

Abstract
Retinopathy of prematurity adversely affects premature infants because of oxygen-induced damage of the immature retinal vasculature, resulting in pathological neovascularization (NV). Our pilot studies using the mouse model of oxygen-induced retinopathy (OIR) showed marked increases in angiogenic mediators, including endothelins and endothelin receptor (EDNR) A. We hypothesized that activation of the endothelin system via EDNRA plays a causal role in pathological angiogenesis and up-regulation of angiogenic mediators, including vascular endothelial growth factor A (VEGFA) in OIR. Mice were exposed to 75% oxygen from post-natal day P7 to P12, treated with either vehicle or EDNRA antagonist BQ-123 or EDNRB antagonist BQ-788 on P12, and kept at room air from P12 to P17 (ischemic phase). RT-PCR analysis revealed increased levels of EDN2 and EDNRA mRNA, and Western blot analysis revealed increased EDN2 expression during the ischemic phase. EDNRA inhibition significantly increased vessel sprouting, resulting in enhanced physiological angiogenesis and decreased pathological NV, whereas EDNRB inhibition modestly improved vascular repair. OIR triggered significant increases in VEGFA protein and mRNA for delta-like ligand 4, apelin, angiopoietin-2, and monocyte chemoattractant protein-1. BQ-123 treatment significantly reduced these alterations. EDN2 expression was localized to retinal glia and pathological NV tufts of the OIR retinas. EDN2 also induced VEGFA protein expression in cultured astrocytes. In conclusion, inhibition of the EDNRA during OIR suppresses pathological NV and promotes physiological angiogenesis.
Retinal angiogenesis has been extensively studied, under both physiological and pathological conditions, to elucidate the molecular mechanisms governing vessel growth and development. Pathological angiogenesis is a common hallmark of ischemic retinopathies, such as diabetic retinopathy and retinopathy of prematurity (ROP). Ischemia triggers hypoxia and improper growth factor production, leading to molecular changes that disrupt normal vessel growth and homeostasis, resulting in pathological angiogenesis. In ROP, which is a leading cause of visual impairment in preterm infants, the immature retina is exposed to high extrauterine oxygen, which causes degeneration of the retinal microvessels (vaso-obliteration) and inhibition of normal vascular development. Thereafter, retinal ischemia occurs, which leads to compensatory pathological neovascularization (NV) with sprouting of abnormal vessels from the retina into the vitreous.1,2 In 2007, 13% of live births in the United States were reported premature (<37 weeks' gestation).3 The oxygen-induced retinopathy (OIR) model of ischemic retinopathy conveniently reproduces the vaso-obliteration and NV phases of ROP. During the hyperoxic phase of the disease, production of key growth factors, such as vascular endothelial growth factor A (VEGFA) and angiopoietins, is suppressed and the developing microvessels regress. In the subsequent phase of the disease, an imbalanced expression of these factors results in aberrant growth of new vessels, leading to pathological NV.4,5 Studies in the OIR mouse model have demonstrated that retinal ischemia triggers release of angiogenic mediators, such as VEGFA, which promotes formation of aberrant leaky vessels. In some cases, this causes retinal detachment and blindness.2,6,7 Current therapies include injections of anti-VEGFA agents and laser photocoagulation,3 but a less invasive and effective therapy is yet to be developed.
The retinal vasculature grows by radial migration of endothelial tip cells. Tip cells lead vascular sprouting by responding to a gradient of VEGFA, produced by the retinal astrocytes, and other guidance cues in the local environment.8 Studies indicate that increased pathological NV during OIR involves a plethora of angiogenic mediators, such as VEGFA, erythropoietin,4,5 angiopoietin-2 (ANGPT2),9 apelin (APLN),10 and delta-like ligand 4 (DLL4).11 An imbalance of these mediators leads to formation of chaotic vessels, which are often misguided into the vitreous and cause retinal detachment and blindness. The role of angiogenic mediators in pathological NV is well established, but the interplay of such mediators with other vasoactive factors is largely unknown.
We conducted mRNA analysis using the OIR model and discovered that expression of endothelins (ETs) and ET receptors is highly increased during the ischemic phase of the disease. The ETs are a family of 21–amino acid peptides with three isoforms, EDN1, EDN2, and EDN3. Each of these is encoded by a different gene and released by proteolysis of precursor proteins to act in an autocrine or paracrine manner. All three are distributed widely throughout various tissues, including kidney, lung, intestines, liver, brain, and retina.12–15 ETs can bind to two distinct G protein-coupled receptors, endothelin receptors (EDNRs) A and B. EDNRA has a higher affinity for EDN1 and EDN2, whereas EDNRB binds to all three ETs equally.16 Activation of both receptors is known to mediate vasoconstriction in vascular smooth muscle cells, whereas EDNRB activation mediates vasodilation in vascular endothelial cells (EC) via endothelial nitric oxide production. The importance of the ET system has been demonstrated in various diseases. Hypoxia up-regulates EDN2, ET receptors, and VEGFA mRNA expression in a hypoxia-inducible factor-1α–dependent manner in murine and human breast tumor cell lines.17 Under normal conditions, astrocytes in the brain express low levels of ETs, but after ischemia, ETs and their receptors are greatly enhanced.18,19 Retinas from diabetic rats show an increase in mRNA for EDN1, EDN3, and their receptors.20 Studies in models of photoreceptor disease/injury have shown that transcripts for EDN2 and EDNRB are increased and are localized to photoreceptors and glial cells, respectively.21 These studies demonstrate that endothelins are involved in pathogenesis of both vascular and nonvascular diseases.
Recent work suggests that overexpression of EDN2 in the mouse neural retina leads to hypoxia, hinders perfusion of the growing vessels, and limits development of both the superficial and deep vessels. Moreover, EDN2-dependent inhibition of angiogenesis in the neural retina is mediated through EDNRA, but not EDNRB.22 These data suggest that EDN2 activation of EDNRA plays a significant role in inhibiting angiogenesis in the developing retina. However, the impact of ETs and their receptors on pathological NV in ischemic retinopathy is completely unknown.
We, therefore, investigated the role of the endothelin system in pathological NV. Transcripts of EDN2 and EDNRA are increased during the ischemic phase of OIR, and inhibition of EDNRA during OIR promoted physiological angiogenesis and suppressed pathological NV. In addition, blockade of EDNRA reversed misdirected vessel growth by enhancing vessel sprouting at the leading edges of neovessels and reduced ischemia-induced angiogenic mediators. Neutralizing actions of endothelins through EDNRA blockade may provide an effective approach to resolving pathological NV and enhancing vision in patients with ROP.
Materials and Methods
Treatment of Animals
All procedures with animals were performed in accordance with The Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the institutional animal care and use committee (Animal Welfare Assurance number A3307-01). A mouse model of OIR was used as previously described.6 Briefly, at post-natal day 7 (P7), C57BL/6 mouse pups, along with nursing mothers, were placed in 75% oxygen. At P12, they were returned to room air (RA; 21% oxygen) for 5 days until P17. By using this model of ischemic retinopathy, obliteration of the retinal vessels is induced during the exposure to 75% oxygen. Immediately on return to RA, the avascular central retina becomes hypoxic, leading to significant vitreous NV that peaks at P17.
