

Abstract
A fundamental feature of immune systems is the ability to distinguish pathogenic from self and commensal elements, and to attack the former but tolerate the latter1. Prokaryotic CRISPR-Cas immune systems defend against phage infection using Cas nucleases and small RNA guides that specify one or more target sites for cleavage of the viral genome2,3. Temperate phages are viruses that can integrate into the bacterial chromosome, and they can carry genes that provide a fitness advantage to the lysogenic host4,5. However, CRISPR-Cas targeting that relies strictly on DNA sequence recognition provides indiscriminate immunity to both lytic and lysogenic infection by temperate phages6—compromising the genetic stability of these potentially beneficial elements altogether. Here we show that the Staphylococcus epidermidis CRISPR-Cas system can prevent lytic infection but tolerate lysogenization by temperate phages. Conditional tolerance is achieved through transcription-dependent DNA targeting, and ensures that targeting is resumed upon induction of the prophage lytic cycle. Our results provide evidence for the functional divergence of CRISPR-Cas systems and highlight the importance of targeting mechanism diversity. In addition, they extend the concept of ‘tolerance to non-self’ to the prokaryotic branch of adaptive immunity.
Clustered, regularly interspaced, short palindromic repeat (CRISPR) loci and their CRISPR-associated (cas) genes function together as a prokaryotic adaptive immune system which can protect bacteria and archaea from invading genetic elements such as viruses (phages)2,3. Repeat elements of the CRISPR locus are intercalated with short “spacer” sequences which typically match phage or plasmid genomes and dictate the targets for immunity based on sequence identity. Active immunity requires transcription of the CRISPR locus, followed by cleavage of the transcript within repeat sequences by Cas endoribonucleases7,8. This liberates small CRISPR RNAs (crRNAs) which specify the target for RNA-guided Cas nucleases that defend the cell from infection by degrading invading genomes2,3. Furthermore, CRISPR-Cas systems can behave in an adaptive fashion through what appears to be direct acquisition of new spacer sequences from invading elements, thereby conferring sequence-specific, heritable immunity. Based on cas gene content and organization, CRISPR-Cas systems have been classified into three main types (I–III) and at least twelve subtypes (A–F)9. A growing body of work indicates a common role for these systems in antiviral defense, but the diversity of crRNA processing and targeting mechanisms2,3 that have been described suggest the potential for differences in their precise functions in vivo.
It is well established that CRISPR-Cas systems can tolerate ‘self’ spacer elements within the CRISPR locus DNA via sequence discrimination at the flanking repeats. For type I and type II systems, this requires that short sequences which license targeting, known as protosopacer adjacent motifs (PAMs), are absent from the repeat sequences flanking each spacer10,11. For type III systems, targeting is prevented by excessive base pairing between the repeat-derived crRNA tag and its corresponding DNA sequence12. Tolerance to ‘non-self’ DNA elements, on the other hand, has yet to be described. Previous reports indicate that active CRISPR-Cas systems and their targets cannot co-exist in the same cell6,13,14. Thus, CRISPR-Cas targeting that relies strictly on DNA sequence recognition does not offer the flexibility to accommodate genetic elements with ambiguous fitness costs, such as temperate phages. Upon infection, temperate phages can kill the host cell by initiating a lytic cycle, but they may also spare the cell from lysis and establish a lysogenic cycle, typically via repression of lytic genes and integration into the host chromosome as a so-called prophage15. In addition to preventing lysis, lysogenization can result in a variety of phenotypic outcomes which can improve host fitness, for example via expression of non-viral ‘moron’ genes carried on temperate phage genomes4,5. The lysis/lysogeny decision is generally governed by a central promoter region which responds to stochastic and environmental factors to control transcription in divergent directions, thereby promoting one or the other infection cycle15. Under certain conditions, the prophage can re-initiate a lytic cycle and excise from the chromosome—a process referred to as prophage induction. Commitment to either the lytic or lysogenic cycle does not involve changes in the viral genome sequence. Hence, it was generally accepted that CRISPR-Cas targeting of temperate phages should exclude both infection outcomes; in addition to preventing lysis, CRISPR attack of an integrated prophage target precludes stable lysogenization. Although this appears to be the case for type I-E (ref. 6) and II-A (ref. 16) CRISPR-Cas systems, the potential for tolerance during type III immunity had not been explored.
In order to investigate the behavior of type III CRISPR immunity during temperate phage infection, we introduced pGG3, a plasmid carrying the type III-A CRISPR-Cas system of Staphylococcus epidermidis RP62a (ref. 17), into Staphylococcus aureus RN4220 (ref. 18). This strain is sensitive to the lambda-like temperate phages of S. aureus Newman, a clinical isolate harboring four heteroimmune prophages (ΦNM1-4) which carry genes that enhance the pathogenicity of their host19. We also identified a spacer in one of the CRISPR loci of S. aureus MSHR1132 (ref. 20) with near-perfect identity to a conserved target sequence present in ΦNM1 (Fig. 1a), ΦNM2, and ΦNM4. This spacer, referred to as 32T (Supplementary Table 1), was added to the CRISPR locus of pGG3. Using ΦNM1, we first established that this spacer prevents lytic infection by showing that plaquing efficiency is reduced approximately seven orders of magnitude when compared to a strain carrying the pGG3 plasmid without the ΦNM1-targeting spacer (Fig. 1b). We then introduced an erythromycin resistance gene (ermC) into ΦNM1 to facilitate quantification of lysogens which have stably integrated a chromosomal prophage (creating ΦNM1-ErmR). Using this system, we expected to find results consistent with a report describing CRISPR-mediated immunity to lysogenization by phage lambda in E. coli6. Surprisingly, we obtained the same efficiency of lysogenization compared to the control strain lacking spacer 32T (Fig. 1c). To test whether the presence of mismatches between the 32T crRNA and its target was influencing this phenomenon, we engineered spacer 32T* with a perfect match to its target, but obtained the same results (Fig. 1b and c). We next sought to determine whether genetic CRISPR-Cas inactivation is responsible for the apparent tolerance of these lysogens by testing them for sensitivity to ΦNM2. All 14 clones maintained resistance to ΦNM2 mediated by spacer 32T (Extended Data Fig. 1a, d). Finally, we demonstrated that spacer 32T tolerance does not result from genetic alteration of the target phage (Extended Data Fig. 1b, c). Tolerance was also observed for ΦNM4 (Extended Data Fig. 2), demonstrating that the tolerance phenomenon is not specific for the ΦNM1-ErmR phage or its integration locus. These results demonstrate that type III-A CRISPR immunity can block lytic infection but tolerate lysogenization without concomitant genetic CRISPR-Cas inactivation or alteration of the phage genome.
Fig. 1. Type III-A CRISPR immunity can block lytic infection but tolerate lysogenization.

a, Base pairing interaction between crRNA 32T and its target in the ΦNM1 genome (highlighted in gray). The crRNA tag is a sequence transcribed from the CRISPR repeat that needs to be unpaired with the flanking region of the target to license immunity. The target gene is transcribed from left to right. b, CRISPR immunity against ΦNM1 infection provided by spacers 32T and 32T* (similar to 32T but without mismatches), measured as a decrease in the number of plaque forming units (pfu) with respect to the non-targeting control pGG3 (C). c, Lysogenization with ΦNM1-ErmR in the presence of spacers 32T and 32T* or the pGG3 control (C), measured as the number of chloramphenicol- and erythromycin-resistant colony forming units (cfu) per ml obtained after infection. Control cells lysogenized with ΦNM1 (C’) lack the ermC insertion and do not yield erythromycin-resistant cfu. Error bars: mean ± s.d. (n=3).
To determine whether prophage tolerance is a spacer-specific phenomenon, we designed a variety of spacers with 100% target identity, targeting different regions of the ΦNM1 genome on both strands (Fig. 2a). We first tested the ability of each spacer to prevent lytic infection (Fig. 2b). Surprisingly, spacer functionality varied with the predicted transcriptional context of each target sequence. Spacers matching putative lytic genes to the right of the central promoter which are predicted to be unidirectionally transcribed were only effective when they targeted the predicted non-template strand (top strand according to our spacer nomenclature). Meanwhile, transcription is predicted to be bi-directional to the left of the central promoter19. Spacers targeting this region prevented plaque formation regardless of the strand targeted. This resembled the activity reported for the type III-B CRISPR-Cas system of the archaeon, Sulfolobus islandicus REY15A, where immunity to plasmid transformation depended on the presence of promoters flanking a target sequence21. We thus reasoned that transcription-dependent targeting could explain the discrepancies in spacer functionality. Indeed, ΦNM1 transcription profiles assessed by RNA-sequencing of RN4220 cultures 6, 15, 30, and 45 min post infection revealed predominantly unidirectional transcription to the right of the central promoter, while bi-directional transcription was detected to the left of the central promoter (Extended Data Fig. 3).
Fig. 2. Transcription of target sequences is required for type III-A CRISPR immunity.

