genome.umin.jp/) or Genotyping Console (Affymetrix).
Inhibitor and semi-solid culture assay
Cells were grown in IMDM media (Gibco, Life Technologies, Grand Island, NY, USA) supplemented with a cocktail of cytokines (100ng of TPO, SCF, IL3, IL6 and FLT3L; PeproTech, Rocky Hill, NJ, USA) and plated in 48 well plates with or without the spliceosome inhibitor meayamycin (CAS# 933474-21-1).15 For human cell colony formation (semi-solid culture) assays, a total of 105 cells (K562 or normal bone marrow) were plated in triplicate in 1mL of methylcellulose medium supplemented with G-SCF, GM-SCF, and EPO cytokines (MethoCult, StemCell Technologies, Vancouver, BC) in 35-mm culture plates at 37°C with 5% CO2.15
Lentiviral infection
Lentiviral shRNA constructs targetingPRPF8 were obtained from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA). Viral packaging cell supernatants containing either the control virus vector (without insert) or shRNA vectors were used to infect 5×105 K562 cells, unfractionated normal bone marrow cells, or CD34+ cells isolated from normal bone marrow using magnetic beads (Miltenyi Biotech Inc, Auburn, CA, USA). Cells were cultured for two days in the presence of puromycin (2μg/μl) to enrich for virus infected cells following which total RNA was extracted for measurement of mRNA levels.
Quantitative RT-PCR
Total RNA was extracted from bone marrow mononuclear cells and cell lines using the Nucleospin RNA II Kit (Macherey-Nagel, Bethlehem, PA). cDNA was synthesized from 100ng total RNA using random hexamer primers (Invitrogen, Carlsbad, CA, USA). Gene expression levels were measured using real-time PCR with the ABI PRISM 7500 Fast Sequence Detection System (Applied Biosystems). Primers and probes were purchased from Life Technologies (PRPF8: Hs00197615_m1 and Hs01556852_m1; GAPDH: Hs99999905_m1). The expression level of target genes was measured in duplicate and normalized to the GAPDH mRNA as previously described.30
Yeast strains and copper assays
The S. cerevisiae strain yJU75, which is deleted for chromosomal Prp8, was transformed with plasmids containing either the yeast wild type Prp8 gene or the yeast Prp8gene carrying each single point mutation corresponding to the human MDS-associated mutations. To monitor splicing efficiency, we used the ACT1-CUP1 reporter gene comprised of the yeast actin (ACT1) intron inserted upstream of the copper resistance gene, CUP1 open reading frame. Growth of the cells in the presence of copper provides a measure of splicing efficiency of the ACT1 intron. To test the function of Prp8 mutants, the ACT1 intron was modified by various 5′ splice site, branch site or 3′ splice site mutations as described33 or prepared by overlapping PCR and in vivo gap-repair cloning.34 For the copper resistance assays, cultures were grown to mid-log phase in –Leu medium, diluted to A600= 0.2, and equal volumes were dropped onto –Leu plates containing 0–2.0mM CuSO4.34 Plates were scored and photographed after 3 days at 30°C.
Caspase assay
The Caspase-glo 9 assay was used to measure caspase-9 activity (Promega, Madison, WI) according to the manufacturer’s recommended protocol. Luciferase activity generated is proportional to the amount of caspase activity present.
Splice site analyses
For analysis of 5′ and 3′ splice site sequences, 30 nucleotides spanning the exon/intron junctions were extracted from the UCSC genome browser (http://www.genome.ucsc.edu/). Splice site strength was scored using MaxEntScan.35 The control set consisted of a randomly selected set of 1000 exons from exons generated by the TCGA dataset used in the analyses. Sequence logos were generated using the WebLogo online application (http://weblogo.berkeley.edu).
Publicly available databases and analytical tools
The February 2009 human reference sequence (GRCh37) produced by the Genome Reference Consortium was used as the reference genome (UCSC genome browser; http://genome.ucsc.edu/cgi-bin/hgGateway). Expression array data was extracted from Oncomine (https://www.oncomine.org/). Somatic mutation data was obtained from the Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Cytogenetic and mutational status was obtained for AML patients (n=197) from The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/).
Results
Identification of PRPF8 mutations and deletions
Based on two index cases of patients with PRPF8 mutations previously published,13,16 we screened for lesions affecting this gene in a larger cohort (N=447) of patients with MDS and related conditions (Supplementary Table 1). We found 15 heterozygous mutations in PRPF8 gene: 13 mutations were somatic missense and 2 somatic nonsense mutations. In total, we characterized 15 samples from 13 patients (2 patients had serial samples prior to and after AML progression; Figure 1A, Supplementary Table 1). The most common mutation was in D1598, found in 4 different patient samples. Two additional mutations from TCGA primary acute myeloid leukemia (pAML) database have been added to the analysis (A687P and G1750E, Figure 1A). The positions and identity of PRPF8 mutations identified in myeloid neoplasms are given in Figure 1A along with the distribution of mutations found in other types of tumors (Figure 1A). The somatic nature of PRPF8 mutations found in our patient cohort was confirmed using CD3+ fraction DNA (Figure 1B).
Figure 1. PRPF8 mutations and deletions in myeloid malignancies.