Intravitreal Injection
P12 mice were anesthetized by i.p. injection of 625 mg/kg 2,2,2-tribromoethanol (Avertin, Sigma-Aldrich, St. Louis, MO) and local administration of proparacaine HCL ophthalmic solution (Akorn, Lake Forest, IL) onto the eye. Intravitreal injections were performed by delivering 1 μL of vehicle or BQ-123 (EDNRA antagonist; Tocris Biosciences, Minneapolis, MN) or BQ-788 (EDNRB antagonist; Tocris Biosciences) into the vitreous space by injecting behind the limbus. BQ-123 was solubilized in 0.9% saline containing 10 mmol/L NaHCO3 and 1% bovine serum albumin. BQ-788 was solubilized in ethanol and further diluted in 0.9% saline. Previous studies in cats have shown that a single injection of BQ-123 (30 μg/50 μL per eye, 1 mmol/L) or BQ-788 (33 μg/50 μL per eye, 1 mmol/L) injected into the vitreous was sufficient to elicit a maximum retinal hemodynamic response.23,24 We tested several concentrations of both receptor blockers (BQ-123: 10, 17, and 34 μg/μL per eye; BQ-788: 1.5, 4.5, and 13.5 μg/μL per eye) to find the appropriate concentrations that would ensure maximum response. We determined that 17 μg/μL per eye of BQ-123 (28 mmol/L) and 4.5 μg/μL per eye of BQ-788 (6.8 mmol/L) were optimal for our studies. We chose to deliver a higher concentration of the drugs into a much smaller mouse vitreous space compared with previous studies, to allow for drug loss due to the fluid leakage from the injection site that often occurs during injections. BQ-123 is a highly selective EDNRA antagonist that has been shown to have high affinity and efficacy for the receptor (KD = 1.18 nmol/L in rat heart tissues), and has shown promising results in small-scale patient studies.25,26 BQ-788 is also specific for EDNRB (KD = 1.98 μmol/L in human left ventricle).25 Vehicle or receptor antagonists were delivered using a 35-gauge needle mounted to a 10-μL Hamilton syringe. The tip of the needle was inserted under the guidance of a dissecting microscope (Leica Wild M650; Leica, Bannockburn, IL) through the dorsal limbus of the eye. Injections were performed slowly over 3 minutes.
Quantitative Real-Time PCR
Total RNA was isolated using an RNA purification kit (RNAqueous 4PCR; Applied Biosystems, Austin, TX) for retinal tissue or with reagent (TRIzol; Invitrogen, Grand Island, NY) for cells, according to the manufacturer's instructions. Total RNA was reverse transcribed with M-MLV Reverse Transcriptase (Invitrogen, Grand Island, NY) to generate cDNA. Gene expression was determined by real-time quantitative PCR system (StepOne Plus; Applied Biosystems, Grand Island, NY) with Power SYBR Green Master Mix (Applied Biosystems, Grand Island, NY). Various genes were studied using primers specific to their cDNA templates (Table 1). PCRs were set up to acquire cycle threshold for each sample, determined as the initial increase in fluorescence above background. After the completion of the PCR cycles, melt curves were generated to check the specificity of the amplicons. The relative difference in various gene expressions was calculated by the ΔΔCT method using hypoxanthine phosphoribosyltransferase as the internal control.
Table 1.
Mouse Primer Sequences
| Gene symbol | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| EDN1 | 5′-GACTTTCCAAGGAGCTCCAGAA-3′ | 5′-CAGCTCCGGTGCTGAGTTC-3′ |
| EDN2 | 5′-GTGGTCGCCCCAAGCA-3′ | 5′-TCCACATCTTGCCAGTCTTTAACA-3′ |
| EDNRA | 5′-CCATCGACCCCCTAATTTGG-3′ | 5′-GGGCAATAGCCGTGCATT-3′ |
| EDNRB | 5′-GCTAGGCATCATCGGGAACTC-3′ | 5′-TTGCGCATGCACTTGTTCTT-3′ |
| DLL4 | 5′-TTTGCTCTCCCAGGGACTCT-3′ | 5′-CTGTGGCAATCACACACTCG-3′ |
| APLN | 5′-GCTCTGGCTCTCCTTGACTG-3′ | 5′-GGTAGCGCATGCTTCCTTCT-3′ |
| ANGPT2 | 5′-ACACCGAGAAGATGGCAGTGT-3′ | 5′-CTCCCGAAGCCCTCTTTGTA-3′ |
| CCL2 | 5′-GGCTCAGCCAGATGCAGTTAA-3′ | 5′-CCTACTCATTGGGATCATCTTGCT-3′ |
| VEGFA | 5′-TACCTCCACCATGCCAAGTG-3′ | 5′-TCATGGGACTTCTGCTCTCCTT-3′ |
Western Blot Analysis
Retinas or cultured cells were homogenized in a radioimmunoprecipitation assay lysis buffer (Millipore, Billerica, MA) supplied with phosphatase inhibitor cocktail (Roche, Indianapolis, IN), 1 mmol/L phenyl methyl sulfonyl fluoride, and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Protein samples (20 μg) were subjected to 10% SDS-PAGE. Proteins were transferred onto a nitrocellulose membrane, and the membrane was blocked (5% milk) and incubated with primary antibodies against rabbit EDN2 (1:60; Sigma-Aldrich), rabbit VEGFA (1:1000; Abcam, Cambridge, MA), rabbit p-Y-STAT3 (1:500; Cell Signaling, Danvers, MA), mouse STAT3 (1:2000; BD Biosciences, San Jose, CA), and mouse β-tubulin (1:10,000; Sigma-Aldrich), followed by horseradish peroxidase–conjugated secondary antibody (1:2000; GE Healthcare, Pittsburgh, PA). Immunoreactive proteins were detected using the chemiluminescence system (GE Healthcare).
Immunostaining of Whole-Mount Retinas
Eyes were enucleated at various time points and fixed in 4% paraformaldehyde overnight. The cornea, sclera, lens, vitreous, and hyaloid vessels were removed, and radial incisions were made at four equal intervals of the retina. Retinas were blocked and permeabilized in phosphate-buffered saline (PBS) containing 10% goat serum and 1% Triton X-100 (Sigma-Aldrich) for 30 minutes. Retinas were then stained with Alex594-labeled Griffonia simplicifolia isolectin B4 (1:200; Invitrogen, Carlsbad, CA) overnight at 4°C. Retinas were washed in PBS three times, 20 minutes each. Retinas were flat mounted in mounting medium (Vectashield; Vector Laboratories, Burlingame, CA) and examined by fluorescence and confocal microscopy. Areas of vaso-obliteration and vitreoretinal neovascular tufts were quantified using ImageJ software version 1.47u (NIH, Bethesda, MD), as previously described.6 To perform vessel sprouting analysis, high-magnification images were acquired and vessel sprouts were counted as previously described.11
Immunofluorescence
Eyes were removed, fixed in 4% paraformaldehyde (overnight, 4°C), equilibrated in 30% sucrose, and embedded in Tissue-Tek Optimum Cutting Temperature compound (Sakura Finetek, Torrance, CA). Frozen sections (10 μm thick) were permeabilized in 1% Triton X-100 (20 minutes) and blocked in 10% normal goat serum containing 1% bovine serum albumin (1 hour). Sections were incubated overnight in primary antibodies against rabbit EDN2 (1:60; Sigma-Aldrich), chicken glial fibrillary acidic protein (GFAP; 1:500; Abcam), and biotin-labeled isolectin B4 (1:100; Vector Laboratories) (4°C), followed by reaction with secondary antibodies goat anti-rabbit (1:400; Molecular Probes, Grand Island, NY), goat anti-chicken (1:500; Jackson Immunoresearch, West Grove, PA), and 7-amino-4-methylcoumarin-3-acetic acid-labeled avidin (1:1000; Vector Laboratories). Sections were washed with PBS, mounted with mounting medium (Vectashield), and examined using confocal microscopy.