a, Schematic diagram of the ΦNM1 genome and the position of targets used in this study. T, crRNA anneals to the top strand; B, bottom strand. Gray arrows represent the ΦNM1 central promoter driving divergent transcription. b, Immunity against ΦNM1 infection provided by spacers targeting the phage regions shown in a. Dotted line indicates the limit of detection for the assay. c, Immunity against ΦNM1γ6 infection. Inset; comparison of lysis phenotypes for ΦNM1 (turbid) and ΦNM1γ6 (clear), representative of four technical replicates. d, Leftward promoter consensus sequences at the ΦNM1 and ΦNM1γ6 central promoter. The ΦNM1γ6 mutation in the −10 element is shown in red. The putative transcription start site is noted (+1). e, Comparison of phage transcription profiles from cells infected with ΦNM1 (gray line) or ΦNM1γ6 (red line), 15 minutes post-infection. Phage-derived transcripts are plotted in reads per million total-mapped reads (RPM) relative to their position on the genome; arrows indicate the direction of transcription plotted in each graph; the vertical dotted line marks the position of the central promoter. Error bars: mean ± s.d. (n=3).
Further evidence for the transcription-dependence of type III-A CRISPR-Cas targeting was obtained via the characterization of a spacer 2B CRISPR-escape mutant phage, ΦNM1γ6, exhibiting a clear plaque phenotype characteristic of phages that cannot establish lysogeny (Fig. 2c, inset). Sanger sequencing of the spacer 2B target sequence did not reveal any mutations in the target or flanking sequences (data not shown), thus, we measured the ΦNM1γ6 plaquing efficiency with other spacers to determine whether it possessed a sequence-independent, general CRISPR-escape phenotype (Fig. 2c). Although most spacers provided immunity against ΦNM1γ6, we identified one additional spacer, 4B, which was escaped by the mutant phage. Both the 2B and 4B spacers target the same strand in the lysogenization operon to the left of the central promoter.
Importantly, the two complementary spacers (2T and 4T) targeting the opposite strand of spacers 2B and 4B were not escaped by ΦNM1γ6, indicating that the 2B/4B escape phenotype did not result from changes to the target DNA per se. Consistent with this, we did not observe differences in the ΦNM1 and ΦNM1γ6 plaquing efficiency when targeting the 4B region via Cas9-mediated type II-A CRISPR immunity (Extended Data Fig. 4a), which was shown to cleave dsDNA even in the absence of target transcription22,23. We thus reasoned that the ΦNM1γ6 type III-A CRISPR-escape and clear-plaque phenotypes could result from a localized, unidirectional reduction in transcription, e.g., leftward from the central promoters. Indeed, de novo sequencing of ΦNM1γ6 revealed a single nucleotide polymorphism in a crucial residue of the central promoters’ leftward −10 element (Fig. 2d), immediately upstream of the SAPPVI_g4 cI-like repressor gene required for lysogenic establishment, and ~1700 bp away from the 2B target sequence. Encouraged by this result, we directly assessed ΦNM1γ6 transcription profiles using RNA-seq, 6 and 15 min post infection (Extended Data Fig. 5a). Consistent with our hypothesis, leftward transcription (Fig. 2e, lower panel) of the lysogenization operon 15 min post infection was strongly reduced, while rightward transcription (Fig. 2e, upper panel) in this region was relatively unchanged. Taken together, these findings suggest that transcription across target sequences is a requirement for type III-A CRISPR immunity. Previously reported24 strand-independent immunity against plasmids in S. epidermidis may also follow this rule, as bi-directional transcription was detected across targets (Extended Data Fig. 6).
Given that temperate phages silence transcription of their lytic genes during lysogeny15, we hypothesized that transcription-dependent targeting would allow a variety of otherwise effective spacers to tolerate prophage target sequences. The corollary to this prediction is that targets which are constitutively transcribed during lysogeny (e.g., leftward from the central promoter, Extended Data Fig. 5b) should not be tolerated. Five additional spacers were tested for their ability to tolerate lysogenization by ΦNM1-ErmR (Fig. 3a). As expected, lysogenization was tolerated by spacers 17T, 43T and 61T-1 targeting lytic genes to the right of the central promoter, but not by spacers 2B and 17B targeting genes constitutively expressed leftward during lysogeny. Consistent with the notion that type III-A tolerance results from differences in transcription at the target, we did not observe tolerance with the transcription-independent type II-A CRISPR-Cas system, even when targeting a lytic gene (Extended Data Fig. 4b–h). In order to rule out the possibility that type III-A tolerance is influenced by processes which occur during phage infection, we corroborated these results using a “reverse” CRISPR immunity assay by electroporating CRISPR plasmids into pre-existing lysogens. In this experiment, transformation with a targeting CRISPR-Cas system results in attack of chromosomal prophage sequences and an inability to form colonies14. Again, results varied with the transcriptional context of target sequences in a manner consistent with our previous findings (Fig. 3b). Collectively, these results demonstrate that prophages are not intrinsically tolerated during CRISPR immunity in staphylococci, and suggest rather that type III-A tolerance is only achieved during lysogeny under the condition that transcription is silenced at target sequences.
Fig. 3. Conditional tolerance is achieved via transcription-dependent CRISPR-Cas targeting.

a, ΦNM1-ErmR lysogenization for additional spacers. C, pGG3 non-targeting control. b, Transformation of ΦNM1-lysogenic competent cells with CRISPR-Cas plasmids containing different spacers (transformation efficiency is measured as cfu/ng of plasmid DNA). C, pGG3 non-targeting control. c, Integration of the 43T/B ΦNM1 target region into the chromosome of S. aureus. Target sequences (inserted in both forward and reverse orientations) are under the control of the tetracycline-inducible promoter Pxyl/tet*. The 43T/B crRNAs are shown annealing to either the top or bottom strands. d, Transformation of both strains shown in c, as well as an isogenic control strain lacking the target insertion, with CRISPR-Cas plasmids containing spacers 43T or 43B. Transformants were plated on selective plates with or without anhydrotetracycline (ATc) for induction of the Pxyl/tet* promoter. e, Growth curve of strains shown in d expressing the spacer 43T CRISPR-Cas system, in the presence or absence of ATc addition at the indicated timepoint (black arrow). Error bars: mean ± s.d. (n=3).
In order to definitively demonstrate that transcription-dependent targeting offers a biological mechanism for conditional tolerance, we integrated the ΦNM1 target sequence for spacers 43T and 43B into the chromosome of S. aureus RN4220 under the control of a tightly regulated tetracycline-inducible promoter, thus emulating target lysogenization. The target was placed in both orientations with respect to the inducible promoter (Fig. 3c) and with respect to the chromosomal origin of replication (Extended Data Fig. 7a). The resulting strains were then transformed with the spacer 43T or 43B plasmids in a reverse CRISPR immunity assay, and plated in the absence or presence of the inducer. CRISPR immunity was only achieved when transcription across the target was induced with anhydrotetracycline in the presence of an antisense crRNA, regardless of the target’s orientation (Fig. 3d and Extended Data Fig. 7a). Once again, we confirmed this finding to be a type III-specific phenomenon by transforming the strains from Fig. 3c with the spacer 43B-tII type II-A CRISPR plasmid targeting the same region (Extended Data Fig. 7b). We corroborated this result by following the growth of spacer 43T transformants in liquid media (Fig. 3e). Upon addition of the inducer, growth was only inhibited for cells with the target in the forward orientation for which spacer 43T produces an antisense crRNA. Importantly, tolerance achieved in the absence of the inducer did not appear to affect growth (Fig. 3e, dotted lines). Finally, having established that type III-A CRISPR-Cas systems can block lytic infection but tolerate lysogenization, we examined the effect of tolerant spacers on prophage induction of ΦNM1 lysogens in culture. Compared to a spacerless lysogen control, the phage titer resulting from spontaneous induction of overnight cultures was significantly lower for lysogens harboring a tolerant spacer (Fig. 4a). We next followed the growth of cultures induced directly with the DNA-damaging agent, mitomycin C (Fig. 4b, solid lines). While the spacerless lysogen control cultures succumbed to prophage induction, the presence of a tolerant spacer prevented lysis.
Fig. 4. Prophage induction is limited by type III-A CRISPR immunity in tolerant lysogens.

a, Plaque-forming potential (measured in pfu/ml) of supernatants from overnight cultures of ΦNM1 lysogens carrying the tolerant spacer 32T or 43T CRISPR plasmids, or the pGG3 non-targeting control (C). b, Growth curve of ΦNM1-ErmR lysogens or a non-lysogen control harboring the pGG3 or spacer 43T CRISPR plasmids as indicated, with or without the addition of the prophage-inducing agent mitomycin C (MMC) at the indicated timepoint (black arrow). Error bars: mean ± s.d. (n=3).