(A) The PRPF8 gene is located on 17p13.3 (red vertical box indicated by an arrow). In this study, 24 patients with 17p deletions were analyzed (green bars). TCGA database contained 12 patients with similar deletions of the 17p region (blue bars). Also shown is the domain structure of the PRPF8 protein including several conserved domains: Reverse Transcriptase (RT fingers/palm), Thumb/X, Endonuclease and RNase H domains. Tumor-associated missense mutations form a variety of tumor types are scattered throughout the protein coding region. Myeloid malignancy missense mutations (shown in black) are located in regions that are conserved from yeast to humans as shown by the sequences of a representative sample of organisms (mutated residues shown in bold, the mutations are listed below the figure). (B) The somatic nature of the mutations was confirmed using either targeted deep sequencing (right) or Sanger sequencing (left) in tumor and CD3+ cells. (C) Expression levels of PRPF8 using RT-PCR (Rq) normalized to normal bone marrow values (NBM). Mutant PRPF8 (blue) and NBM samples (yellow) are in the diploid 17p column (left) and deletion 17p samples (n=6) are in right column (white, p<.001). (D) Disease-associated mutations (black) are mapped onto the yeast Prp8 protein structure.
PRPF8 mutations are generally missense and scattered throughout the gene (Figure 1A,D). The high level of conservation between the yeast Prp8 and human PRPF8 proteins allows for precise mapping of homologous mutations (Figure 1D). In addition, 24 cases in our cohort and 12 cases in the TCGA pAML dataset contained deletions of one copy of the PRPF8 locus (Figure 1A) and exhibited PRPF8 haploinsufficiency (Figure 1C and Supplementary Figure 1A, Supplementary Table 2, Supplementary Table 3). A further analysis of a cohort of 447 cases of MDS and related conditions showed that 20% contain mutations in various spliceosomal protein genes, including SF3B1, SRSF2, U2AF1, ZRSR2 and LUC7L2 (Supplementary Figure 1B). Interestingly, in general, the various spliceosomal factor mutations were mutually exclusive (Figure 2A, B). Other mutations coinciding with PRPF8 such as TET2, CBL and TP53 were identified (Figure 2A).
Figure 2. Morphological characteristics of PRPF8 mutants and deletions.

(A) Concomitant mutational spectrum of PRPF8 mutation and deletion cases. Included are other clinical features of the screened cohort (cytogenetics: complex (yes or no); deletion 7q; deletion 5q) and the presence of RS (red), no increase in RS (white) and unknown (grey). Deletion cases are indicated with half-blue rectangles. (B) The cases with PRPF8 mutations and deletions are not concomitant with other spliceosomal mutations except for two cases of SF3B1 and one case of SRSF2 mutations. Deletion cases are indicated with half-blue rectangles. (C) Etiology of PRPF8 mutation (left), deletion (center), and low expression cases (right, Oncomine database), all which are significantly associated with aggressive disease course (secondary AML or complex karyotype). The pAML cases with mutations or deletions are taken from the TCGA. For del(17p) deletion cases, 450 cases were analyzed by SNP-A. (D) Two exemplary mutant PRPF8 cases and two deletion cases confirm a RS phenotype with Prussian blue staining (left panels, blue staining surrounding nuclei). Quantification of RS in each analyzed case is given in Supplementary Table 3. H&E staining (right panels) shows pseudo Pelger-Huet anomaly (bi- or hypo-lobular nuclei), characteristic for 17p deletion cases.
PRPF8 defects were most frequently identified in pAML and sAML (primary and secondary acute myeloid leukemia; Figure 2C), suggesting an association with more aggressive disease phenotypes as opposed to low-risk MDS (Figure 2C; p<0.01, Supplementary Table 3). Serial sample sequencing in two exemplary cases showed that PRPF8 mutations were present at disease onset as assessed by comparison of variant allelic frequencies obtained for mutant and wild type alleles at sequential time points (Supplementary Figure 2A and 2B).
Phenotypic/genotypic associations of PRPF8 mutations
In 60% of cases (54% of mutants and 65% of deletions), an increased presence of RS was noted by Prussian blue staining (Figure 2A,D; Supplementary Figure 1C and Table 2). The RS phenotype without PRPF8 mutation or deletion was also found in 24 additional cases out of 447, 16 of which harbored SF3B1 mutations.
In most of the PRPF8 mutant and deleted samples, pseudo Pelger-Huet anomaly (PHA), characterized by bilobed or hypolobular nuclei, were noted (Figure 2D; Supplementary Table 3, 4). Since PRPF8 is located on 17p13.3 and TP53 is on 17p13.1, some of the del(17p) cases also involve the TP53 locus (Figure 1A). However, increased RS and PHA were also found in cases without TP53 deletion.
The gene expression changes associated with RS phenotype with defective PRPF8 function are shown in Supplementary Figure 3. When we analyzed the 35 samples from TCGA database for which PRPF8 mutational status was available (mutation and low expression as compared to controls with normal PRPF8 expression, p<0.01), 20% of the 200 most differentially expressed genes were associated with mitochondrial function. In addition, when we analyzed the overlap between differentially expressed genes in RS cases with and without PRPF8 mutations, several genes involved in the electron transport chain complex were found (Supplementary Figure 3).
