

Abstract
Background
The emergence of precision medicine allowed the incorporation of individual molecular data into patient care. Indeed, DNA sequencing predicts somatic mutations in individual patients. However, these genetic features overlook dynamic epigenetic and phenotypic response to therapy. Meanwhile, accurate personal transcriptome interpretation remains an unmet challenge. Further, N-of-1 (single-subject) efficacy trials are increasingly pursued, but are underpowered for molecular marker discovery.
Method
‘N-of-1-pathways’ is a global framework relying on three principles: (i) the statistical universe is a single patient; (ii) significance is derived from geneset/biomodules powered by paired samples from the same patient; and (iii) similarity between genesets/biomodules assesses commonality and differences, within-study and cross-studies. Thus, patient gene-level profiles are transformed into deregulated pathways. From RNA-Seq of 55 lung adenocarcinoma patients, N-of-1-pathways predicts the deregulated pathways of each patient.
Results
Cross-patient N-of-1-pathways obtains comparable results with conventional genesets enrichment analysis (GSEA) and differentially expressed gene (DEG) enrichment, validated in three external evaluations. Moreover, heatmap and star plots highlight both individual and shared mechanisms ranging from molecular to organ-systems levels (eg, DNA repair, signaling, immune response). Patients were ranked based on the similarity of their deregulated mechanisms to those of an independent gold standard, generating unsupervised clusters of diametric extreme survival phenotypes (p=0.03).
Conclusions
The N-of-1-pathways framework provides a robust statistical and relevant biological interpretation of individual disease-free survival that is often overlooked in conventional cross-patient studies. It enables mechanism-level classifiers with smaller cohorts as well as N-of-1 studies.
Software
http://lussierlab.org/publications/N-of-1-pathways
Keywords: N-of-1, Single Subject Design, Precision Medicine, Personalized Medicine, Personal Transcriptome, Geneset
Introduction
The adoption of precision medicine is regarded as one of the most significant changes in healthcare due to its ability to dramatically improve diagnosis, prognosis, and patient treatment procedures. While DNA polymorphisms can be ascertained as private variants1 by using a reference genome, individualized interpretation of the epigenome, transcriptome, and proteome remains challenging. Since purely DNA sequence-based associations to diseases, such as those found in genome-wide association studies (GWAS),2 are generally insufficient to unveil the biological underpinning mechanisms,3 4 it is necessary for gene expression and transcriptomic profiling to bridge this mechanistic gap.5–7 Further, 99% of individual molecular biomarkers derived from large patient sample predictors fail to be reproducible.8 Even though the simplicity of a single marker is the correct paradigm for Mendelian diseases, it fails in complex phenotypes. Indeed, different proteins jointly participating in a mechanism (eg, pathway) may alternately be deregulated in different individual patients, yet contribute similarly to the disease pathophysiology. A combination of modestly deregulated molecules can lead to similar phenotypes, which suggests that more complex models of bimolecular expression are required than the conventional single gene/protein marker paradigm.
In the absence of individual interpretation of the ’omics scales, clinical trials must be designed over cohort-level features (case and control populations); however, patients with a similar clinical history and environmental background will respond differentially to an identical therapy. We propose that individual transcriptome interpretation will enable stratification of clinical trial populations or better, novel clinical trial designs.
Single-subject designs, also known as N-of-1 clinical trials, were first introduced by RA Fisher in 1935.9 This type of studies aim to extract information from the pattern of variation of one or several observed variables over time, derived from a single sample (patient, cell, etc).10 Despite their long existence, N-of-1 trials rely on time series analyses (≥3 patients) and remain underpowered for genomics studies. The advent of the increased dynamic range and accuracy of RNA-sequencing over expression arrays11 12 provides an unparalleled opportunity for studying single subject transcriptomes.13 While molecular biomarker discovery in N-of-1 studies may appear unfeasible, we and others have recently shown that highly reproducible multi-gene signatures can be directly calculated using mechanism-associated genesets leveraged from transcriptomes based on expression array14–17 or RNA-Seq technologies.18 Moreover, these geneset classifiers outperform gene-level classifiers and provide biological context,19 20 in addition to computing geneset scores on each sample. However, these approaches require at least three samples and two groups to generate p values.