We report here that type III-A immunity can offer conditional tolerance to ‘non-self’ genetic elements, in this case, temperate phages. This has several important implications for the CRISPR-Cas system and its host population. Tolerance helps ensure the genetic stability of the CRISPR-Cas system, since selective pressure to integrate prophages in the presence of intolerant spacers can drive genetic CRISPR-Cas inactivation (Extended Data Fig. 4c–e)—similar to what occurs during plasmid uptake25. In other words, tolerant spacers ensure that a population can sample potentially beneficial phenotypes that result from prophage integration without compromising their CRISPR-mediated immunity. Tolerance may also be particularly vital for type III systems, which were recently shown to provide immunity in spite of up to 15 mismatches with their spacer26. Thus, without the potential for phages to readily evade targeting via point mutation that is seen for type I and type II systems10,11, the transcriptional dependence of type III-A targeting offers temperate phages an alternative route to lysogenization that need not provide selection for mutants. Furthermore, tolerant lysogens had the added potential of resisting lysis via prophage induction. Although a few spacers targeting the lysogenization functions did not provide tolerance, it is important to note that these genes only constitute a small portion of the phage genome. Hence, spacers targeting this region should be acquired less frequently even if spacer acquisition occurs randomly without an additional mechanism for distinguishing tolerant from intolerant spacers during acquisition.
The requirement for transcription across target sequences during type III-A immunity contrasts with the transcription-independent targeting reported for type I (ref. 27) and type II (ref. 22,23) CRISPR-Cas systems. Given the temporal pattern of target transcription observed during the phage lytic cycle, it might be expected that CRISPR targeting of late genes would not provide immunity if the cell’s survival is already compromised at the onset of targeting. Indeed, we observed some differences in spacer effectiveness when infecting cells in liquid culture at very high MOI (~100): spacers targeting late genes were less protective (Extended Data Fig. 8). However, this effect was not pronounced at a MOI of 10, suggesting that the system is generally robust to delays in target transcription, in accordance with what we observed in efficiency of plaquing assays. Consistent with our findings at large, our survey of sequenced staphylococcal type III spacers showed that naturally acquired spacers with known target sequences produced crRNAs complementary to the non-template strand of predicted ORFs in 9/10 cases (Supplementary Table 2). This bias suggests negative selection of non-functional spacers targeting template strands. Alternatively, type III systems may utilize an unknown mechanism to discriminate template and non-template strands during spacer acquisition. The molecular details of the transcription-dependent targeting mechanism remain unclear. Preliminary experiments indicate that the presence of a transcript provided in trans is not sufficient to license DNA targeting (Extended Data Fig. 9). Consistent with this, induction of transcription across a plasmid-borne target results specifically in loss of the targeted plasmid (Extended Data Fig. 10). Hence, transcription in cis is probably required for DNA targeting. One possibility is that negative supercoiling generated in the wake of a passing transcription bubble could facilitate target DNA melting and improve crRNA recognition27. This hypothesis, however, would not account for the template and non-template strand asymmetry observed in our system. Another possibility is that exposure of the target non-template strand within the transcription bubble is required for annealing of a crRNA21. In this scenario, the observed asymmetry might be explained by occlusion of base-pairing to the template strand by either the nascent transcript or the RNA polymerase28. However, given the small size of the transcription bubble, this explanation may also be incomplete. Alternatively, transcription may be required to activate a targeting mechanism rather than facilitating target recognition or binding per se. In this case, effective targeting could require base pairing potential between the crRNA and the nascent transcript in cis, which would be absent for crRNAs with complementarity to the template strand. Previous reports29,30 have revealed specific RNA target cleavage activity for type III(−B/C) systems that could be relevant to the transcription-dependent DNA targeting observed in staphlyococci. Further experimentation will be needed to clarify these points. In summary, our work expands the repertoire of CRISPR-based immune functions to include a novel capacity for conditional tolerance of foreign elements, and establishes distinct genetic outcomes resulting from immunity to temperate phages via divergent CRISPR-Cas targeting mechanisms.
Methods
Bacterial strains and growth conditions
Cultivation of S. aureus RN4220 (ref. 18), TB4 (ref. 19), and derivative strains was carried out in TSB media (BD) at 37 °C, except when phage infections were performed, or when otherwise noted (see below). Whenever applicable, media were supplemented with chloramphenicol at 10 µg/ml to ensure CRISPR plasmid maintenance. RN4220 strains harboring pCL55-derived insertion vectors were grown similarly, but kanamycin was provided at 25 µg/ml except during reculture for competent cell preparation. E. coli DH5α was grown in LB media (BD) supplemented with kanamycin at 25–50 µg/ml to maintain pCL55-derived plasmids. Selection for ΦNM1-Erm lysogens with resistance to erythromycin (10 µg/ml) was only applied during the lysogenization protocol as described below, and, where applicable, during the subsequent ΦNM2 sensitivity assays.
Estimation of phage lysate titers
Serial dilutions were prepared in triplicate and plated on soft agar lawns of RN4220 in HIB-agar (BD) supplemented with 5 mM CaCl2 (technical replicates). Plates were incubated at 37 °C for 16–24 hr after drying at room temperature.
DNA preparation and cloning
Plasmid DNA was purified from 2–6 ml of E. coli DH5α or S. aureus RN4220 overnight cultures. For preparation from S. aureus cultures, cells were pelleted, resuspended in 100µl TSM buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 0.5 M sucrose) and then treated with 5 µl Lysostaphin (2 mg/ml) at 37 °C for 1.5 hr before treatment with plasmid miniprep reagents from Qiagen. Purification was carried out using Qiagen or EconoSpin columns.
Cloning was performed using RN4220 electrocompetent cells unless otherwise stated. For most type III CRISPR plasmids, scarless addition of repeat-spacer units to the pGG3 parent vector was accomplished by ‘round-the-horn PCR (ref. 31) followed by blunt ligation, using common primer oGG12 and spacer-specific oligos listed in Supplementary Table 3. The pGG3 vector was itself constructed by ‘round-the-horn PCR using primers L55 and A10 to remove extraneous repeat-spacer elements from the pWJ30β (ref. 32) CRISPR array. For construction of the remaining type III CRISPR plasmids, a modified parent vector (pGG3-BsaI) was created by introducing a placeholder spacer harboring two BsaI restriction sites, to facilitate scarless cloning of spacers by replacement with annealed oligo pairs possessing BsaI-compatible overhangs. Type III-A CRISPR arrays were amplified with primers L50/L6 and sequenced by Sanger using either forward or reverse primers. The BsaI cloning method was also used to construct type II CRISPR plasmids from the pDB184 parent vector, a modified version of pWJ40 with only the single placeholder spacer. The pC194-derived pWJ40 vector contains the full S. pyogenes M1GAS type II CRISPR-Cas system and was constructed by amplifying S. pyogenes genomic DNA with oligos L362/W278 and pC194 with oligos W270/ W282, followed by digestion of the PCR products with BglII and BssSI and a subsequent ligation. Type II CRISPR arrays were amplified with primers L448 and W176, and sequenced by Sanger using L448. After the cloning of each spacer, plasmid sizes were verified by restriction digest with BssSI for type III plasmids or BtgI for type II plasmids. pDB184 was created via Gibson assembly of two PCR fragments: a pWJ40 backbone amplified using primers B220/B334, and a CRISPR array amplified from pCas9 (ref. 14) using primers L448/B333.
For construction of pCL55-derived inducible target vectors, cloning was performed using chemically-competent DH5α cells. Briefly, the chloramphenicol resistance cassette was first replaced with a kanamycin resistance cassette amplified from strep LAM202-3 using primers L484/L485. This was accomplished by ‘round-the-horn PCR on the pCL55-iTET parent vector using primers L482/L483, followed by blunt ligation with the PCR-amplified resistance cassette to create the new pKL55-iTET-B parent vector. Directionality of the insertion was verified afterwards by restriction digest with BtgI. Modification of the Pxyl/tet promoter in accordance with pRAB12 (ref. 33) architecture was achieved via two consecutive overlap PCR steps to introduce point mutations using oligo pairs oGG108/oGG109 and oGG110/oGG111, followed by a ‘round-the-horn PCR step and blunt ligation to introduce the downstream operator sequence using oligos oGG112 and oGG113. The resulting pKL55-iTET-RC12 vector haboring the Pxyl/tet* modifications was used for downstream manipulations, as well as integration into the RN4220 chromosome to create the “Targetless” control strain. For forward and reverse target insertions, annealed oligo pairs (oGG124/oGG125 and oGG126/oGG127, respectively) with appropriate overhangs were ligated into the multiple cloning site after digesting the vector with BglII and SacII restriction enzymes (NEB). Target insertions were verified by PCR amplification and Sanger sequencing using the primers oGG64 and oGG88. Inversion of the attP motif for both forward and reverse target vectors was achieved by Gibson assembly of two PCR fragments, using oligos oGG102/oGG103 for the attP motif and oGG104/oGG105 for the backbone. Directional integration into the RN4220 chromosome was verified by amplification of either the attL or attR junctions using primer pairs oGG50/oGG96 and oGG51/oGG96, respectively. The pWJ153 inducible target vector is a pKL55-iTET-RC12- and pE194 (ref. 34)-derived plasmid constructed via multiple steps of either ‘round-the-horn PCR (ref. 31) followed by blunt ligation or Gibson assembly. The full sequence is provided as Supplementary Sequence 1.