Primary Astrocyte Culture
Primary mouse astrocytes were isolated and cultured according to a published method.27 Briefly, cerebral cortices of 1- to 2-day-old CD-1 mice were mechanically and enzymatically disrupted. After centrifugation, cells were plated in modified Eagle's medium (Hyclone, Logan, UT), supplemented with 2 mmol/L glutamine, 20 mmol/L glucose, 5% fetal bovine serum, 5% bovine growth serum, 10% calf serum, 10 ng/mL epidermal growth factor, and 50 IU/mL penicillin/50 μg/mL streptomycin (Hyclone) at 2.5 hemispheres per 6-well plate. This procedure routinely produces confluent monolayers of protoplasmic-like cells 10 to 12 days after plating.
On reaching confluence, cells were exposed to 8 μmol/L cytosine arabinoside for 5 to 7 days to eliminate the growth of rapidly dividing cells, including microglia, macrophages, and oligodendrocytes. Astrocyte cultures were maintained in modified Eagle's medium supplemented with 5% fetal bovine serum, 5% bovine growth serum, and 50 IU/mL penicillin/50 μg/mL streptomycin until used for experimentation between days 28 and 35 in vitro. Cultures were serum starved for 12 hours before the experiment. Astrocyte cultures are routinely >98% pure, as assessed by immunostaining of the glial-specific marker, GFAP. For hypoxia experiments, cell cultures were placed in a 37°C humidified chamber, which was supplied with 95% N2 and 5% CO2. Astrocytes were either stimulated with 0.5% hypoxia or kept at RA conditions inside a 37°C humidified chamber for 24 hours. In separate experiments, cells were stimulated with 100 nmol/L recombinant EDN2 (Sigma-Aldrich) for a time-course analysis. Cell lysates were collected for RT-PCR and Western blot analysis.
Statistical Analysis
Data are presented as means ± SEM. Group differences were evaluated by using Student's t-test or one-way analysis of variance, followed by Tukey's post hoc test for statistical analysis. P < 0.05 was considered significant. Animal studies were performed in groups of 6 to 18 mice. Tissue culture studies were performed in groups of four to six cultures, and each experiment was repeated at least twice.
Results
Endothelin Signaling During OIR
To evaluate the potential impact of endothelins in mediating the damaging effects seen in OIR, we first determined the presence of endothelin transcripts in the OIR retinas. We conducted a whole genome microarray analysis of the RA control and OIR retinas obtained on post-natal day 17 (P17) and evaluated the angiogenic genes that were highly up-regulated in OIR compared with RA controls. The results indicated that EDN2 (42-fold), EDN1 (twofold), and EDNRA (twofold) receptors were among the many genes that were increased on P17 (data not shown).
To confirm these findings and further assess the expression profile of endothelin system components, we performed a time-course analysis of mRNA levels of EDN1, EDN2, EDNRA, and EDNRB starting from the onset of the ischemic phase (P12) to P17 of RA and OIR retinas using RT-PCR. Relative mRNA expression of each gene was quantified by normalizing it to the expression of internal control and to its respective RA control and plotted as fold of each RA control. Our data showed a time-dependent increase in the transcripts of EDN2, EDN1, and EDNRA from P12-P17 (Figure 1). EDN2 levels were significantly increased from P13 to P17 (100-fold on P13, 138-fold on P14, and 130-fold on P17 versus P12), indicative of a sustained increase in the message of EDN2 throughout the ischemic phase of OIR (Figure 1A). EDN1 levels were oscillating from P12 to P17, but were increased on P17 compared with P12 to P16 (Figure 1B). In addition, expression of EDNRA steadily increased from P15 to P17 (0.7-fold on P15, 1.4-fold on P16, and 2.3-fold on P17 versus P12), whereas expression of EDNRB was only modestly increased from P13 to P17 (Figure 1C).
Figure 1.

Oxygen-induced retinopathy (OIR) increases endothelins and endothelin receptor expression. Mice were sacrificed at the times indicated, and EDN2, EDN1, EDNRA, and EDNRB mRNA expression in the whole retina was determined using RT-PCR. Data were normalized to both the expression of internal control HPRT and to gene mRNA expression of each RA control and plotted as fold of RA at each time point. A: Relative mRNA expression of EDN2 increases time dependently and is sustained throughout ischemic phase of OIR. B: Relative mRNA expression of EDN1 oscillates during ischemia, but is significantly high on P17 compared with all other time points. C: Relative mRNA expression of EDNRA increases time dependently from P15 to P17, whereas EDNRB expression is significant at P13 to P14 and P17 compared with P12. D and E: Relative protein expression of EDN2 is significantly induced during the ischemic phase of OIR at both P15 and P17. ∗P < 0.05 versus P12 (n = 3 retinas; A); ∗P < 0.05 versus P12, †P < 0.05 versus P13, and ‡P < 0.05 versus P12 to P16 (n = 3 retinas; B); ∗P < 0.05 versus P12, †P < 0.05 versus P12 to P14 (n = 3 retinas; C); ∗P < 0.05 versus RA (n = 3 to 4 retinas; D and E).
To ascertain that results from mRNA expression correlate with functional protein expression of EDN2, we performed Western blot analysis of EDN2. The results showed that EDN2 expression is significantly induced at both P15 and P17 (Figure 1, D and E). Altogether, these data suggested that endothelin signaling is significantly increased during OIR and increased action of endothelins, predominantly via the EDNRA, could be implicated in the pathogenesis of OIR.
Role of the Endothelin System in Pathological Angiogenesis
To investigate the functional role of endothelin signaling in OIR, we performed intravitreal injections of EDNRA antagonist BQ-123 (17 μg/μL per eye) and EDNRB antagonist BQ-788 (4.5 μg/μL per eye) on P12 (immediately after the hyperoxic vaso-obliterative phase), and processed the retinas on P17. The dose concentrations of the antagonists were chosen on the basis of previous literature and our pilot experiments. The inhibitors were injected on P12 to neutralize the actions of EDN2, which was dramatically increased starting at P13 (Figure 1A). We performed analysis to examine potential effects of BQ-123 on EDN1, EDN2, and EDNRA mRNA levels on P17. This analysis showed no significant change in their expression, suggesting that BQ-123 treatment did not modulate mRNA expression of the endothelins or their receptors.
We performed flat mounting, visualized the blood vessels using isolectin B4 staining, and observed that blockade of EDNRA using BQ-123 significantly enhanced the regrowth of normal vessels into the avascular central retina, compared with the vehicle control. Conversely, the areas of pathological NV were significantly reduced by the BQ-123 treatment (Figure 2, A–C). Inhibition of EDNRB slightly reduced NV tuft formation and modestly improved vascular repair, but the latter change was not statistically significant (Figure 2, D–F). These findings suggest that increased endothelin signaling, predominantly via EDNRA activation, limits vascular repair and promotes pathological angiogenesis during OIR.
Figure 2.

Endothelin receptor blockade suppresses pathological NV and promotes physiological angiogenesis. P12 Oxygen-induced retinopathy (OIR) mice were injected intravitreally with vehicle (Veh) or BQ-123 (17 μg/μL per eye) and sacrificed on P17. Retinas were dissected, flat mounted, and stained with isolectin B4 (red, blood vessels), and images were taken using fluorescent microscope. A: Treatment of OIR mice with BQ-123 enhances vascular repair by promoting physiological angiogenesis (indicated by reduced vessel dropout area within the white borders) and suppresses pathological NV tuft formation (white arrows). The vessel dropout area was quantified using ImageJ software version 1.47u as percentage of whole retinal area, normalized to the average of the OIR + Veh, and plotted as fold of OIR + Veh. B: OIR mice treated with BQ-123 show significant reduction in vessel dropout area compared with vehicle. The pathological NV tuft formation was measured using semi-automated thresholding technique in ImageJ software version 1.47u. The total area of NV tufts was calculated as percentage of whole retinal area, normalized to the average of the OIR + Veh, and plotted as fold of OIR + Veh. C: OIR mice treated with BQ-123 show significant reduction in NV tuft area compared with vehicle. D–F: Treatment of OIR mice with BQ-788 modestly increases vascular repair by promoting physiological angiogenesis (indicated by reduced vessel dropout area within the yellow borders) and suppresses pathological NV tuft formation (yellow arrows). Quantification shows modest reduction in vessel dropout area and a small, but significant, reduction in NV tuft area compared with vehicle. ∗P < 0.05 versus OIR + Veh. n = 12 retinas, three litters of mice (B and C); n = 6 retinas, two litters of mice (E and F). Scale bar = 500 μm (A and D).