N-of-1-pathways aims at uncovering deregulated mechanisms at the single patient level, and highlighting both the individuality and commonality of a patient trait. The approach consists of (i) identifying deregulated mechanisms in a patient sample(s) by pooling genes in genesets, (ii) comparing deregulated mechanisms between patients using information theory similarity, and (iii) patient-level visualization or representation of deregulated mechanisms for clinical interpretation (eg, diagnosis, prognosis, and therapeutics). The first N-of-1-pathways component (i) consists of selecting the most appropriate statistical analysis given the patient samples (paired, time series, etc), with the fundamental principle that a single patient is the statistical universe where genesets are sampled. We quantitatively and qualitatively evaluated the pathways revealed in individual patients and show comparable accuracies with conventional methods at the cohort level.
The novelty of our approach consists of the reformulation of the problem to identify deregulated mechanisms across patients (‘population-based’ or ‘cohort-based’) into a statistic pertaining to each individual patient. We hypothesize that we can transfer these previously described geneset-level methodologies in paired sample analyses, thus enabling N-of-1 trials with two paired samples. To our knowledge, very few methodologies exist at the level of a single patient that are able to provide geneset-level information. We reference the FAIME methodology15 that we developed in 2012 and ssGSEA (single sample GSEA),21 a variant of genesets enrichment analysis (GSEA)22 designed for single sample analyses, which both transform gene-level expression data into pathway-level scores. However, the transformations to geneset-level expressions require additional analyses and statistical interpretation, while the proposed N-of-1-pathways framework provides directly interpretable results.
Altogether, the need for precision medicine and novel N-of-1 trial designs substantiate the development of robust analytic methods that are individual-centered with the patient as the statistical universe (both the control-subject and the case-subject). This consideration enables current advances in genomic technologies to provide individualized biological interpretations and precise actionable deliverables.
We conducted these studies using RNA-Seq-based transcriptomes of patients with lung adenocarcinoma published in The Cancer Genome Atlas (TCGA). The classification of this type of cancer remains a challenge as less than 20% of patients with stages III and above survive more than 5 years.23
Methods
Dataset and preprocessing
Four datasets dedicated to lung adenocarcinoma were used: one exploration dataset and three external validation datasets (table 1). The exploration dataset consisted of normalized RSEM (RNA-Seq by Expectation Maximization) gene expression profiles for 55 paired uninvolved and tumoral lung samples (downloaded on March 15, 2013). All measurements were log2 transformed. If several alternative transcripts referring to the same HGNC gene name were present, only that with maximum expression was considered for further analysis. In our efforts to minimally transform or bias the data, we processed all the mRNAs transcripts regardless of their expression levels. In other words, while mRNAs with low expression levels are routinely filtered out in expression analysis pipelines,24 our proposed method is inclusive of all mRNA expression. The three external validation datasets25–27 were derived from microarray expression profiles (table 1), and only reported deregulated pathways or differentially expressed gene lists were used in our studies.
Table 1.
Dataset description
| Dataset | Exploration study | External validation | ||
|---|---|---|---|---|
| Study I* | Study II | Study III | ||
| Authors | NA | Yap et al25 | Xi et al26 | Kim et al27 |
| Source | TCGA | Nucleic Acids Res | Nucleic Acids Res | Nat Commun |
| Gene expression profile | ||||
| Date | Download March 2013 | 2005 | 2008 | 2013 |
| Type | RNA-Seq | Microarray | Microarray | Beadchip |
| Platform | Illumina RNA-Seq V.2 | Affymetrix HG-U133A | HuEx-1_0-st | Illumina human-6 V.2.0 |
| Genes measured | 20 502 | 22 283 | 17 800 | NA |
| Deregulated genes | NA | 3442† | 2369 | 804 |
| Provided GO terms | NA | 67 | NA | NA |
| Patients | ||||
| Total | 110 | 58 | 40 | 184 |
| Normal | 55‡ | 9 | 20‡ | 92‡ |
| Tumor | 55‡ | 49 | 20‡ | 92‡ |
| Men | 22 (40%) | 32 (55.2%)§ | 14 (35%) | NA |
| Women | 32 (58.2%) | 17 (29.3%)§ | 22 (55%) | NA |
| Age | ||||
| Median | 66 | 61 | 69 | NA |
| Range | 42–86 | 38–81 | 42–86 | NA |
| Disease stage | ||||
| I | 28 (50.9%) | 25 (43.1%) | 23 (57.5%) | NA |
| II | 14 (25.5%) | 8 (13.8%) | 5 (12.5%) | NA |
| III | 11 (20%) | 14 (24.1%) | 7 (17.5%) | NA |
| IV | 1 (1.8%) | 2 (3.4%) | 1 (2.5%) | NA |
*Only reported deregulated pathways were used as part of the lung adenocarcinoma signature for external validation.