Decreased expression of PRPF8 leads to increased cellular proliferation
To evaluate the effect of reduced PRPF8 expression on cellular proliferation, we knocked down PRPF8 mRNA levels using two shRNA lentivirus constructs (Supplementary Figure 4). Decreased expression levels of PRPF8 were associated with increased cellular proliferation (Figure 3A) and increased clonogenicity (Figure 3B). Control and knockdown cells showed indistinguishable activity in a caspase 9 assay indicating no difference in apoptosis (Figure 3C). When PRPF8 mutant and deletion cells were treated with meayamycin, a potent pre-mRNA splicing inhibitor which targets the splicing factor 3b complex,15 growth of PRPF8 defective cells were more susceptible to inhibition than normal bone marrow cells (Figure 3D). The AML cell line KG1 that is monosomic for chromosome 17 is also more sensitive to meayamycin than normal bone marrow cells (Figure 3D).
Figure 3. Cell proliferation and viability following PRPF8 knockdown.

(A) Proliferation of cells with reduced PRPF8 levels. Growth curve axis (number of viable cells in 106/ml) is shown on left y-axis and days on x-axis. In both types of cells, knockdown of PRPF8 leads to increased viable-cell counts. Error bars are calculated as one standard error. (B) Colony formation of K562 and CD34+ cells with PRPF8 knockdown showing increased colony counts. Cells, 104 K452 and 105 CD34+, were plated in semi-solid culture supplemented with cytokines and counted after 4 and 10 days, respectively. (C) Caspase 9 activity assay shows that there is no increase in apoptotic activity in PRPF8 knockdown cells. (D) Effect of treatment with the pre-mRNA splicing inhibitor meayamycin on patient cells with deletion 17p (two primary samples, Del17p AML #1 and #2) and mutant PRPF8 patient cells as well as normal bone marrow cells (NBM) and the KG1 cell line.
Functional consequences of homologous mutations
An important question arises as to whether the PRPF8 mutations confer an altered function or if they are loss of function alleles and thus merely recapitulate the haploinsufficiency of the deletion cases. Since the PRPF8 protein is exceptionally conserved between yeast and humans (Figure 1A), we created homologous mutations in yeast Prp8 to determine if the human disease-associated PRPF8 mutations cause splicing defects. Nine different yeast mutations: N760P (hA687P), V1088N (hV1015N), E1364K (hE1292K), M1379I (hM1307I), D1670Y/N (hD1598Y/N), G1822E (hG1750E), H1947R (h1875R), corresponding to the human somatic mutations were constructed and introduced into cells in which the chromosomal Prp8 gene was deleted, thus making the mutant Prp8 the only source of activity. In all cases, cell growth at all tested temperatures was indistinguishable from wild type suggesting that these mutations do not grossly affect function. To detect more subtle defects, we used a modification of the classical yeast splicing suppressor assay (Figure 4A, B). Briefly, we used a reporter plasmid that contains the ACT1–CUP1 reporter gene in which the ACT1 exon 1 and intron are inserted into the CUP1 gene. such that If splicing function is normal, the ACT1 intron is spliced out allowing the production of active CUP1 protein which confers a level of copper resistance to the cells that is proportional to the level of pre-mRNA splicing. If splicing is aberrant because of mutations in the ACT1 intron splice sites or Prp8 mutations, copper resistance is lowered. Previous studies have shown that certain splice site mutations in the ACT1 intron can be suppressed by second site mutations in splicing factors, particularly Prp8.34,49
Figure 4. Yeast Saccharomyces cerevisiae in vivo Prp8 splicing assay.
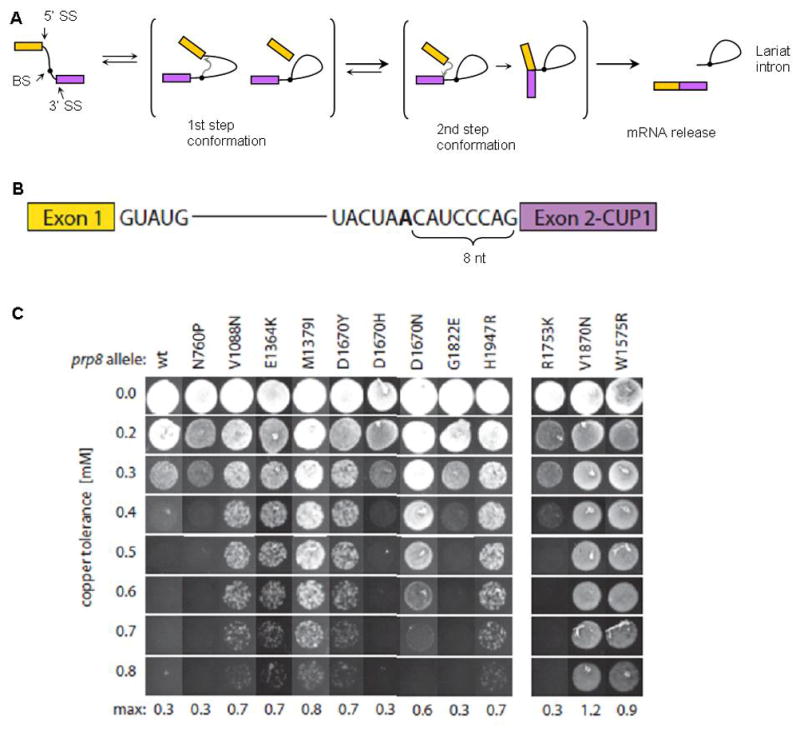
(A) Schematic representation of the two-step splicing pathway (SS, splice site; BS, branch site). Briefly, two consecutive trans-esterifications involve three sites of the intron: in the first step, the BS attacks the 5′ SS, producing a lariat intermediate and the cleaved 5′ exon. In the second step, the 5′ exon attacks the 3′ SS, resulting in spliced mRNA and the lariat intron. Prp8 is required for both chemical steps and has been shown to regulate the conformational changes in the spliceosome that accompany the process. (B) Diagram of the ACT1-CUP1 reporter gene. The CUP1 gene allows growth in copper-containing media and is interrupted with an intron (originally from ACT1 gene) such that increased splicing results in increased resistance to copper. The