In addition, we further analyzed the hypoxic avascular regions for angiogenic vessel sprouts, which normally lead the growing vascular plexus toward the avascular regions of the retina. The OIR retinas treated with vehicle had limited sprouting into the avascular zone and instead the growing vessels were composed of misdirected and disorganized NV tufts. On the contrary, BQ-123–treated retinas showed increased frequency of healthy sprouting and improved organization of the vessel network (Figure 3, A and B), which correlate with the above evidence showing increased vascular coverage and reduced NV. These findings demonstrate a causal role of EDNRA activation in governing pathological angiogenesis during OIR.
Figure 3.

Endothelin receptor A blockade promotes vessel sprouting in oxygen-induced retinopathy (OIR) retinas. P12 OIR mice were injected intravitreally with vehicle (Veh) or BQ-123 (17 μg/μL per eye) and sacrificed on P17. Retinas were dissected, flat mounted, and stained with isolectin B4 (red, blood vessels), and images were taken at high magnification using confocal microscope. A: Two representative images of retinal vessel sprouting (white arrowheads) are shown for each group. B: Quantification of number of vessel sprouts reveals that OIR mice treated with BQ-123 show more vessel sprouting compared with OIR + Veh. ∗P < 0.05. n = 6 retinas, two litters of mice (B). Scale bar = 20 μm (A).
Endothelin Signaling Regulates VEGFA Production and Other Angiogenic Mediators
To study the molecular signaling associated with EDNRA activation and pathological angiogenesis, we analyzed production of pro-angiogenic mediators. We measured activation of STAT3 and VEGFA expression in RA control, vehicle, and BQ-123–treated retinas using Western blot analysis. EDN1 acts to increase STAT3 activation via EDNRA in adipocytes.28 During OIR, STAT3 activation has been shown to be correlated with increased VEGFA expression and pathological angiogenesis.29 OIR triggered a significant increase in VEGFA production (Figure 4A) and phosphorylation of STAT3 (Figure 4B) compared with RA control, which was abrogated by BQ-123 treatment. This suggests that endothelin signaling via EDNRA mediates VEGFA production, possibly through STAT3 activation during OIR.
Figure 4.
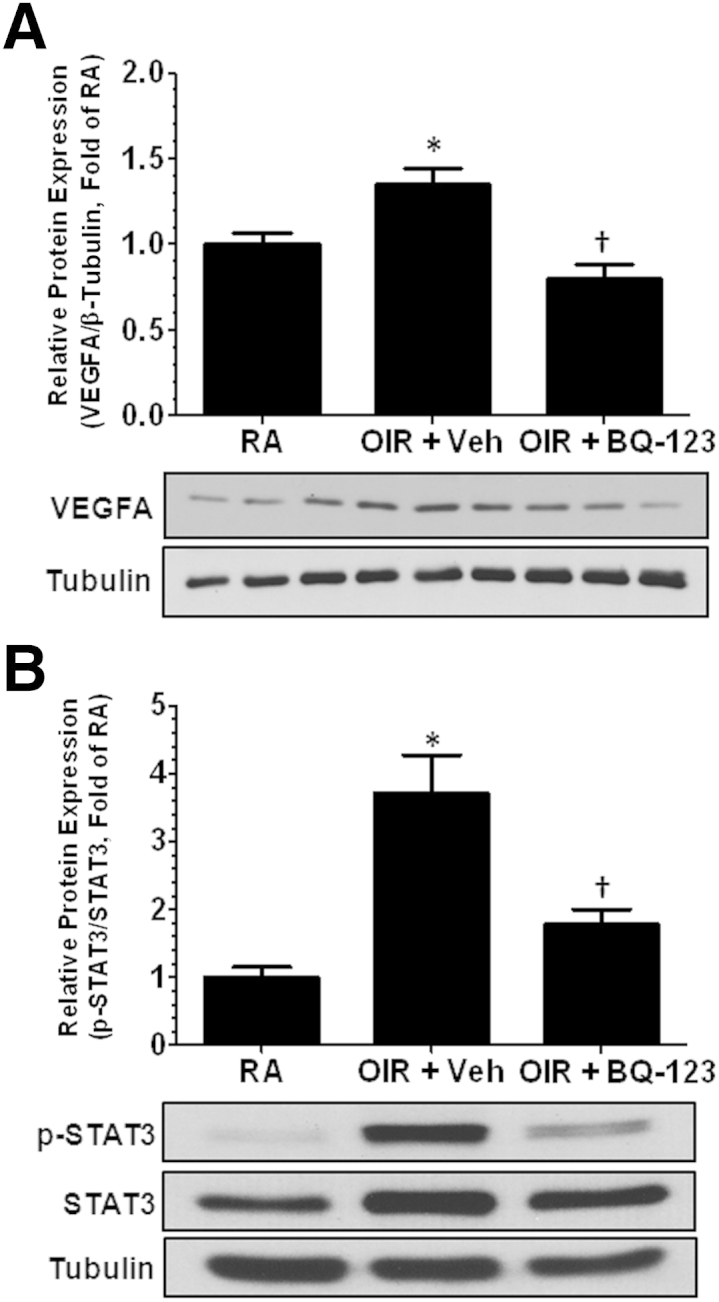
Endothelin receptor A blockade prevents oxygen-induced retinopathy (OIR)-induced increase in VEGFA. P12 OIR mice were injected intravitreally with vehicle (Veh) or BQ-123 (17 μg/μL per eye) and sacrificed on P17. Retinas were collected and processed for Western blot analysis. VEGFA (A) and p-STAT3 (B) protein expression is significantly increased in OIR + Veh group compared with RA control; however, EDNRA blockade prevents such increase in VEGFA and STAT3 activation. Densitometry analysis reveals statistical differences between the groups. ∗P < 0.05 versus RA, †P < 0.05 versus OIR + Veh. n = 6 to 9 retinas, two to three litters of mice (A and B).
In addition, we analyzed several genes previously linked to pathological angiogenesis during OIR. OIR significantly increased mRNA expression of DLL4, APLN, ANGPT2, and monocyte-chemoattractant protein-1 (CCL2). These were reduced by blockade of EDNRA, suggesting that EDNRA activation leads to increased production of pro-angiogenic mediators (Figure 5). Together, these findings support our hypothesis that endothelins play a role in pathological angiogenesis by increasing production of VEGFA and other angiogenic molecules. However, blockade of EDNRA prevents overexuberant angiogenic sprouting by limiting production of angiogenic mediators and promoting physiological vascular repair.
Figure 5.