Construction of the ΦNM1-ErmR lysogen was achieved via pKOR allelic exchange (ref. 35). ~1 kb homology arms were amplified from the chromosome of S. aureus RN4220::ΦNM1 using primer pairs oGG181/oGG182 and oGG185/oGG186, while the ~1.25 kb ermC resistance cassette was amplified from a pE194 plasmid preparation using primers oGG183 and oGG184. A ~3.25 kb fragment was assembled by SOEing PCR (ref. 36) using external primers oGG181 and oGG186 with clonase (QuikChange) attB adapters that allow directional integration into the pKOR vector35. Sequence integrity of the ~3.25 kb insertion was verified by Sanger using primers L29, oGG191, oGG192, W277, and L325.
Preparation of electrocompetent S. aureus cells
S. aureus RN4220, TB4, or derivative strains were grown overnight in TSB medium, diluted 1:100 in fresh medium without antibiotics, then allowed to grow to an OD600 reading of 0.8–1.0 for RN4220 or 0.7–0.9 for TB4. Measurements were taken using a NanoDrop 2000c Spectrophotometer (Thermo Scientific) and disposable polystyrene cuvettes. Following reculture, cells were pelleted at 4 °C, and 2–3 washes were carried out using chilled, sterile dH20 or 10 % glycerol. Cells were ultimately resuspended in 1/100th volume of chilled, sterile 10 % glyercol and 50 µl aliquots were distributed for storage at −80 °C.
Efficiency of plaquing assays
High titer lysates (~1012 pfu/ml) of either ΦNM1, ΦNM1γ6, or ΦNM2 were serially diluted in triplicate and applied to soft agar lawns of RN4220 strains harboring CRISPR plasmids, including pGG3 or pDB184 spacerless control lawns infected in parallel (technical replicates). Plates were incubated at 37 °C for 18 hr. Following incubation, plates were monitored at bench top for up to 24 hr to facilitate quantification of plaque forming units.
Quantification of erythromycin-resistant lysogens
Overnight cultures of RN4220 with respective CRISPR plasmids were inoculated in triplicate from single colonies in HIB medium supplemented with chloramphenicol (biological replicates). After chilling at 4 °C, 1:10 dilutions were prepared in 1 ml fresh HIB supplemented with chloramphenicol and 5 mM CaCl2. Diluted cultures were infected with ΦNM1-Erm at ~MOI 10 and incubated on ice for 30 min. Following incubation on ice, cultures were transferred to a 37 °C incubator for 30 min with shaking. Serial dilutions from each culture were then applied to HIB-agar plates supplemented with chloramphenicol, erythromycin, and 5 mM CaCl2 for quantification of lysogenic colony forming units. In selected cases, type III-A CRISPR locus and target sequence integrity was verified by colony PCR after re-streaking single colonies using primer pairs L6/L50 (CRISPR array) and oGG25/oGG26 (ORF 2) or oGG38/oGG39 (ORF 32). Where applicable, Sanger sequencing of PCR products was also performed using these primers. When verifying type II lysogenization isolates, the spacer 43B-tII target region was amplified using primers oGG233 and oGG234, and the type II CRISPR array was amplified using L448 and W176. The presence of integrated ΦNM1 or ΦNM1-ErmR prophages was confirmed by colony PCR using primer pairs oGG191/W277 and oGG206/W276 to amplify the attL and attR junctions, respectively. In order to estimate the total number of recipient cells, serial dilutions of untreated overnight cultures were plated on TSB- or HIB-agar supplemented with chloramphenicol.
ΦNM2-sensitivity assay
High-titer lysate of ΦNM2 (~1012 pfu/ml) was applied to the surface of a pre-dried HIB-agar plate supplemented with 5 mM CaCl2 and appropriate antibiotics, then allowed to dry for an additional ~30 min at room temperature. Single colonies isolated from Erm lysogeny experiments or CRISPR plasmid transformations were streaked through the ΦNM2-seeded region using a sterile plastic loop and then incubated for ~12 hr at 37 °C.
Enumeration of pfu liberated from lysogen cultures
Overnight cultures of either RN4220::ΦNM1-Erm or RN4220::ΦNM1 lysogens harboring targeting CRISPR plasmids or non-targeting control plasmids were inoculated in triplicate from single colonies in HIB media supplemented with chloramphenicol (biological replicates). Following overnight growth, cells were transferred to 4 °C and then pelleted by centrifugation at 5000 RPM for 5 min. Supernatants were filtered, and 100 µl from each lysate was mixed with 100 µl of either an indicator strain or targeting strain overnight culture for plating by the soft agar method. After drying at room temperature, plates were incubated 18 hr at 37 °C.
Screen for lipase-negative ΦNM4 lysogens
An overnight culture of S. aureus TB4 harboring the spacer 32T CRISPR plasmid was recultured to log phase growth in HIB medium supplemented with 5 mM CaCl2. After measurement of OD600, cells were treated with ΦNM4 at ~MOI 50. Following incubation for 1 hr, cells were plated on TSA supplemented with 5 % egg yolk emulsion. After ~24 hr incubation at 37 °C, approximately 1000 colonies were inspected for lipase secretion. Two lipase-negative candidates were re-streaked to single colonies, and the presence of an integrated ΦNM4 prophage was confirmed by colony PCR using primers oGG50 and oGG96 to amplify the attL junction.
Phage DNA isolation and deep sequencing
Samples of high titer phage lysates (~1012 pfu/ml) were treated with DNase and RNase to a final volume of 150 µl for 1 hr at 37 °C. Samples were treated with EDTA (pH 8.0) to a final concentration of 20 mM, followed by treatment with SDS to a final concentration of 0.5% and 2 µl proteinase K. Samples were incubated for 1 hr at 65 °C, and then subjected to a PCR purification protocol (Qiagen). Paired-end library preparation was performed on purified phage DNA using a Nextera Tagmentation protocol (Illumina), and samples were pooled for multiplexed sequencing on a MiSeq (Illumina). De novo assembly of phage genomes was performed using ABySS (ref. 37).
RNA preparation for RT-PCR and RNA-seq
For RT-PCR, overnight cultures were diluted 1:20 in 25 ml fresh media and grown for 2.5 hr at 37 °C with shaking. Following reculture, cells were pelleted and washed twice in 1 ml ice cold TSM buffer, and then treated with 3 µl Lysostaphin (2 mg/ml) for 20 min at 37 °C in 500 µl TSM buffer. Treated cells were pelleted and then resuspended in 750 µl cold TRIzol Reagent (Life Technologies) after discarding of the supernatant. The following chloroform extraction and precipitation was carried out according to the manufacturer’s protocol. After resuspension in dH2O, samples were treated with Qiagen DNase I for 45 min at 30 °C, and then re-purified using RNeasy cleanup columns (Qiagen). In some cases, it was necessary to repeat this step a second time in order to ensure the complete removal of DNA. Following cleanup, all samples were again treated with DNase I (Sigma-Aldrich) for 30–45 min at 30 °C, prior to use in the reverse transcription reaction.
For RNA-seq, overnight cultures were diluted 1:100 in fresh HIB supplemented with chloramphenicol and 500 µM CaCl2, and grown for 1.5 hr (approximately mid-log phase) at 37 °C with shaking. Cultures were removed, infected at MOI ~20, and then split into 10 ml portions for an additional 6, 15, 30, or 45 min of growth. Immediately following incubation, samples were mixed with 10 ml of a 1:1 acetone/ethanol solution and transferred to −80 °C. The ΦNM1 lysogen was grown similarly, except without antibiotics, and harvested immediately after the 1.5 hr reculture at 37 °C. After at least one overnight at −80 °C, samples were thawed on ice and pelleted by centrifugation at 5000 RPM for 10 min. After two washes of 1 ml TE buffer, cells were resuspended in 1 ml RLT buffer (Qiagen) supplemented with BME, and transferred to 2-ml tubes pre-loaded with ~0.5–1cc of 0.1mm glass beads (BioSpec). Samples were processed in a Mini-Beadbeater instrument (BioSpec) three times for 10 sec at 4200 oscillations/min, with 40 sec of chilling on ice between runs. After beadbeating, samples were spun down for 2 min at >13,000 RPM in a refrigerated microcentrifuge. 750 µl of supernatant was transferred to a clean tube for mixing with 500 µl of 100% ethanol, and the following RNeasy purification was carried out according to the manufacturer’s protocol (Qiagen). After elution, samples were treated with either Qiagen or Sigma-Aldrich DNase I for 30–45 min at 30 °C, and then re-purified using RNeasy cleanup columns. In some cases, it was necessary to repeat this step a second time to ensure the complete removal of DNA. rRNA-depleted samples were subsequently generated using the RiboZero™ Magnetic Kit for bacteria (Epicentre), according to the manufacturer’s protocol.