intron tested here has been mutated so that the BS adenosine (bold) is placed 8 nucleotides away from exon 2, instead of 43 nucleotides as found in the wild type ACT1 intron. (C) Yeast growth on different copper concentrations (Y-axis). Different columns represent wt and disease-associated mutant alleles in the left panel, and control mutant alleles in the right panel. Amino acid designations are from yeast Prp8. Control p.R1753K is a first–step suppressor mutant, while p.V1870N and p.W1575R are second–step suppressor mutants. Mutations p.V1088D/I/N (hV1015), p.E1364K (hE1292), p.M1379I (hM1307I), p.D1670Y/H/N (hD1598Y) and p.H1947R (hH1875R) show a second-step suppressor phenotype, while p.N760P (hA687P), and p.G1822E (hG1750E) show a wild type phenotype.
We therefore tested the Prp8 mutants for their ability to suppress splicing defects and thus restore copper resistance in strains containing reporter genes with several different intron mutations known to inhibit splicing in Prp8 wild-type strains. These mutations were located at the 5′ splice site (e.g. U2A and G5A of the GUAUG consensus), the branch site (e.g. A>G at the branch point), the 3′ splice site (e.g. GAG/instead of CAG/), or an intron containing a sub-optimally short (8 nt vs. 43 nt in wild type) distance between the branch site and the 3′ splice site (Figure 4B).
Previous work has shown that different intron mutations block splicing at different steps in the reaction (Figure 4A).34,49 Based on this paradigm, we tested the disease-associated Prp8 mutants with several types of intron mutations. Splicing of introns with 5′ splice site mutations (defective in the 1st step of splicing) was not rescued by disease-associated Prp8 alleles. However, we did observe improvement of splicing for intron mutants defective in the 2nd step, in particular for the short branch site-3′SS distance mutant, as indicated by the improved growth in the presence of copper (Figure 4C). In this experiment, the previously described Prp8 alleles, Prp8-p.R1753K (known to remove first step blocks), and Prp8-p.V1870N and p.W1575R (known to remove second step blocks) were used as controls. As expected, splicing of the short branch to 3′ splice site intron mutant (a prototypical 2nd step defect mutant), is not improved by the first step Prp8-p.R1753Ksuppressor allele, but only by the 2nd step Prp8-p.V1870N and p.W1575R suppressor alleles (Figure 4C). Among the disease-associated Prp8 mutations, changes at 5 different positions (p.V1088, p.E1364, p.M1379, p.D1670, and p.H1947) all improved splicing of the short branch-3′SS intron mutant in the yeast model (Figure 4C). At position p.V1088, three different substitutions (to N, D, and I) had a similar effect (not shown), suggesting that transition to the second step may be favored by any mutation at this position. By contrast, p.D1670Y and p.D1670N but not p.D1670H, exhibited the suppressor effect (Figure 4C). Only two of the tested disease-associated Prp8 mutants, p.N769P and p.G1822E, showed little or no suppression in the yeast assay. These results suggest that the PRPF8 mutations in MDS may alter the internal dynamics of the spliceosome leading to changes in splicing patterns.
PRPF8 mutations and low PRPF8 expression affect splicing patterns in human cells
Since disease-associated PRPF8 mutations showed splicing alterations when tested in yeast, we hypothesized that they might have an effect on alternative splicing in human AML samples. Using deep RNA sequencing data in cases of pAML from TGCA, alternative exon usage patterns were analyzed with SpliceTrap software (Supplementary Table 4) in PRPF8 mutants (n=2, A687P and G1750E), del(17p)/low-expression cases (n=17) and wild-type PRPF8 cases (n=14). PRPF8 defect-specific alternative splicing patterns were categorized into two groups: those genes with increased exon exclusion (lower exon usage) and genes with increased exon inclusion (higher exon usage; Figure 5A). Globally, we observed that hundreds of exons are affected in both groups and in both types of PRPF8 defects (Figure 5B and C). PRPF8 low expressers displayed modestly increased exon exclusion as compared to controls, while in mutant cases, more robust results were found in that roughly twice as many exons are affected and far more of these are altered to a greater extent than seen in PRPF8 low expressers (Figure 5B). These results indicate that PRPF8 mutations (and likely haploinsufficient expression) result in widespread changes in alternative splicing.