Endothelin receptor A blockade prevents oxygen-induced retinopathy (OIR)-induced increase in angiogenic mediators. P12 OIR mice were injected intravitreally with vehicle (Veh) or BQ-123 (17 μg/μL per eye) and sacrificed on P17. Retinas were collected and processed for RT-PCR analysis. OIR induces mRNA expression of DLL4, APLN, ANGPT2, and CCL2 compared with RA control; however, EDNRA blockade limits the increase in the angiogenic genes. ∗P < 0.05 versus RA, †P < 0.05 versus OIR + Veh. n = 6 to 9 retinas, two to three litters of mice.
Endothelin-2 Is Localized to Retinal Glia and NV Tufts
To localize the expression of endothelin-2, we performed immunofluorescence staining of frozen sections of P17 RA and OIR retinas. In the RA control retinas, EDN2 immunoreactivity was present in the outer plexiform layer (OPL), inner nuclear layer, and inner plexiform layer (IPL), whereas in OIR retinas, it was present in the OPL and strongly expressed in the ganglion cell layer (GCL) (Figure 6).
Figure 6.

EDN2 colocalizes in retinal glia and blood vessels in the oxygen-induced retinopathy (OIR) retinas. P17 frozen cross sections (10 μm thick) from RA and OIR were processed for imaging analysis and reacted with anti-GFAP (marker of retinal glia, including astrocytes and Müller cells, red), anti-EDN2 (green), and isolectin B4 (marker of blood vessels, blue). A: Confocal microscopy reveals that in RA retinas, EDN2 is expressed in the OPL, inner nuclear layer (INL), and IPL, whereas in OIR retinas, it is strongly expressed in the GCL. Within the GCL, EDN2 is present in retinal glia (yellow) and in blood vessels (turquoise) (white arrows). B: High-magnification confocal microscopy of the same region (white boxed areas) reveals a close-up view of EDN2 in the retinal glia and blood vessels of the NV tufts (white arrows). (Representative images shown, n = 6 to 9). Primary cortical astrocytes from mouse were serum starved for 12 hours and cultured in normoxia (21% oxygen) and hypoxia (0.5% oxygen) for 24 hours at 37°C. RNA from the cells was extracted using the TRIzol reagent and processed for RT-PCR analysis. C: Hypoxia significantly increases mRNA expression of EDN2 and VEGFA in cortical astrocytes compared to normoxia controls. D: Stimulation of astrocytes with 100 nmol/L EDN2 induces time-dependent changes in the VEGFA protein levels and significant increase at 24 hours after treatment compared with zero-time point. ∗P < 0.05 versus normoxia (n = 4, two independent experiments; C); ∗P < 0.05 versus baseline (n = 4; D). Scale bar = 20 μm (A and B). ONL, outer nuclear layer.
The lack of staining of EDN2 in the IPL of OIR retina can be attributed to the thinning of retina and IPL that typically occurs during OIR.30,31 In the GCL and nerve fiber layer, EDN2 had a strong presence within and around the NV tufts, particularly in reactive astrocytes and possibly in Müller cell end feet and both in and around ECs, as indicated by its colocalization with retinal glia and blood vessels (Figure 6, A and B).
In OIR retinas, GFAP immunoreactivity was much stronger than in the RA control retinas and extended into the inner layers of the retina as well, suggesting its localization in Müller cells and astrocytes (Figure 6A). Many of the GFAP-positive processes were also positive for EDN2, suggesting that EDN2 is expressed in both astrocytes and Müller cells. In addition, primary cultures of mouse cortical astrocytes exposed to hypoxia (0.5% oxygen) for 24 hours exhibited a significant increase in mRNA expression of EDN2 compared with normoxia control (Figure 6C), confirming that astrocytes are a source of EDN2 under hypoxic conditions. Levels of mRNA for EDN1, EDNRA, and EDNRB were not significantly changed (data not shown). Increases in VEGFA mRNA served as a positive control for the experiment, because astrocytes are a major source of VEGFA in the retina.8
A further study testing the effects of EDN2 on VEGFA protein levels showed a small, but statistically significant, increase after 24 hours of treatment (Figure 6D). The delay in onset of this increase suggests that the EDN2 effect is indirect. The modest increase in VEGFA protein levels indicates that EDN2 is not the predominant driver of hypoxia-induced increases in VEGFA expression in astrocytes. These data suggest that EDN2 from the astrocytes may initiate signaling events on astrocytes in an autocrine manner or on neighboring cells in a paracrine manner.
Discussion
Therapies for ROP are currently limited to laser photocoagulation and anti-VEGFA injections. Both have off-target effects and are invasive.1 Blockade of VEGFA can affect survival and function of retinal neurons32 and may impair retinal development. Therefore, there is a dire need for novel targets. To our knowledge, the data presented herein provide the first evidence that vasoactive endothelins and their receptors are involved in pathological angiogenesis during OIR. Although the function of endothelins has been extensively studied in models of diabetes,33,34 hypertension,35 cancer,36–38 and other diseases, their role during pathological angiogenesis has not been demonstrated previously. Herein, we show that expression of EDN2 and EDNRA is highly increased in a mouse model of OIR and that blockade of EDNRA reduces pathological NV, promotes physiological vascular repair, and reduces expression of angiogenic mediators, including VEGFA.
Endothelins are classically described as potent vasoconstrictors that regulate blood pressure and volume homeostasis, but they can act on virtually any cell type in the body.35 In isolated porcine retinal arterioles, EDN1 stimulation causes vasoconstriction through the activation of EDNRA, suggesting that endothelins also play a significant role in maintaining retinal homeostasis.39 A recent study by Rattner et al22 suggests that overexpression of EDN2 in the neural retina provokes hypoxia and inhibits developmental angiogenesis via activation of EDNRA and not EDNRB. To our knowledge, our studies provide the first evidence of endothelin system activation during ischemic retinopathy and that pathological angiogenesis is reduced predominantly through inhibition of EDNRA, and not EDNRB, further extending previous findings.
EDNRA blockade did not modulate mRNA expression of EDN1, EDN2, or EDNRA. Blockade of EDNRA function does not require any effect on EDNRA expression. In some instances, receptor blockade causes an increase in expression of the receptor. In addition, blockade of EDNRB using BQ-788 resulted in modest improvement in OIR-induced vaso-obliteration and a small, but significant, decrease in NV. Thus, it is possible that activation of EDNRB plays a modest role in the pathological features. Alternately, with the high concentrations of the antagonists used in this study, it is also possible that the EDNRB blocker effects are due, in part, to inhibition of EDNRA and that the EDNRA blocker might also have had some effect on EDNRB function. It is well known that EDNRA and EDNRB act differently and may have antagonistic effects, depending on their location. In smooth muscle cells, EDNRA and EDNRB mediate vasoconstriction, proliferation, and fibrosis, whereas in ECs, EDNRB mediates vasodilation. EDNRB also appears to serve as a clearance receptor to regulate plasma concentrations of endothelins.35 It is plausible that blockade of EDNRA may lead to a compensatory increase in EDNRB and a subsequent reduction in retinal endothelin levels and signaling. On the other hand, blockade of EDNRB may lead to a compensatory increase in EDNRA, leading to elevation in retinal endothelin levels and inhibition of vascular repair. This phenomenon could explain the predominance of EDNRA-mediated protection versus modest EDNRB-mediated protection in the above findings.
Our studies clearly illustrate the role of endothelins in pathological angiogenesis, as indicated by formation of NV tufts. Previous work has shown that endothelins play a role in regulating growth of ovarian cancer cell lines and tumors.40,41 EDN1 induces an angiogenic phenotype in cultured ECs and stimulates NV in a Matrigel plug assay in vivo.42 Solid tumors contain regions of hypoxia, which induces ET expression. Hypoxia-induced increases in EDN2 expression and signaling have been shown to promote tumor cell survival during hypoxic stress.17 These data suggest that endothelins are induced by hypoxia and could be involved in regulating angiogenesis. Our studies confirm these findings in a model of ischemic retinopathy and demonstrate that activation of the endothelin system via EDNRA increases pathological NV while reducing physiological angiogenesis.