RT-PCR
Reverse transcription was performed using M-Mulv Reverse Transcriptase (NEB), with DNA-free total RNA isolated from RN4220 cultures harboring either the pNes(wt-d) or pNes(wt-i) plasmids as templates for cDNA synthesis. For pNes(wt-d), reverse transcription was performed with either the L8 or L86 primers in two separate 30 µl reactions, alongside mock reactions (−RT enzyme). For pNes(wt-i), the same was carried out using primers L8 or L87. Following incubation, 1 µl of each reaction was used as a template for PCR, with respective primer pairs for each sample.
Phage transcriptome analysis and visualization
Reads were aligned to reference genomes using Bowtie and sorted using Samtools. Using a custom script, sorted reads were accessed via Pysam, normalized as RPM values, and plotted in log scale as the average over consecutive windows of 500 base pairs using matplotlib tools for IPython.
Transformation assays
S. aureus RN4220 plasmid preps were dialyzed on 0.025 µm nitrocellulose filters (Millipore) and then quantified using a NanoDrop 2000c Spectrophotometer (Thermo Scientific). 50 µl aliquots of electrocompetent cells were transformed in triplicate with 80 ng dialyzed DNA per transformation using a GenePulser Xcell (BioRad) with the following parameters: 2900 V, 25 µF, 100 Ω, 2 mm (technical replicates). After electroporation, cells were immediately resuspended in TSB to a final volume of 200 µl and recovered at 30 °C for 2 hr with shaking. Serial dilutions were then prepared before plating with appropriate antibiotics. For reverse CRISPR immunity assays targeting insertion vectors, additional plating in the presence of ATc at a final concentration of 0.5 µg/ml was performed in parallel using the same dilutions. Plates were incubated at 37 °C for 18–24 hr.
Plate reader growth curves
For ATc induction experiments, overnight cultures were launched from single colonies in triplicate and diluted 1:200 in TSB broth (biological replicates). Following 1 hr of growth, ATc was added at a final concentration of 0.5 µg/ml where applicable. Measurements were taken every 5 minutes. For mitomycin C induction experiments, overnight cultures were launched from single colonies in duplicate and diluted 1:100 in HIB broth (biological replicates). Following 1.5h of growth, mitomycin C was added at a final concentration of 0.5 µg/ml where applicable. Measurements were taken every 10 minutes. For ΦNM1 infections, overnight cultures were launched from single colonies in triplicate and diluted 1:100 in HIB broth supplemented with CaCl2 5 mM (biological replicates). After 1 hr 25 min of growth, OD600 was measured for 3 representative cultures in order to estimate MOI. Aliquots were then loaded into 96-well plates along with ΦNM1 at the appropriate MOI (10 or 100), where applicable. Measurements were taken every 5 minutes. For ΦNM1γ6 infections, overnight cultures were launched from single colonies in triplicate and diluted 1:200 in HIB supplemented with CaCl2 5 mM (biological replicates). An average OD600 was measured after 1 hr of growth, and ΦNM1γ6 was added at a MOI of 10 based on this value, where applicable. Measurements were taken every 5 minutes.
Plasmid curing assay
RN4220 cells harboring both the pGG3 CRISPR-Cas plasmid and the pWJ153 target plasmid were cultured in TSB supplemented with chloramphenicol (10 ug/ml) to an OD600 of 0.45. After splitting the culture in two, transcription across the target was induced for one of the cultures via the addition of anhydrotetracycline (ATc) to a final concentration of 0.25 ug/ml. Aliquots of cells were harvested before (0) and after (1, 2, 3, 4, 5 and 6 hours) the time of induction. Following purification of DNA, plasmids were linearized with the common single cutter BamHI and subjected to agarose gel electrophoresis. In parallel, serial dilutions of both cultures were prepared in triplicate for each time point and plated on TSA plates supplemented with chloramphenicol and erythromycin or chloramphenicol alone, for quantification of antibiotic-resistant cfu (technical replicates).
Extended Data
Extended Data Figure 1. Characterization of spacer 32T isolates lysogenized with ΦNM1-ErmR.
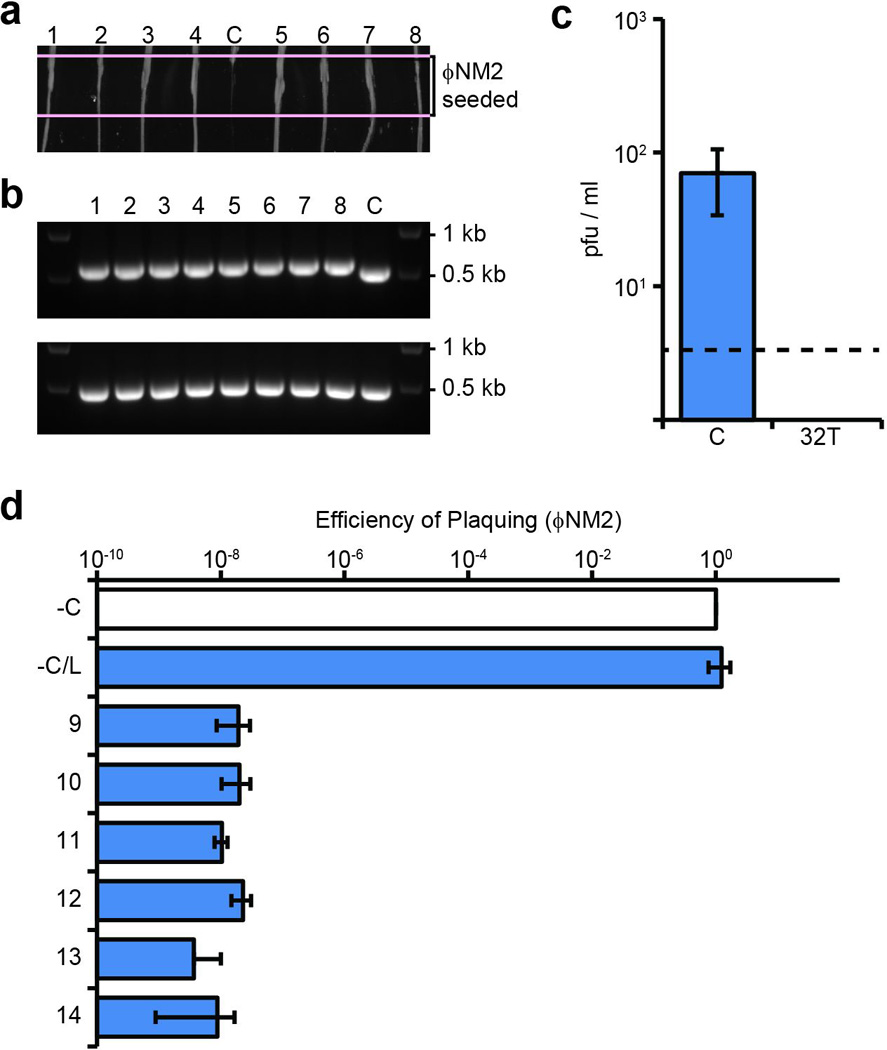
a, ΦNM2 sensitivity assay. Eight randomly selected ΦNM1-ErmR lysogen clones were re-streaked through the indicated ΦNM2-seeded region from top to bottom (1–8); C, sensitive ΦNM1-ErmR lysogen harboring the pGG3 control plasmid. b, PCR amplification of the CRISPR array (upper panel) and spacer 32T target region (lower panel) for the strains tested in a. The pGG3 control lysogen (C) lacks a phage-targeting spacer in its CRISPR array. 1 kb and 0.5 kb size markers are indicated. All 8 PCR products for the target region were sequenced by the Sanger method and no mutations were found (data not shown). c, Plaque-forming potential of filtered supernatants from spacer 32T lysogen overnight cultures inoculated in triplicate. Plaque-forming units (pfu) were enumerated on soft agar lawns of RN4220 harboring either the pGG3 control (C) or spacer 32T CRISPR plasmids. Dotted line represents the limit of detection for this assay. d, ΦNM2 plaquing efficiency on soft agar lawns of an additional six randomly selected ΦNM1-ErmR lysogen clones isolated during infection of RN4220/spacer 32T (9–14); a ΦNM1-ErmR lysogen harboring the pGG3 control plasmid was also tested (−C/L). Plaquing efficiency on the non-lysogenic indicator strain harboring pGG3 is shown for comparison (−C). Error bars: mean ± s.d. (n=3). Panels a and b represent single experiments performed for 8 of 32 isolates.
Extended Data Figure 2. Characterization of spacer 32T isolates lysogenized with ΦNM4.