Figure 5. Aberrant alternative splicing in PRPF8 mutants and deletions.

(A) Schematic representation of alternative splicing (inclusion or exclusion) of the middle exon. (B) Number of genes whose splicing is affected in the RNA sequencing analysis of low expressers (left) and mutants (right). The x-axis shows the percentage difference of low expressers or mutants to control ‘wild type’ expressers of PRPF8 of either exon inclusion or exclusion, binned as 5–10%, 10–15% and >15% absolute change. The number of genes that show alternative exon exclusion is higher in all categories, in low expressers as well as in mutants. Overall, the number of genes with either inclusion or exclusion is higher in mutants. (C) The overlap of affected exons between mutants (M) and low expressers (LE) in either included (top) or excluded exons (bottom) using a threshold of >10% absolute change. (D) The splice site sequences of the 20 most affected exons in each category are compared. The average strength of each set of splice sites was determined using the MaxEntScan program, represented by the number above each splice site. The first box represents control, the middle box mutant PRPF8, and the lower box the low expressers. The graphical representation of nucleotide frequencies, calculated by Weblogo, at each splice site is shown for each case. (E) Using the data shown in (D), the deviation of the MaxEnt scores from the control values for each set of exons is plotted.
We also compared the overlaps of the sets of exons excluded or included in mutant and low expression samples. If the PRPF8 mutations resulted simply in a loss of function, the effects on splicing should be similar in mutant and deletion cases. While there was significant overlap (>50%) between mutant and low expresser cases in the spectrum of more skipped and more retained exons (Figure 5C), there were far more exons affected by PRPF8 mutations and these were less likely to be altered in low expression samples. In addition, the magnitude of exon splicing alterations was higher for the mutant cases than the low expression cases (Figure 5B). These results support the hypothesis that the PRPF8 mutant proteins have a neomorphic functional phenotype rather than simply a defective function.
The yeast Prp8 mutant phenotype suggested that mutations might affect the ability of the spliceosome to reject poor splice sites. To determine whether PRPF8 mutants and low expressers are more permissive for poor sequences surrounding the 3′ and 5′ splice sites of the alternatively spliced exons, we selected the 20 exons with the greatest changes from control samples (either exon exclusion or inclusion) and calculated the frequencies of nucleotides at each position within the consensus 5′ and 3′ splice site sequences (Figure 5D). In addition, we computed the average splice site strength as measured by MaxEnt scores35 for each splice site of each set. The results show that there are significant differences in splice site strength adjacent to affected exons (Figure 5E). The top section of Figure 5D shows that the scores for splice sites adjacent to a random control set of exons are in the narrow range of 8.0–8.5. The center section shows the splice sites adjacent to exons affected by PRPF8 mutations. In these cases, the splice sites adjacent to the more included exons are quite weak compared to the flanking splice sites. Thus, these exons would be expected to be poorly included under normal conditions. The fact that they are activated by the PRPF8 mutations suggests that splice site recognition has been altered and, specifically, that poor 3′ splice sites adjacent to the regulated exons are most affected (Figure 5E). In contrast, the 3′ splice sites adjacent to exons that are skipped more frequently in PRPF8 mutants are stronger than average (8.8 compared to 8.3) while the downstream 3′ splice sites are relatively weak. These differences again suggest that mutant PRPF8 is affecting splice site strength or efficiency. The changes seen in PRPF8 low expression samples are less dramatic and more consistent between more and less included exons supplying further evidence that the disease-associated PRPF8 mutants are functionally different from loss of function mutations.
These results are consistent with the phenotype of the homologous yeast mutants in which a suboptimal 3′ splice site was activated by the mutations (Figure 4). We conclude that the PRPF8 mutants facilitate splicing of the upstream 5′ splice sites to weak 3′ splice sites preceding the alternative exon. As a result, the subsequent splicing of the downstream pair of splice sites would be forced, leading to enhanced inclusion of alternative exons. Similarly, in cases where the alternative exon is more frequently skipped in the mutant samples, the downstream 3′ splice site, which can be thought of as competing for the first 5′ splice site with the alternative 3′ splice site, is weaker than average. Again, the mutant PRPF8 protein could be promoting splicing to the weaker 3′ splice site, in this case leading to enhanced exon exclusion.
Validation of aberrant splicing in candidate target genes
To validate the above splicing analysis in primary samples, we first tested the twenty most misspliced genes using RT-PCR gel band intensity and selected 5 genes for further analysis (GATA1, NDUFAP6, SLC25A19, SFXN2, and RPS24). We asked whether similar splicing patterns are seen in PRPF8 mutant and deletion cases and in K562 cells in which PRPF8 expression was knocked down using shRNA. Normal bone marrow cells were used as controls. Alternative exon usage was measured in two ways: 1) RT-PCR was performed on cellular RNA samples using primers located in the two exons flanking the alternative exon (Figure 6A). The percentages of exon included to exon excluded products were determined by measurement of gel band intensity. 2) Targeted deep RNA sequencing (Figure 6B and C). The results were also compared to the ratios obtained from the TCGA data set.
Figure 6. Confirmation of alternatively spliced genes.