It is of great interest to determine the cell type(s) responsible for endothelin secretion and its actions. In situ hybridization studies have shown that EDN1 is expressed in cells of the human retina, including the retinal pigment epithelium, photoreceptors, IPL, GCL, and astrocytes.43 Various stimuli, such as increased intraocular pressure in models of glaucoma, hypoxia in models of brain ischemia, and tumor necrosis factor (TNF)-α in various retinal disease models, have been shown to induce EDN1 release from astrocytes, and this has been implicated in astrogliosis.44 Studies have shown that endothelins are up-regulated in injured or diseased photoreceptors. Light-induced degeneration of photoreceptors leads to increases in EDN1, EDNRA, and EDNRB in the retinal pigment epithelium and astrocytes.45 In a mouse model of retinal photoreceptor degeneration, expression of EDN2, along with other stress response genes, was highly increased.46 In addition, studies in models of photoreceptor disease/injury have shown that EDN2 transcripts are increased in photoreceptors. It serves as a general stress signal and activates EDNRB on neighboring Müller cells, which increase EDNRB expression in response to increased EDN2 levels.21 In our studies, we also observed a dramatic increase in EDN2 transcripts and a modest increase in EDNRB transcript levels in OIR conditions.
Rattner et al22 recently showed that EDN2 acting on EDNRA, but not EDNRB, on retinal neurons and/or glia induces hypoxia and production of one or more factors that arrest migration of the developing vasculature. Their studies suggest that paracrine or autocrine signaling of endothelins must limit their diffusion and that local action of endothelins must take place in the close proximity with retinal neurons and ECs. We are showing that transcripts of EDN2 are highly increased in OIR and expression of EDN2 is localized to retinal glia and hypertrophic blood vessels or NV tufts. From previous studies described above and on the basis of our data, it can be projected that EDN2 is produced by retinal glia and ECs in NV tufts within the hypoxic regions of the retina and activates signaling events in these cells. The same cells then may produce factor(s) that are ultimately responsible for pathological angiogenesis during OIR. In addition, the presence of EDN2 in the tufts implies that EDN2 may directly regulate formation of abnormal vessels, inhibit physiological angiogenesis, and play a causal role in pathological angiogenesis.
To further confirm the localization of EDN2, we showed through cell culture that hypoxia increases EDN2 mRNA in primary cortical astrocytes from mice. We measured mRNA expression of EDN1, EDNRA, and EDNRB in cultured astrocytes and found no significant differences between normoxic and hypoxic astrocytes. Although EDN1 mRNA levels did not change, it is possible that EDN1 protein released into the media is increased. A previous study has shown that TNF-α–mediated EDN1 synthesis and release from human optic nerve head astrocytes are augmented after hypoxia.47 However, there was no difference in basal release of EDN1 in control cells exposed to normoxia or hypoxia. This study suggests that TNF-α is required to enhance release of EDN1 from astrocytes. Future studies are needed to investigate the direct or indirect contributions by EDN1 or EDN2 in retinal glia cells.
Our studies also revealed that hypoxia increases VEGFA mRNA in astrocytes. In addition, EDN2 stimulation modestly increased VEGFA protein expression in astrocytes. Thus, it is likely that EDN2 produced by hypoxic stimulation of astrocytes contributes to the hypoxia-induced increase in VEGFA mRNA. However, EDN2 is only one of the many factors that could directly or indirectly influence VEGFA production during hypoxia. Moreover, in our studies, the 24-hour delay in onset of the VEGFA protein increase after EDN2 treatment strongly suggests an indirect effect involving activation of other factors. Therefore, the involvement of EDNRA activation in hypoxia-induced VEGFA production could be minimal or small. There are other potential sources of VEGFA in the retina in addition to astrocytes, including the Müller cells48 and ganglion cells.49,50 Altogether, it is possible that endothelins indirectly control VEGFA expression through an unknown factor and, therefore, we observed secondary effects of endothelins in our in vitro and in vivo studies. In our in vivo studies, EDNRA blockade effects on VEGFA may be indirect due to enhanced vascular repair and recovery of normal vascular function, which would reduce relative hypoxia and normalize VEGFA levels.
More important, our analysis showed that EDN2 mRNA and protein were highly increased during the ischemic phase of OIR. This strengthens support for EDN2-induced activation of EDNRA. Nevertheless, EDN1 mRNA expression was also increased, and the blockers we used do not discriminate between EDN1 and EDN2. Thus, we do not discount the role of EDN1-induced activation of EDNRA. On the basis of the literature, EDN1 and EDN2 have nearly the same affinity for EDNRA,35 suggesting that activation of EDNRA by EDN1 or EDN2 may contribute to the observed pathological effects.
The mechanisms by which EDN2 modulates pathological angiogenesis in OIR are not yet clear, but appear to involve regulation of the angiogenic mediators. Previous study indicates that overexpression of EDN2 leads to increased production of VEGFA, ANGPT2, DLL4, and APLN transcripts in the superficial layer of the developing retina via activation of EDNRA. Consequently, this leads to inhibition of physiological angiogenesis, localized increases in vascular sprouting, and overgrowth of hypoxic, misguided, and nonperfused vessels.22 Such vessels are morphologically similar to NV tufts observed during pathological angiogenesis. We find that in a disease model, ischemia-induced endothelin system activation via EDNRA leads to increased production of VEGFA protein and ANGPT2, DLL4, APLN, and CCL2 transcripts. These changes are correlated with increased pathological NV, due to localized overgrowth of immature and misdirected vessels, and decreased physiological angiogenesis, due to reduced vessel sprouting into the zone of vascular repair. Studies suggest that the above alterations can be attributed to disruption of the balance of signaling molecules, such as DLL4, APLN, and ANGPT2. DLL4 is induced by VEGFA as a negative regulator of vascular sprouting. Attenuation of its signaling during OIR enhances angiogenic sprouting, promotes rapid regrowth of vessels into the hypoxic regions, and reduces pathological NV.11 In addition, it has been shown that APLN knockdown during OIR prevents NV and promotes maturation of ECs by recruiting pericytes.10 ANGPT2 sensitizes retinal vessels to VEGFA and causes NV.51 Moreover, ANGPT2 is expressed in NV tufts under hypoxia, and its heterozygous deletion abrogates pathological NV during OIR.52 Therefore, it can be inferred that OIR-induced VEGFA up-regulates DLL4, which leads to inhibition of angiogenic sprouting. In addition, OIR-induced APLN and ANGPT2 are responsible for pathological NV and misdirected vascular growth. Our studies show that blockade of the EDNRA-normalized VEGFA protein, along with DLL4, APLN, and ANGPT2 transcripts, enhanced angiogenic sprouting and reduced NV. Collectively, our studies illustrate that critical signaling events that govern vessel maturation and physiological angiogenesis were restored through blockade of EDNRA activation. This further supports our hypothesis that the endothelin system plays a critical role in pathological angiogenesis in OIR by modulating production of angiogenic mediators.
Our study shows that endothelin signaling contributes to pathological angiogenesis. This work has taken the next step in identifying EDN2 as a target and a player that has an influence on angiogenic mediators, such as VEGFA, DLL4, and APLN, during OIR. It also builds on previous findings and sets forth a new concept that key factors upstream and downstream of VEGFA play a crucial role in determining the outcome of angiogenesis. We can conclude from our data that in ischemic retinopathies, endothelins, predominantly via activation of EDNRA, influence VEGFA production, which increases DLL4, ANGPT2, and APLN expression and inhibits directional vessel sprouting. These actions further increase tissue hypoxia, leading to formation of NV tufts and inhibition of physiological angiogenesis. To better understand the mechanism of actions of endothelins, future studies should investigate the direct role of EDN1 or EDN2 using tissue-specific expression of endothelins and their receptors in models of ischemic retinopathies.