†and NA indicate data not available.
‡Indicates paired samples derived from lung tumor tissues with matched normal lung tissues.
§Indicates significantly different from the exploration study (FET p≤0.05).
FET, Fisher's exact test; GO, Gene Ontology; TCGA, The Cancer Genome Atlas.
Gene Ontology annotations of biological processes
We aggregated genes into pathway-level mechanisms using Gene Ontology annotations of Biological Processes (GO-BP)28 29 (see online supplementary methods). These GO annotations were used for four types of GO enrichment analyses: GSEA,30 differentially expressed gene (DEG) enrichment, ssGSEA,21 and N-of-1-pathways analyses.
N-of-1-pathways framework
Within-patient analyses
N-of-1-pathways was performed on the exploration dataset independently for each patient, using gene expression profiles from two paired samples: uninvolved and tumoral lung tissues, in the context of lung adenocarcinoma. The proposed statistical analysis method consisted of a non-parametric paired Wilcoxon test (Wilcoxon signed-rank test) performed within each patient on the paired gene expression profiles restricted to a given pathway. Wilcoxon statistics, W+ and W−, provided a direction of deregulation for each geneset, as overall ‘up-regulated’ or ‘down-regulated’, respectively. Both false discovery rate (FDR) and Bonferroni corrections were applied to adjust p values for multiple comparisons. In each paired sample, only deregulated pathways with adjusted p values with FDR ≤5% and Bonferroni ≤1% were retained for further analysis. These p values are transformed into z scores using an inverse standard normal distribution (z score=qnorm(abs(p value/2) in R) for ulterior analyses (eg, heatmaps).
Cross-patient analyses
Each GO-BP mechanism had an associated FDR for each patient. The GO-BP terms were then ranked according to the total number of patients sharing a given GO term that reached significance at FDR ≤5%. The prioritized GO-BP terms were listed from the most commonly to the least observed in lung adenocarcinoma patients, yet significant in at least one patient. The N-of-1-pathways statistical analysis component is available in R and Java at http://Lussierlab.org/publications/N-of-1-pathways.
Theoretical results: validation using synthetic data
A synthetic geneset that contains a percentage of concordant deregulated genes was generated using the exploration dataset. Each point of figure 1 represents one geneset size of a simulated pathway varying from 15 to 500 genes by increments of 5 and generated by randomly selecting ‘n’ genes among the 20 502 reported in the exploration study. Further, a proportion of genes ‘r’ (ratio represented in %) involved in this synthetic geneset was considered deregulated. The expression of genes of the normal sample included in that ratio r was then artificially increased by a twofold change and assigned to the tumoral sample. This ratio was varied by 5% to 100% with increments of 5%. The null hypothesis was stated as H0: ‘The geneset is not deregulated.’ For each pair (n, r), we applied the N-of-1-pathways statistical analysis component 1000 times in order to estimate the false negative rate (type II error β). This resampling was repeated for 1960 combinations of n and r. The type II errors reported in figure 1 were computed as the number of times the truly deregulated pathway of the simulation is not found deregulated (false negative) divided by 1000 (1 960 000 calculations of N-of-1-pathways calculated using the 150 teraflops, 18 000-core Beagle Cray XE6 supercomputer of the Computation Institute located at the Argonne National Laboratory). Since figure 1 focuses on cataloging the type II error according to one specifically sized pathway at a time, we considered a pathway significantly found deregulated by N-of-1-pathways when the unadjusted p value was ≤0.05.
Figure 1.
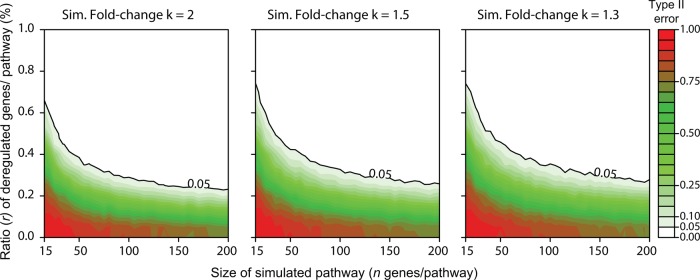
Synthetic data: evaluation of the size and ratio of concordant deregulated genes within a pathway required to be found deregulated in the N-of-1-pathways statistical analysis component. Each point represents one size of a simulated pathway generated by randomly selecting n genes and a ratio r of the deregulated genes within the pathway. The ratio r is artificially increased by a k-fold change in a simulated pathway seeded in the exploration dataset (k∈{1.3, 1.5, 2}). We then applied the N-of-1-pathways statistical analysis component to verify if the simulated pathway was found deregulated with a significance threshold (type I error) chosen as an unadjusted p value of ≤0.05. For each value (n, r, k), we repeated this procedure 1000 times in order to estimate the false negative rate (type II error β). Sim., simulated (see the ‘Theoretical results: validation using synthetic data’ section in the Methods).