(A) Gene specific primers were designed to flank the alternative exon. The size of each PCR product was between 100–200 bp. DeltaPsi value is calculated as the difference in the percent of intron retention of the wild type as compared to the percent of intron retention in cells with aberrant PRPF8. (B) GATA1 exon2 inclusion as determined by gel analysis of RT-PCR of RNA from K562 cells with shRNA knockdown of PRPF8, patient cells with a PRPF8 mutation, and patient cells with a 17p deletion. The results are presented as the change in the percent of intron retention compared to control K562 cells or normal bone marrow cells respectively. (C) Four genes were selected for validation of RNA sequencing results. RPS24, SLC25A19, SFXN2, and NDUFAF6, were analyzed for changes in alternative splicing using data from TCGA database, deep sequencing or gel analysis of RT-PCR products. The schematic representation of each gene is given on the right, with the two alternative isoforms given below each other. The alternative exon in each case is colored yellow. Exon inclusion values for PRPF8 knock-down, low-expresser or mutant (p.A687P and p.G1750E) samples were subtracted from the corresponding wild type values for each group (i.e. positive values represent increased inclusion and negative values represent increased exon exclusion). For the deep sequencing analysis, at least 3 primary samples were used for each category.
Overall, the results show that samples from PRPF8 knock down cells, low expression cells (n=5) and mutant patient cells (n=3) all had similar exon splicing ratios to those obtained from the TCGA data set (Figure 6C). For the SFXN2 gene, exon inclusion percentages were similar in mutant and low expressers. In the case of the NDUFAF6 gene, all samples showed similar changes in exon exclusion. A different pattern is seen in the RPS24 gene where knockdown of PRPF8 in K562 cells showed increased inclusion of the exon, while the low expressers and mutants showed preferential exon exclusion. In concordance with a recent report,36 we also found that GATA1, a transcription factor that plays a critical role in hematopoiesis, is misspliced in human PRPF8 mutants, low expressers, and knockdown cells (Figure 6B).
Discussion
Spliceosomal protein genes have been recently identified as an important class of genes affected by somatic mutations in MDS.3,4,15–17,37 Here we present novel evidence that the spliceosomal gene PRPF8 plays a direct role in MDS. PRPF8 mutations and haploinsufficiency lead to distinct oncogenic and phenotypic features including the presence of RS and the PHA phenotype. The association of PHA with del(17p) has been reported previously.38 RS are observed in both SF3B1 and PRPF8 mutations. However, SF3B1 mutations are associated with a mild disease phenotype while PRPF8defects are associated with a more aggressive phenotype. Furthermore, knockdowns of these two factors have different effects on cells. Unlike SF3B1,4,15 PRPF8 knockdown leads to increased cellular proliferation and increased clonogenicity. Nevertheless, the association of SF3B1 and PRPF8 mutations with the RS phenotype, suggests that there may be common genes or pathways affected by both SF3B1 and PRPF8.
PRPF8 is located on chromosome 17p13.3, close to the TP53 locus (17p13.1). While precise mapping of 17p deletions did show two minimally deleted regions centered on TP53 and PRPF8, both genes were affected by a deletion in multiple cases (data not shown). Frequent cases were also identified with deletion or mutation of PRPF8 concomitant with TP53 mutations. Mechanistically, loss of function mutations in zebrafishPRPF8 have been shown to result in missplicing of TP53,36 while PRPF8 knockdown in mammalian cells led to increased compensatory transcriptional activity of TP53,39 suggesting another possibility of synergetic interaction between PRPF8 defects and TP53.
While PRPF8 has not been previously implicated in the regulation of mitochondrial genes, our analysis of patients with RS and PRPF8 defects demonstrated changes in the expression profiles of several genes involved in electron transport chain complexes. Some of the most misspliced genes seen in our RNA sequencing analysis are involved in mitochondrial function, e.g., NDUFAF6, SFXN2, and SLC25A19. NDUFAF6 plays a crucial role in the assembly of complex I (NADH-ubiquinone oxidoreductase) of the mitochondrial respiratory chain.40,41 Sideroflexin-2, SFXN2, is a putative mitochondrial iron transporter.42,43 SLC25A19 is a mitochondrial thiamine pyrophosphate transporter.44,45 RPS24, another highly misspliced gene that we analyzed in primary patient samples, is mutated in Diamond-Blackfan anemia.46,47 A further connection between PRPF8 and hematopoiesis is seen in zebrafish where a PRPF8 mutation resulted in accumulation of many aberrantly spliced transcripts along with the impairment of myeloid differentiation.36 This study reported the missplicing of GATA1 in zebrafish. In agreement with this observation, we found that human GATA1 is aberrantly spliced in association with in vivo and in vitro lesions in PRPF8. All of these results support the notion that PRPF8 mutations or haploinsufficiency affects the alternative splicing of genes involved in mitochondrial metabolism and hematopoietic differentiation.
We found large scale alterations in alternative splicing patterns in samples from patients with PRPF8 mutations and deletions. Several studies in yeast have shown that Prp8 mutations can improve splicing of sub-optimal splice sites by modulating spliceosome conformation.48,49 In agreement with this notion, our study demonstrated that mutations in yeast Prp8 alter the protein’s propensity to allow splicing of certain sub-optimal 3′ splice sites normally blocked in the second step. Alternative splice sites can also be activated at a late stage of spliceosome assembly after mutant PRPF8 has joined the complex.50 In human cells, we can detect enhanced activation of sub-optimal alternative splice sites when PRPF8 is mutated or haploinsufficient. These findings suggest that the deregulated selection of splice sites might be one of the significant pathophysiologic mechanisms in PRPF8 defects.