a, Visualization of TB4-derived strains grown on egg-yolk agar. Integration of ΦNM4 within the geh locus of TB4 results in strongly reduced lipase secretion, enabling a screen for ΦNM4 lysogenization with spacer 32T. Right-most lanes display two lipase-negative isolates from the lysogenization screen; picture is representative of five technical replicates for each isolate. b, ΦNM2 sensitivity assay. Strains shown in panel a were re-streaked through the indicated ΦNM2-seeded region from top to bottom. The pGG3 lysogen and spacer 32T non-lysogen in the two left-most lanes serve as sensitive and insensitive controls, respectively. Picture is representative of three technical replicates for each isolate. c, ΦNM2 plaquing efficiency on soft agar lawns of the strains analyzed in panels a and b. 32T(L1) and 32T(L2) refer to the two ΦNM4 lysogens isolated during the spacer 32T egg-yolk screen. Error bars: mean ± s.d. (n=3).
Extended Data Figure 3. Visualization of ΦNM1 transcription profiles 6, 15, 30, and 45 min post infection (MOI 20).
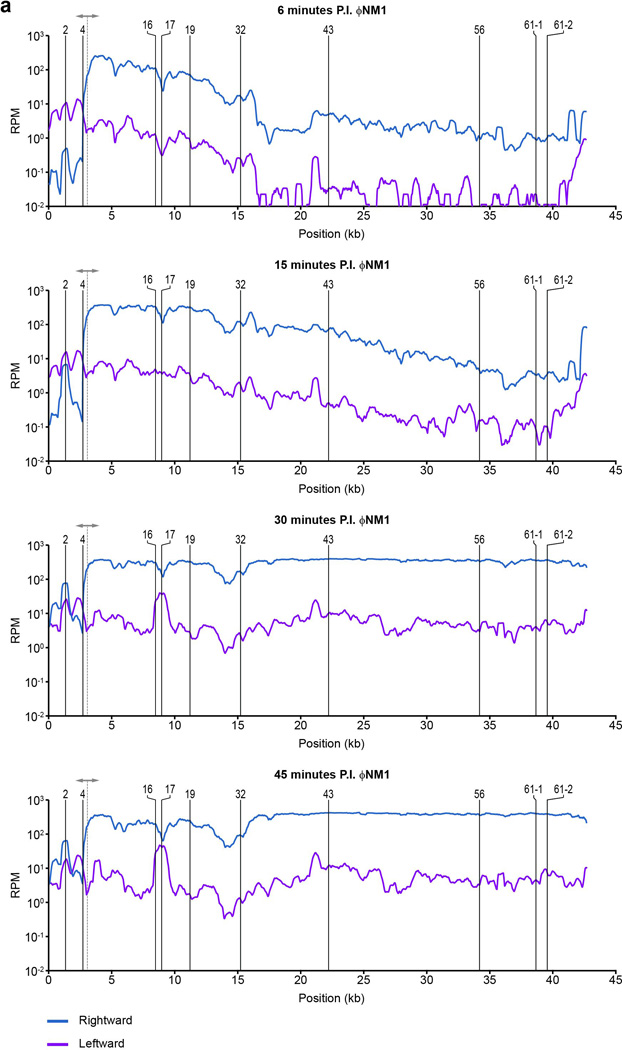
Rightward and leftward expression values are plotted as blue and fuchsia lines, respectively, in reads per million (RPM). Position of relevant spacer targets are indicated with vertical solid lines. The dotted line with arrowheads marks the position of the central promoter. To improve readability, all curves were smoothened by plotting the average RPM values over a 500 bp sliding-window. To the left of the central promoter, rightward expression is comparable to leftward expression by 30 min post infection, consistent with the strand-independent targeting observed for this region.
Extended Data Figure 4. Type II CRISPR-Cas targeting in S. aureus prevents both lytic and lysogenic infection.
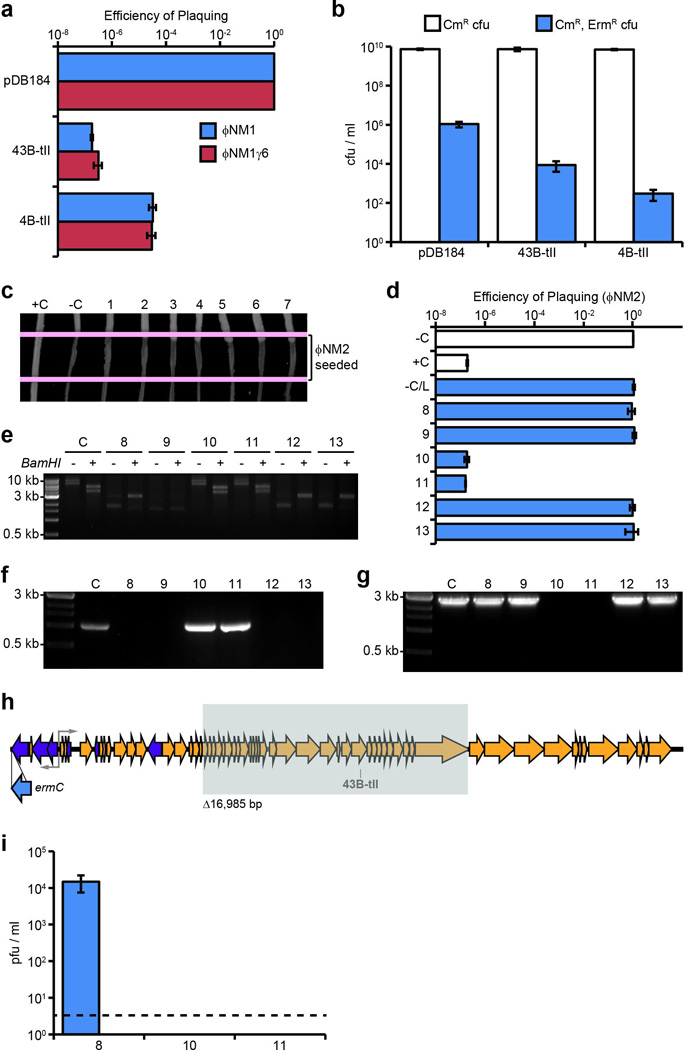
a, Plaquing efficiency of ΦNM1 and ΦNM1γ6 on lawns of RN4220 harboring type II-A CRISPR-Cas plasmids as indicated. The parental vector, pDB184, serves as a non-targeting control. b, ΦNM1-ErmR lysogenization of RN4220 harboring either the spacer 43B-tII, 4B-tII, or non-targeting type II-A CRISPR plasmids. c, ΦNM2 sensitivity assay for seven randomly selected ΦNM1-ErmR lysogen clones isolated during infection of RN4220/spacer 43B-tII (1–7). For comparison, a resistant non-lysogen harboring the spacer 43B-tII plasmid and a sensitive lysogen harboring the pDB184 plasmid were included as controls (respectively, C+ and C−). Picture represents a single experiment for 7 of 22 isolates. d, ΦNM2 plaquing efficiency on soft agar lawns for an additional six randomly selected ΦNM1-ErmR lysogen clones isolated during infection of RN4220/spacer 43B-tII (8–13); a ΦNM1-ErmR lysogen harboring the pDB184 plasmid is also tested (−C/L). For comparison, plaquing efficiency of ΦNM2 on the non-lysogenic indicator strain harboring pDB184 or the targeting spacer 43B-tII plasmid are also shown (−C and +C, respectively). e, Agarose gel electrophoresis of plasmid DNA purified from isolates 8–13 and the parental spacer 43B-tII strain (C). +/− indicate the presence or absence of treatment with the BamHI restriction enzyme which produces 2 bands for the wild type spacer 43B-tII plasmid: 5367 bp and 3972 bp. Size markers correspond to 10 kb, 3 kb, and 0.5 kb bands of the 1kb DNA ladder from NEB. f, Colony PCR spanning the type II CRISPR array for isolates 8–13. Spacer 43B-tII plasmid DNA was used as a template for the control (C). 3 kb and 0.5 kb size markers are indicated. g, Colony PCR spanning the target region for isolates 8–13 and a ΦNM1-ErmR lysogen harboring the pDB184 control plasmid (C). Isolates # 10 and 11 harbor identical deletions within the prophage that remove the target region (see below). 3 kb and 0.5 kb size markers are indicated. The presence of attL and attR prophage integration arms was also verified independently for each isolate using PCR (data not shown). h, Location of the 16,985 bp deletion identified within the prophage harbored by isolates # 10 and 11 (shaded gray box). The location and orientation of the ermC insertion cassette is also shown (blue arrow). Deletion was mapped by primer walking. An ~9.1 kb product spanning the deletion was ultimately amplified using primers oGG6 and oGG241, and the deletion junction was sequenced by the Sanger method using oGG245. A perfect 14 bp direct repeat micro-homology flanks the deletion. i, Plaque-forming potential of overnight culture supernatants from isolates # 8, 10, and 11. Supernatants were plated by the soft agar method with RN4220 cells harboring the non-targeting pDB184 control plasmid as an indicator strain. Supernatants were also plated with spacer 43B-tII targeting lawns, yielding no detectable pfu. Isolate 8 appears to exhibit wild type levels of spontaneous prophage induction (compare to pGG3 control in Fig. 4a). No plaque-forming units were detected from the supernatants of isolates # 10 and 11 whatsoever, presumably resulting from their deletion of genes essential for prophage induction, including the ORF 43 major capsid protein. Dotted line represents the limit of detection for this assay. Error bars: mean ± s.d. (n=3). Panels e through g represent single experiments for 6 of 22 isolates.