Acknowledgments
C.P. designed and performed the experiments and wrote the manuscript; S.P.N. helped design experiments and edited the manuscript; W.Z. mentored and helped design experiments; Z.X. assisted with animal studies; S.S.-R. and K.M.D. provided astrocytes; R.W.C. helped design experiments and edited the manuscript; R.B.C. mentored and helped design experiments and edited the manuscript.
Footnotes
Supported by NIH grants R01-EY11766 and R01-EY04618, the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development grant BX001233, American Heart Association grant 11SDG744008, and the Culver Vision Discovery Institute at Georgia Regents University.
The contents do not represent the views of the Department of Veterans Affairs or the US government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosures: None declared.
References
- 1.Sapieha P., Joyal J.S., Rivera J.C., Kermorvant-Duchemin E., Sennlaub F., Hardy P., Lachapelle P., Chemtob S. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J Clin Invest. 2010;120:3022–3032. doi: 10.1172/JCI42142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith L.E. Pathogenesis of retinopathy of prematurity. Acta Paediatr Suppl. 2002;91:26–28. doi: 10.1111/j.1651-2227.2002.tb00157.x. [DOI] [PubMed] [Google Scholar]
- 3.Salvin J.H., Lehman S.S., Jin J., Hendricks D.H. Update on retinopathy of prematurity: treatment options and outcomes. Curr Opin Ophthalmol. 2010;21:329–334. doi: 10.1097/ICU.0b013e32833cd40b. [DOI] [PubMed] [Google Scholar]
- 4.Kermorvant-Duchemin E., Sapieha P., Sirinyan M., Beauchamp M., Checchin D., Hardy P., Sennlaub F., Lachapelle P., Chemtob S. Understanding ischemic retinopathies: emerging concepts from oxygen-induced retinopathy. Doc Ophthalmol. 2010;120:51–60. doi: 10.1007/s10633-009-9201-x. [DOI] [PubMed] [Google Scholar]
- 5.Smith L.E. Through the eyes of a child: understanding retinopathy through ROP the Friedenwald lecture. Invest Ophthalmol Vis Sci. 2008;49:5177–5182. doi: 10.1167/iovs.08-2584. [DOI] [PubMed] [Google Scholar]
- 6.Stahl A., Connor K.M., Sapieha P., Chen J., Dennison R.J., Krah N.M., Seaward M.R., Willett K.L., Aderman C.M., Guerin K.I., Hua J., Lofqvist C., Hellstrom A., Smith L.E. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813–2826. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan C.K., Pham L.N., Zhou J., Spee C., Ryan S.J., Hinton D.R. Differential expression of pro- and antiangiogenic factors in mouse strain-dependent hypoxia-induced retinal neovascularization. Lab Invest. 2005;85:721–733. doi: 10.1038/labinvest.3700277. [DOI] [PubMed] [Google Scholar]
- 8.Gerhardt H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 2008;4:241–246. doi: 10.4161/org.4.4.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsauer M., D'Amore P.A. Getting Tie(2)d up in angiogenesis. J Clin Invest. 2002;110:1615–1617. doi: 10.1172/JCI17326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasai A., Ishimaru Y., Higashino K., Kobayashi K., Yamamuro A., Yoshioka Y., Maeda S. Inhibition of apelin expression switches endothelial cells from proliferative to mature state in pathological retinal angiogenesis. Angiogenesis. 2013;16:723–734. doi: 10.1007/s10456-013-9349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lobov I.B., Renard R.A., Papadopoulos N., Gale N.W., Thurston G., Yancopoulos G.D., Wiegand S.J. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A. 2007;104:3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubanyi G.M., Polokoff M.A. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- 13.Masaki T. Endothelins: homeostatic and compensatory actions in the circulatory and endocrine systems. Endocr Rev. 1993;14:256–268. doi: 10.1210/edrv-14-3-256. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K., Jones P.M., Kanse S.M., Lam H.C., Spokes R.A., Ghatei M.A., Bloom S.R. Endothelin in the gastrointestinal tract: presence of endothelinlike immunoreactivity, endothelin-1 messenger RNA, endothelin receptors, and pharmacological effect. Gastroenterology. 1990;99:1660–1667. doi: 10.1016/0016-5085(90)90472-d. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K., Ghatei M.A., Jones P.M., Murphy J.K., Lam H.C., O'Halloran D.J., Bloom S.R. Endothelin in human brain and pituitary gland: presence of immunoreactive endothelin, endothelin messenger ribonucleic acid, and endothelin receptors. J Clin Endocrinol Metab. 1991;72:693–699. doi: 10.1210/jcem-72-3-693. [DOI] [PubMed] [Google Scholar]
- 16.Lam H.C., Lee J.K., Lu C.C., Chu C.H., Chuang M.J., Wang M.C. Role of endothelin in diabetic retinopathy. Curr Vasc Pharmacol. 2003;1:243–250. doi: 10.2174/1570161033476600. [DOI] [PubMed] [Google Scholar]
- 17.Grimshaw M.J., Naylor S., Balkwill F.R. Endothelin-2 is a hypoxia-induced autocrine survival factor for breast tumor cells. Mol Cancer Ther. 2002;1:1273–1281. [PubMed] [Google Scholar]
- 18.Jiang M.H., Hoog A., Ma K.C., Nie X.J., Olsson Y., Zhang W.W. Endothelin-1-like immunoreactivity is expressed in human reactive astrocytes. Neuroreport. 1993;4:935–937. doi: 10.1097/00001756-199307000-00024. [DOI] [PubMed] [Google Scholar]
- 19.Nie X.J., Olsson Y. Endothelin peptides in brain diseases. Rev Neurosci. 1996;7:177–186. doi: 10.1515/revneuro.1996.7.3.177. [DOI] [PubMed] [Google Scholar]
- 20.Chakrabarti S., Gan X.T., Merry A., Karmazyn M., Sima A.A. Augmented retinal endothelin-1, endothelin-3, endothelinA and endothelinB gene expression in chronic diabetes. Curr Eye Res. 1998;17:301–307. doi: 10.1076/ceyr.17.3.301.5216. [DOI] [PubMed] [Google Scholar]
- 21.Rattner A., Nathans J. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J Neurosci. 2005;25:4540–4549. doi: 10.1523/JNEUROSCI.0492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rattner A., Yu H., Williams J., Smallwood P.M., Nathans J. Endothelin-2 signaling in the neural retina promotes the endothelial tip cell state and inhibits angiogenesis. Proc Natl Acad Sci U S A. 2013;110:E3830–E3839. doi: 10.1073/pnas.1315509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izumi N., Nagaoka T., Sato E., Sogawa K., Kagokawa H., Takahashi A., Kawahara A., Yoshida A. Role of nitric oxide in regulation of retinal blood flow in response to hyperoxia in cats. Invest Ophthalmol Vis Sci. 2008;49:4595–4603. doi: 10.1167/iovs.07-1667. [DOI] [PubMed] [Google Scholar]
- 24.Nakabayashi S., Nagaoka T., Tani T., Sogawa K., Hein T.W., Kuo L., Yoshida A. Retinal arteriolar responses to acute severe elevation in systemic blood pressure in cats: role of endothelium-derived factors. Exp Eye Res. 2012;103:63–70. doi: 10.1016/j.exer.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Peter M.G., Davenport A.P. Characterization of the endothelin receptor selective agonist, BQ3020 and antagonists BQ123, FR139317, BQ788, 50235, Ro462005 and bosentan in the heart. Br J Pharmacol. 1996;117:455–462. doi: 10.1111/j.1476-5381.1996.tb15212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goddard J., Johnston N.R., Hand M.F., Cumming A.D., Rabelink T.J., Rankin A.J., Webb D.J. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation. 2004;109:1186–1193. doi: 10.1161/01.CIR.0000118499.69469.51. [DOI] [PubMed] [Google Scholar]