We have argued above that 1) PRPF8 mutations and haploinsufficiency produce similar pathophysiologies in myeloid neoplasms yet 2) PRPF8 mutations produce a molecular phenotype distinct from haploinsufficiency suggesting that the mutations have a neomorphic character. To reconcile these findings, we first note that there is significant overlap between the gene sets whose alternative splicing is affected by mutations and haploinsufficiency. This suggests that one or more downstream genes involved in neoplastic progression are similarly affected by both defects. Second, the mutational phenotype may also encompass some degree of reduced PRPF8 function in that the proposed defects in proof-reading might also lead to increased lifetimes of nonfunctional spliceosomal complexes. This would sequester much of the mutant PRPF8 and partially phenocopy haploinsufficiency.
In conclusion, the core spliceosomal protein PRPF8 is frequently mutated or deleted in myeloid neoplasms and joins other spliceosomal factors as potential driver genes. We show that PRPF8 lesions lead to neomorphic spliceosomal activity and increased cellular proliferation resulting in a distinct phenotype of aggressive myeloid malignancies with increased RS. Our alternative splicing analyses demonstrate that these effects are likely mediated through missplicing of genes involved in iron accumulation in mitochondria and abnormal hematopoiesis. Our functional studies further suggest that PRPF8 mutations confer a distinct gene expression phenotype that only partially overlaps that of the haploinsufficient state.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 HL082983 (J.P.M), RO1 GM093074 (R.A.P.), and RO1 GM049044 (M.M.K.), Scott Hamilton CARES Initiative (H.M.), Aplastic Anemia & MDS International Foundation (H.M.), Fulbright Scholar Fellowship, US Department of State (A.K). We thank Naoko Hosono, Kathryn Guinta and Brittney Dienes for experimental and technical assistance.
Footnotes
Conflict of interests
Authors declare no competing financial interests.
Authorship
A.K., J.P.M., R.A.P., M.M.K. and H.M. designed research, performed research, collected data, performed statistical analysis, and wrote the manuscript; B.P. and M.C. designed research, performed statistical analysis, contributed analytical tools, interpreted data and wrote the manuscript. J.S., M.M.K., M.N., Z.K.O., E.D.H. K.Y., S.O. and J.B. collected and interpreted data.
Supplementary information is available at Leukemia’s website.
References
- 1.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2013 doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–1282. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 9.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 10.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24:1799–1804. doi: 10.1038/leu.2010.167. [DOI] [PubMed] [Google Scholar]
- 12.Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27:1401–1403. doi: 10.1038/leu.2013.35. [DOI] [PubMed] [Google Scholar]
- 13.Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45:942–946. doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez-Segui I, Makishima H, Jerez A, Yoshida K, Przychodzen B, Miyano S, et al. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia. 2013;27:1943–1946. doi: 10.1038/leu.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26:542–545. doi: 10.1038/leu.2011.232. [DOI] [PubMed] [Google Scholar]
- 16.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–3218. doi: 10.1182/blood-2011-12-400994. [DOI] [PubMed] [Google Scholar]
- 18.Hoskins AA, Moore MJ. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci. 2012;37:179–188. doi: 10.1016/j.tibs.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maciejewski JP, Padgett RA. Defects in spliceosomal machinery: a new pathway of leukaemogenesis. Br J Haematol. 2012;158:165–173. doi: 10.1111/j.1365-2141.2012.09158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grainger RJ, Beggs JD. Prp8 protein: at the heart of the spliceosome. Rna. 2005;11:533–557. doi: 10.1261/rna.2220705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–643. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konforti BB, Konarska MM. U4/U5/U6 snRNP recognizes the 5′ splice site in the absence of U2 snRNP. Genes Dev. 1994;8:1962–1973. doi: 10.1101/gad.8.16.1962. [DOI] [PubMed] [Google Scholar]
- 23.Luo HR, Moreau GA, Levin N, Moore MJ. The human Prp8 protein is a component of both U2- and U12-dependent spliceosomes. Rna. 1999;5:893–908. doi: 10.1017/s1355838299990520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schellenberg MJ, Wu T, Ritchie DB, Fica S, Staley JP, Atta KA, et al. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol. 2013;20:728–734. doi: 10.1038/nsmb.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 26.Towns KV, Kipioti A, Long V, McKibbin M, Maubaret C, Vaclavik V, et al. Prognosis for splicing factor PRPF8 retinitis pigmentosa, novel mutations and correlation between human and yeast phenotypes. Hum Mutat. 2010;31:E1361–1376. doi: 10.1002/humu.21236. [DOI] [PubMed] [Google Scholar]
- 27.Shaffer LG, Tommerup N. An International System for Human Cytogenetics Nomenclature. Basel: Karger; 2009. ISCN 2009. [Google Scholar]