Extended Data Figure 5. Visualization of transcription profiles for ΦNM1γ6 and the ΦNM1 prophage.

Graphical presentation is the same as in Fig. S3. a, ΦNM1γ6 transcription profiles 6 and 15 min post infection (MOI 20). Comparison with ΦNM1 samples at equivalent time points (Fig. S3) reveals a marked decrease in leftward transcription to the left of the central promoter region. We calculated the fold-change in RPM between ΦNM1 and ΦNM1γ6 samples 15 min post infection. Leftward expression within the region bounded by the start of the genome and the central promoter was reduced 32-fold, while only a 4-fold reduction in leftward expression was observed overall. Meanwhile, rightward expression was reduced 4-fold both overall and in this region. This suggests an ~8-fold net reduction in leftward transcription originating from the central promoter. b, ΦNM1 prophage transcription profiles. Strong leftward transcription originates from the central promoter and a few upstream regions which are presumed to be important for lysogenic maintenance. Rightward transcription was weaker than leftward transcription as expected, but not absent. Given the strength of rightward transcription observed during the lytic cycle (Fig. S3), however, this transcription may originate from a subpopulation of cells undergoing prophage induction, rather than the stable lysogen majority.
Extended Data Figure 6. Detection of transcription across target insertions for the pNes(wt-d) and pNes(wt-i) plasmids.
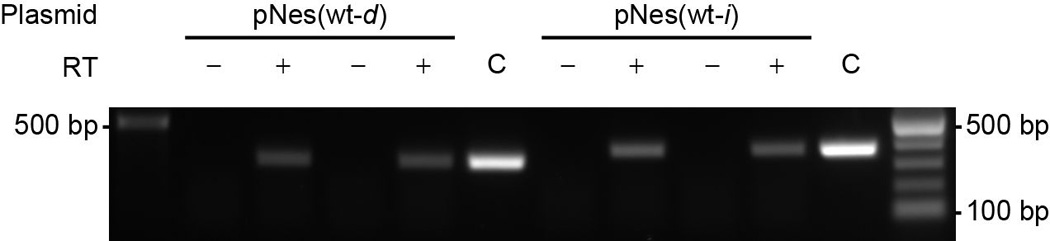
For each target plasmid (ref. 24), reverse transcription was performed in both directions with DNase-treated total RNA from RN4220 cells harboring the indicated plasmids, using either forward or reverse primers for cDNA synthesis in two separate reactions. PCR was performed on cDNA products, or plasmid DNA templates for control (C) lanes. +/− indicate the presence or absence of reverse transcriptase enzyme in the RT reaction mixture used for PCR. 500 bp and 100 bp size markers are indicated. Picture is representative of a single technical replicate.
Extended Data Figure 7. Reverse CRISPR-immunity assays using inverted chromosomal target insertions or type II CRISPR-Cas plasmids.
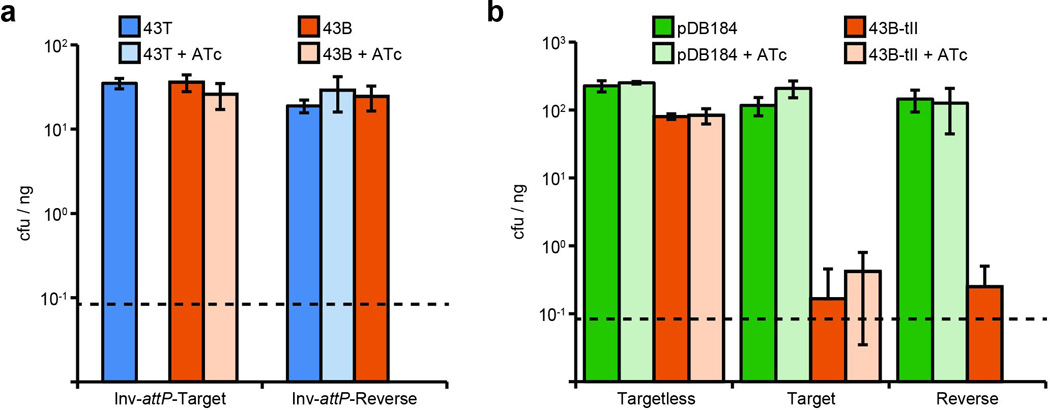
Values represent the average transformation efficiency of three transformations in colony forming units (cfu) per microgram (µg) of plasmid DNA transformed. ATc, anhydrotetracycline at 0.5 µg/ml. Dotted lines represent the limit of detection for these assays. a, Reverse CRISPR-immunity assays using inverted target vector insertions and spacer 43T or 43B plasmid DNA. Inversion of the attP motif (‘Inv-attP-’) for forward and reverse insertion vectors causes integration in the opposite orientation relative to the chromosomal origin of replication. b, Reverse CRISPR-immunity assays using type II-A CRISPR plasmid DNA to transform strains from Fig 3b. The pDB184 parent vector serves as a non-targeting control. Error bars: mean ± s.d. (n=3).
Extended Data Figure 8. Infection with ΦNM1 in liquid culture.
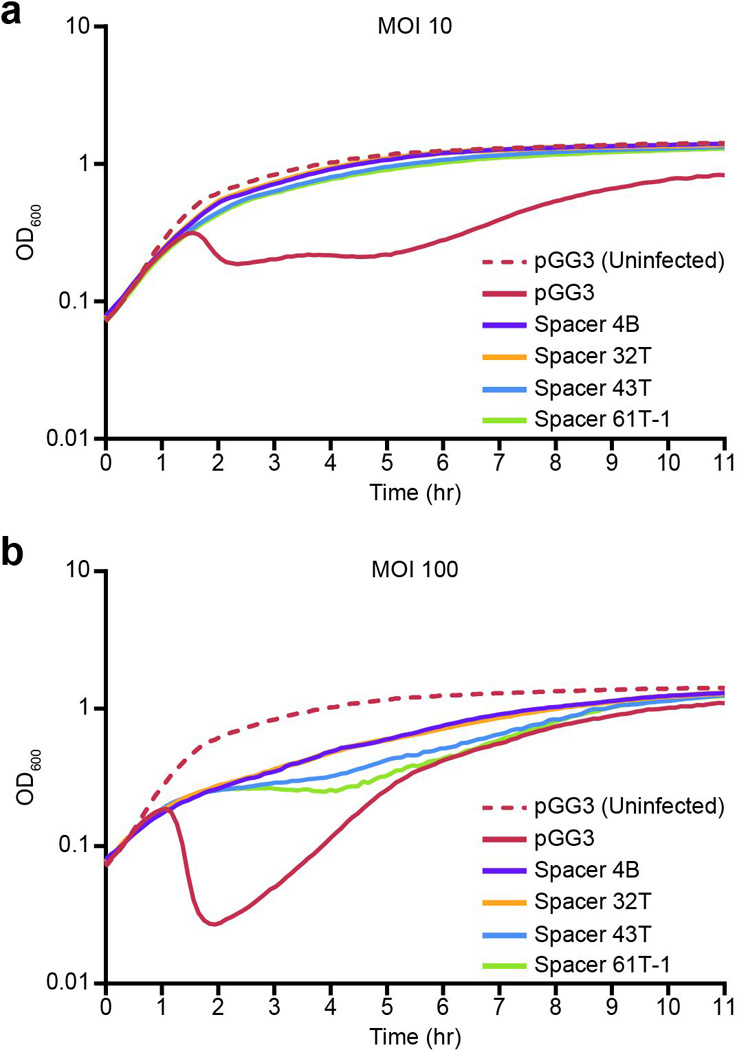
Growth curves of RN4220 cells harboring the indicated CRISPR plasmids were infected at time zero with ΦNM1 at a MOI of 10 (a) or 100 (b). Growth of uninfected RN4220/pGG3 cultures is also shown (dotted red lines).
Extended Data Figure 9. Immunity to ΦNM1γ6 in liquid culture is unaffected by the presence of a tolerated chromosomal target.

Growth curves of the indicated chromosomal insertion strains from Fig. 3 harboring either spacer 43T or pGG3 CRISPR plasmids, in the absence (dotted lines) or presence (solid lines) of ΦNM1γ6 addition at a MOI of 10. Black arrow denotes the time of phage addition; no ATc induction is utilized in this assay. The presence of a chromosomal target for spacer 43T has no discernable effect on culture growth during spacer 43T-mediated immunity to ΦNM1γ6 (compare solid green and blue lines).
Extended Data Figure 10. Inducible curing of a target plasmid.