- 27.Laird M.D., Wakade C., Alleyne C.H., Jr., Dhandapani K.M. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Tang Y.C., Liu C.W., Chang H.H., Juan C.C., Kuo Y.C., Kao C.C., Huang Y.M., Kao Y.H. Endothelin-1 stimulates resistin gene expression. Endocrinology. 2014;155:854–864. doi: 10.1210/en.2013-1847. [DOI] [PubMed] [Google Scholar]
- 29.Bartoli M., Al-Shabrawey M., Labazi M., Behzadian M.A., Istanboli M., El-Remessy A.B., Caldwell R.W., Marcus D.M., Caldwell R.B. HMG-CoA reductase inhibitors (statin) prevents retinal neovascularization in a model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2009;50:4934–4940. doi: 10.1167/iovs.08-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vessey K.A., Wilkinson-Berka J.L., Fletcher E.L. Characterization of retinal function and glial cell response in a mouse model of oxygen-induced retinopathy. J Comp Neurol. 2011;519:506–527. doi: 10.1002/cne.22530. [DOI] [PubMed] [Google Scholar]
- 31.Narayanan S.P., Suwanpradid J., Saul A., Xu Z., Still A., Caldwell R.W., Caldwell R.B. Arginase 2 deletion reduces neuro-glial injury and improves retinal function in a model of retinopathy of prematurity. PLoS One. 2011;6:e22460. doi: 10.1371/journal.pone.0022460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishijima K., Ng Y.S., Zhong L., Bradley J., Schubert W., Jo N., Akita J., Samuelsson S.J., Robinson G.S., Adamis A.P., Shima D.T. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fineberg D., Jandeleit-Dahm K.A., Cooper M.E. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol. 2013;9:713–723. doi: 10.1038/nrendo.2013.184. [DOI] [PubMed] [Google Scholar]
- 34.Motawi T.K., Rizk S.M., Ibrahim I.A., El-Emady Y.F. Alterations in circulating angiogenic and anti-angiogenic factors in type 2 diabetic patients with neuropathy. Cell Biochem Funct. 2014;32:155–163. doi: 10.1002/cbf.2987. [DOI] [PubMed] [Google Scholar]
- 35.Kohan D.E. Role of collecting duct endothelin in control of renal function and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2013;305:R659–R668. doi: 10.1152/ajpregu.00345.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson J., Bagnato A., Battistini B., Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer. 2003;3:110–116. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- 37.Murdoch C., Giannoudis A., Lewis C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 38.Rosano L., Spinella F., Bagnato A. The importance of endothelin axis in initiation, progression, and therapy of ovarian cancer. Am J Physiol Regul Integr Comp Physiol. 2010;299:R395–R404. doi: 10.1152/ajpregu.00304.2010. [DOI] [PubMed] [Google Scholar]
- 39.Hein T.W., Ren Y., Yuan Z., Xu W., Somvanshi S., Nagaoka T., Yoshida A., Kuo L. Functional and molecular characterization of the endothelin system in retinal arterioles. Invest Ophthalmol Vis Sci. 2009;50:3329–3336. doi: 10.1167/iovs.08-3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Battistini B., Chailler P., D'Orleans-Juste P., Briere N., Sirois P. Growth regulatory properties of endothelins. Peptides. 1993;14:385–399. doi: 10.1016/0196-9781(93)90057-n. [DOI] [PubMed] [Google Scholar]
- 41.Bagnato A., Salani D., Di Castro V., Wu-Wong J.R., Tecce R., Nicotra M.R., Venuti A., Natali P.G. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res. 1999;59:720–727. [PubMed] [Google Scholar]
- 42.Salani D., Taraboletti G., Rosano L., Di Castro V., Borsotti P., Giavazzi R., Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000;157:1703–1711. doi: 10.1016/S0002-9440(10)64807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ripodas A., de Juan J.A., Roldan-Pallares M., Bernal R., Moya J., Chao M., Lopez A., Fernandez-Cruz A., Fernandez-Durango R. Localisation of endothelin-1 mRNA expression and immunoreactivity in the retina and optic nerve from human and porcine eye: evidence for endothelin-1 expression in astrocytes. Brain Res. 2001;912:137–143. doi: 10.1016/s0006-8993(01)02731-7. [DOI] [PubMed] [Google Scholar]
- 44.Prasanna G., Krishnamoorthy R., Yorio T. Endothelin, astrocytes and glaucoma. Exp Eye Res. 2011;93:170–177. doi: 10.1016/j.exer.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torbidoni V., Iribarne M., Ogawa L., Prasanna G., Suburo A.M. Endothelin-1 and endothelin receptors in light-induced retinal degeneration. Exp Eye Res. 2005;81:265–275. doi: 10.1016/j.exer.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 46.Swiderski R.E., Nishimura D.Y., Mullins R.F., Olvera M.A., Ross J.L., Huang J., Stone E.M., Sheffield V.C. Gene expression analysis of photoreceptor cell loss in bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage. Invest Ophthalmol Vis Sci. 2007;48:3329–3340. doi: 10.1167/iovs.06-1477. [DOI] [PubMed] [Google Scholar]
- 47.Desai D., He S., Yorio T., Krishnamoorthy R.R., Prasanna G. Hypoxia augments TNF-alpha-mediated endothelin-1 release and cell proliferation in human optic nerve head astrocytes. Biochem Biophys Res Commun. 2004;318:642–648. doi: 10.1016/j.bbrc.2004.04.073. [DOI] [PubMed] [Google Scholar]
- 48.Pierce E.A., Avery R.L., Foley E.D., Aiello L.P., Smith L.E. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A. 1995;92:905–909. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stone J., Chan-Ling T., Pe'er J., Itin A., Gnessin H., Keshet E. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1996;37:290–299. [PubMed] [Google Scholar]
- 50.Sapieha P., Sirinyan M., Hamel D., Zaniolo K., Joyal J.S., Cho J.H., Honore J.C., Kermorvant-Duchemin E., Varma D.R., Tremblay S., Leduc M., Rihakova L., Hardy P., Klein W.H., Mu X., Mamer O., Lachapelle P., Di Polo A., Beausejour C., Andelfinger G., Mitchell G., Sennlaub F., Chemtob S. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med. 2008;14:1067–1076. doi: 10.1038/nm.1873. [DOI] [PubMed] [Google Scholar]
- 51.Oshima Y., Deering T., Oshima S., Nambu H., Reddy P.S., Kaleko M., Connelly S., Hackett S.F., Campochiaro P.A. Angiopoietin-2 enhances retinal vessel sensitivity to vascular endothelial growth factor. J Cell Physiol. 2004;199:412–417. doi: 10.1002/jcp.10442. [DOI] [PubMed] [Google Scholar]
- 52.Feng Y., Wang Y., Pfister F., Hillebrands J.L., Deutsch U., Hammes H.P. Decreased hypoxia-induced neovascularization in angiopoietin-2 heterozygous knockout mouse through reduced MMP activity. Cell Physiol Biochem. 2009;23:277–284. doi: 10.1159/000218174. [DOI] [PubMed] [Google Scholar]