- 28.Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makishima H, Jankowska AM, McDevitt MA, O’Keefe C, Dujardin S, Cazzolli H, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117:e198–206. doi: 10.1182/blood-2010-06-292433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan SN, Jankowska AM, Mahfouz R, Dunbar AJ, Sugimoto Y, Hosono N, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013;27:1301–1309. doi: 10.1038/leu.2013.80. [DOI] [PubMed] [Google Scholar]
- 31.Umen JG, Guthrie C. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev. 1995;9:855–868. doi: 10.1101/gad.9.7.855. [DOI] [PubMed] [Google Scholar]
- 32.Badhai J, Frojmark AS, Razzaghian HR, Davey E, Schuster J, Dahl N. Posttranscriptional down-regulation of small ribosomal subunit proteins correlates with reduction of 18S rRNA in RPS19 deficiency. FEBS Lett. 2009;583:2049–2053. doi: 10.1016/j.febslet.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesser CF, Guthrie C. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics. 1993;133:851–863. doi: 10.1093/genetics/133.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Query CC, Konarska MM. Suppression of multiple substrate mutations by spliceosomal Prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell. 2004;14:343–354. doi: 10.1016/s1097-2765(04)00217-5. [DOI] [PubMed] [Google Scholar]
- 35.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 36.Keightley MC, Crowhurst MO, Layton JE, Beilharz T, Markmiller S, Varma S, et al. In vivo mutation of pre-mRNA processing factor 8 (Prpf8) affects transcript splicing, cell survival and myeloid differentiation. FEBS Lett. 2013;587:2150–2157. doi: 10.1016/j.febslet.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012;26:2447–2454. doi: 10.1038/leu.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soenen V, Preudhomme C, Roumier C, Daudignon A, Lai JL, Fenaux P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood. 1998;91:1008–1015. [PubMed] [Google Scholar]
- 39.Allende-Vega N, Dayal S, Agarwala U, Sparks A, Bourdon JC, Saville MK. p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene. 2012;32:1–14. doi: 10.1038/onc.2012.38. [DOI] [PubMed] [Google Scholar]
- 40.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKenzie M, Tucker EJ, Compton AG, Lazarou M, George C, Thorburn DR, et al. Mutations in the gene encoding C8orf38 block complex I assembly by inhibiting production of the mitochondria-encoded subunit ND1. J Mol Biol. 2011;414:413–426. doi: 10.1016/j.jmb.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 42.Yoshikumi Y, Mashima H, Ueda N, Ohno H, Suzuki J, Tanaka S, et al. Roles of CTPL/Sfxn3 and Sfxn family members in pancreatic islet. J Cell Biochem. 2005;95:1157–1168. doi: 10.1002/jcb.20481. [DOI] [PubMed] [Google Scholar]
- 43.Ye X, Xu J, Cheng C, Yin G, Zeng L, Ji C, et al. Isolation and characterization of a novel human putative anemia-related gene homologous to mouse sideroflexin. Biochem Genet. 2003;41:119–125. doi: 10.1023/a:1022026001114. [DOI] [PubMed] [Google Scholar]
- 44.Lindhurst MJ, Fiermonte G, Song S, Struys E, De Leonardis F, Schwartzberg PL, et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci U S A. 2006;103:15927–15932. doi: 10.1073/pnas.0607661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spiegel R, Shaag A, Edvardson S, Mandel H, Stepensky P, Shalev SA, et al. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66:419–424. doi: 10.1002/ana.21752. [DOI] [PubMed] [Google Scholar]
- 46.Badhai J, Frojmark AS, EJD, Schuster J, Dahl N. Ribosomal protein S19 and S24 insufficiency cause distinct cell cycle defects in Diamond-Blackfan anemia. Biochim Biophys Acta. 2009;1792:1036–1042. doi: 10.1016/j.bbadis.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boria I, Quarello P, Avondo F, Garelli E, Aspesi A, Carando A, et al. A new database for ribosomal protein genes which are mutated in Diamond-Blackfan Anemia. Hum Mutat. 2008;29:E263–270. doi: 10.1002/humu.20864. [DOI] [PubMed] [Google Scholar]
- 48.Collins CA, Guthrie C. Allele-specific genetic interactions between Prp8 and RNA active site residues suggest a function for Prp8 at the catalytic core of the spliceosome. Genes Dev. 1999;13:1970–1982. doi: 10.1101/gad.13.15.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu L, Query CC, Konarska MM. Opposing classes of Prp8 alleles modulate the transition between the catalytic steps of pre-mRNA splicing. Nat Struct Mol Biol. 2007;14:519–526. doi: 10.1038/nsmb1240. [DOI] [PubMed] [Google Scholar]
- 50.House AE, Lynch KW. Regulation of alternative splicing: more than just the ABCs. J Biol Chem. 2008;283:1217–1221. doi: 10.1074/jbc.R700031200. [DOI] [PubMed] [Google Scholar]
- 51.Yang K, Zhang L, Xu T, Heroux A, Zhao R. Crystal structure of the beta-finger domain of Prp8 reveals analogy to ribosomal proteins. Proc Natl Acad Sci U S A. 2008;105:13817–13822. doi: 10.1073/pnas.0805960105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahn D, Kudla G, Tollervey D, Beggs JD. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev. 2012;26:2408–2421. doi: 10.1101/gad.199307.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.