Proxy gold standard for the internal and external validations
Since a gold standard (GS) for lung adenocarcinoma does not exist, we generated proxy GSs15 17 18 in order to objectively assess the accuracy of the significantly deregulated mechanisms identified by N-of-1-pathways (see online supplementary methods and figures 2–5).
Figure 2.
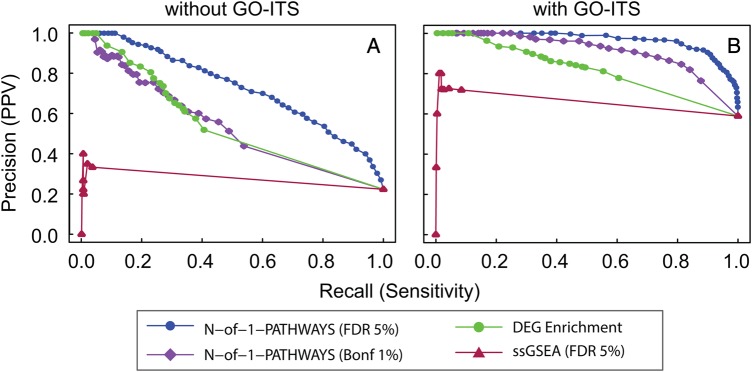
Concordant deregulated pathways (genesets) uncovered by N-of-1-pathways, ssGSEA, differentially expressed gene (DEG) enrichment, and genesets enrichment analysis (GSEA) methods within the exploration dataset (internal validation). To evaluate the Gene Ontology annotations of Biological Process (GO-BP) associated terms yielded by the N-of-1-pathways method, we compared these pathways to those found by a single sample method: ssGSEA, and two well-established cohort-based methods: differentially expressed gene (DEG) enrichment and GSEA. We then generated precision-recall curves based on the perfect GO overlap (A), and GO semantic similarity overlap (B; GO-Information Theoretic Similarity (GO-ITS) ≥0.7; see the ‘Information Theory Similarity’ section in the Methods). When GSEA is chosen as the proxy gold standard (see the ‘Proxy gold standard for the internal and external validations’ section in the Methods), the N-of-1-pathways method uncovered deregulated pathways comparable to, or better than, those of DEG enrichment analysis, with or without GO-ITS analysis, respectively. When DEG enrichment is chosen as the proxy gold standard, N-of-1-pathways performed marginally better than GSEA (see online supplementary figure S2). Bonf, Bonferroni; FDR, false discovery rate; PPV, positive predictive value.
Figure 3.

Concordant deregulated pathways between exploration and external validation studies. To establish that N-of-1-pathways yielded relevant Gene Ontology (GO) terms associated pathways, we compared the deregulated pathways to two conventional enrichment methods in three independent lung adenocarcinoma studies (see Methods and table 1). In (A), the Venn diagram corresponds to the overlap of deregulated GO annotations of Biological Process (GO-BP) terms associated pathways between the three external validation studies I, II and III (deregulated pathways were directly retrieved from the ones published in study I, while they were enriched using DEG enrichment from the DE genes list published in studies II and III, at FDR≤5%). In (B–F), the unveiled pathways of the exploration set were compared to those discovered in the independent validation studies using either genesets enrichment analysis (GSEA) (C and E), DEG enrichment as a proxy gold standard (D and F), or pathways reported in study I (B). Bonf., Bonferroni; FDR, false discovery rate; PPV, positive predictive value.
Figure 4.