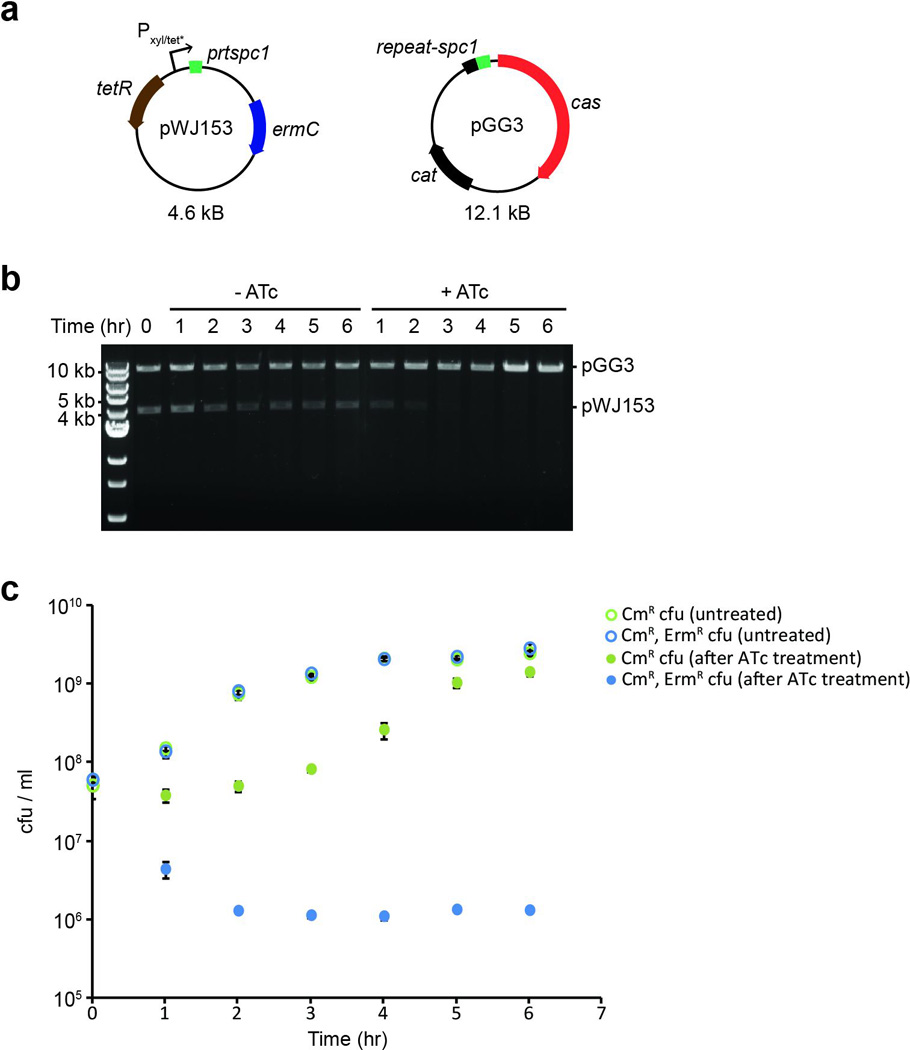
a, Schematic diagram of plasmids utilized in the plasmid curing experiment. The pGG3 CRISPR plasmid harbors a single spacer (“spc1”) targeting a sequence (“prtspc1”) inserted downstream of the Pxyl/tet* inducible promoter in pWJ153. b, Agarose gel electrophoresis of linearized plasmid DNA purified from both anhydrotetracycline-treated (+ATc) and untreated (−ATc) cultures at the indicated timepoints. 10 kb, 5 kb, and 4 kb size markers are indicated. Picture is representative of a single technical replicate. c, Colony forming units (cfu) recovered from cultures analyzed in panel (b) at each time point. Cells were plated with selection for either CmR cfu (green) or CmR, ErmR cfu (blue). Targeting of the pWJ153 plasmid via induction with ATc (filled circles) is accompanied by a severe drop in erythromycin-resistant cfu relative to untreated cultures (open circles). Error bars: mean ± s.d. (n=3).
Supplementary Material
Acknowledgements
The authors would like to acknowledge Ted Bae for providing strains; Bruce Levin, Josh Modell and Poulami Samai for critical discussion of the paper, and Asma Hatoum-Aslan, as well the Torres Lab (New York University), for their optimization of RNA preparation protocols for S. aureus. Additional thanks to John Chen of the Novick lab for advice on the construction of temperate phages with a selectable marker. Finally, we acknowledge The Rockefeller University Genomics Resource Center core facility for performing the next-generation sequencing in this work. L.A.M is supported by the Searle Scholars Program, the Rita Allen Scholars Program, an Irma T. Hirschl Award, a Sinsheimer Foundation Award and a NIH Director’s New Inovator Award (1DP2AI104556-01).
Footnotes
Supplementary information.
Supplementary Tables 1–3.
Supplementary Sequence 1.
Author contributions. GWG and LAM designed experiments. Research was executed by GWG. WJ constructed the pWJ40 and pWJ153 plasmids and performed the plasmid curing experiment. DB constructed plasmid pDB184, assisted with phage de novo assembly, and provided the RPM normalization script for RNA-seq data. GWG and LAM wrote the paper.
The authors have no conflicting financial interests.
References
- 1.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrangou R. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip. Rev. RNA. 2013;4:267–278. doi: 10.1002/wrna.1159. [DOI] [PubMed] [Google Scholar]
- 3.Sorek R, Lawrence CM, Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013;82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- 4.Brüssow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cumby N, Davidson AR, Maxwell KL. The moron comes of age. Bacteriophage. 2012;2:225–228. doi: 10.4161/bact.23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edgar R, Qimron U. The Escherichia coli CRISPR system protects from lambda lysogenization, lysogens, and prophage induction. J. Bacteriol. 2010;192:6291–6294. doi: 10.1128/JB.00644-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brouns SJ, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deveau H, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenova E, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe. 2012;12:177–186. doi: 10.1016/j.chom.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson AD, et al. lambda Repressor and cro--components of an efficient molecular switch. Nature. 1981;294:217–223. doi: 10.1038/294217a0. [DOI] [PubMed] [Google Scholar]
- 16.Nozawa T, et al. CRISPR inhibition of prophage acquisition in Streptococcus pyogenes. PLoS One. 2011;6:e19543. doi: 10.1371/journal.pone.0019543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatoum-Aslan A, Samai P, Maniv I, Jiang W, Marraffini LA. A ruler protein in a complex for antiviral defense determines the length of small interfering CRISPR RNAs. J. Biol. Chem. 2013;288:27888–27897. doi: 10.1074/jbc.M113.499244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kreiswirth BN, et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 19.Bae T, Baba T, Hiramatsu K, Schneewind O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006;62:1035–1047. doi: 10.1111/j.1365-2958.2006.05441.x. [DOI] [PubMed] [Google Scholar]
- 20.Holt DC, et al. A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin. Genome Biol. Evol. 2011;3:881–895. doi: 10.1093/gbe/evr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng L, Garrett RA, Shah SA, Peng X, She Q. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol. Microbiol. 2013;87:1088–1099. doi: 10.1111/mmi.12152. [DOI] [PubMed] [Google Scholar]
- 22.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang W, et al. Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 2013;9:e1003844. doi: 10.1371/journal.pgen.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manica A, Zebec Z, Steinkellner J, Schleper C. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2013;41:10509–10517. doi: 10.1093/nar/gkt767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westra ER, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nudler E. RNA polymerase active center: the molecular engine of transcription. Annu. Rev. Biochem. 2009;78:335–361. doi: 10.1146/annurev.biochem.76.052705.164655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hale CR, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, et al. Structure and Mechanism of the CMR Complex for CRISPR-Mediated Antiviral Immunity. Mol. Cell. 2012;45:303–313. doi: 10.1016/j.molcel.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 31.Moore SD, Prevelige PE., Jr A P22 scaffold protein mutation increases the robustness of head assembly in the presence of excess portal protein. J. Virol. 2002;76:10245–10255. doi: 10.1128/JVI.76.20.10245-10255.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatoum-Aslan A, Maniv I, Samai P, Marraffini LA. Genetic Characterization of Antiplasmid Immunity through a Type III-A CRISPR-Cas System. J. Bacteriol. 2014;196:310–317. doi: 10.1128/JB.01130-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helle L, et al. Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus. Microbiology. 2011;157:3314–3323. doi: 10.1099/mic.0.052548-0. [DOI] [PubMed] [Google Scholar]
- 34.Horinouchi S, Weisblum B. Nucleotide sequence and functional map of pE194, a plasmid that specifies inducible resistance to macrolide, lincosamide, and streptogramin type B antibodies. J. Bacteriol. 1982;150:804–814. doi: 10.1128/jb.150.2.804-814.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Horton RM. In vitro recombination and mutagenesis of DNA: SOEing together tailor-made genes. Methods Mol. Biol. 1993;15:251–261. doi: 10.1385/0-89603-244-2:251. [DOI] [PubMed] [Google Scholar]
- 37.Simpson JT, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.