Unveiling the individuality and commonality of the deregulated mechanisms (exploration dataset). (A) Heatmap generated by the list of z scores for each patient (in rows) and each Gene Ontology annotation of Biological Process (GO-BP) term found significantly deregulated in at least one of the 55 patients (in columns). Below the heatmap, three subpanels show additional details for each GO-BP term: (i) the number of patient sharing the given pathway (patient count), (ii) the curated categorization of the GO-BP terms into 10 classes, and (iii) the similarity with the external gold standard (GS) (see the ‘Information theoretic similarity’ section in the Methods). (B) Kaplan–Meier survival curve of the three Partitioning Around Medoids (PAM) clusters derived from the GO-BP z score without clinical information (see ‘The Kaplan–Meier survival curve’ section in the Methods). When only the two most extreme clusters are considered, there is a statistically significant difference in survival (p=0.03), while the difference is just a trend (p=0.09) when the three clusters are considered together. (C, D) Clustering of distinct patients according to the two principal components (see the ‘Principal component analysis of individual GO-BPs’ section in the Methods) of individual GO-BP terms performed on the exploration set. Two diametric extreme survival phenotypes are annotated: ‘death of disease <1 yr’ (red) and ‘disease-free survival >5 yr’ (blue). PCA, principal component analysis. *t test, p<0.05.
Figure 5.

Comparison of the Gene Ontology annotations of Biological Process (GO-BP) terms predicted by N-of-1-pathways in the exploration dataset and their similarity to those of the external gold standard (combination of three validation studies). (A) Global comparison of the 55 patients’ results taken together. Overall, 92% of GO-BP terms of the external gold standard (GS) are found to be predicted by the N-of-1-pathways (0.7<ITS<1 and ITS=1). Conversely, 61% of the predicted and related GO-information theoretic similarity (GO-ITS) overlap with the GS (0.7<ITS<1 and ITS=1) and 10% of GO-BP terms remain unrelated (0.3<ITS) (see online supplementary tables S1 and S2 which provide a subset of results). (B) The level of similarity of individual deregulated mechanisms with the external GS (as a ratio) for each patient. The two diametric extreme prognosis phenotypes show significant differences in their shared GO-BP terms with the GS (in bold) (Wilcoxon test, p=0.03). The two diametric extreme survival phenotypes are annotated: ‘disease-free survival >5 years’ (blue) and ‘death of disease <1 yr’ (red) (figures 4C and 6C).
Information theoretic similarity
We calculated the similarity between GO-BP terms using Jiang's information theoretic similarity (GO-ITS)31 that ranges from 0 (no similarity) to 1 (perfect match) (figures 2–6).
Figure 6.

N-of-1-pathways representation (star plot) of individual Gene Ontology annotations of Biological Process (GO-BPs) of diametric extreme patients. Most significantly deregulated GO-BP terms were identified (Wilcoxon test, p<0.05; fold change >4) between the two groups of patients with diametric extreme phenotypes (death of disease in less than 1 year and at least 5 years of disease-free survival (red and blue dots in figure 4C); n=8 patients; figure 5B). The top 15 deregulated mechanisms between these groups were calculated using the N-of-1-pathways statistical analysis component in the exploration dataset. (A) Hierarchical clustering of the 15 GO terms using the GO-information theoretic similarity (GO-ITS) metric (see the Methods section). (B) Legend of the star plots, each edge corresponding to one GO-BP term. Each star reflects a single patient's deregulation of GO-BP terms (see the end of the first paragraph of the ‘N-of-1-pathways framework’ section in Methods). (C) Each extreme patient's own star plot representation of the 15 GO-BP terms. The green zone represents up-regulated pathways (given the N-of-1-pathways direction of deregulation), while the gray zone indicates down-regulation. The non-deregulated zone (z score=0) is represented by a dotted line dividing the two colored zones. We also applied the same representation framework to the GO-BP terms whose FAIME scores were significantly deregulated (online supplementary figure S3).
Heatmap
The heatmap drawn in figure 4A was computed using the default clustering parameters of the HeatPlus package in R (the Euclidean metric and complete aggregation method were used). z Scores were computed for each pathway of each individual patient (see the ‘N-of-1-pathways framework’ section in the Methods). These individual patient lists of p values (vectors) were then annotated side-by-side as a matrix for creating a heatmap of the 55 TCGA samples.
The Kaplan–Meier survival curve
The Kaplan–Meier survival curve (figure 4B) was computed using GraphPad Prism V.6.02 software using the survival data associated to the exploration dataset (table 1). Patients who died of non-cancer causes in the first 12 months were excluded (defined as ‘tumor-free’ or ‘unknown tumor recurrence status’; exploration dataset). Three clusters of GO-BP z scores were obtained using the Partitioning Around Medoids (PAM) clustering method (cluster package in R; see the ‘N-of-1-pathways framework’ section in the Methods).
Principal component analysis of individual GO-BP terms
Principal component analysis (PCA) of individual GO-BP terms (figure 4C, D) was carried out using FactoMineR (default parameter in R) on the exploration dataset matrix of z scores (see the ‘Heatmap’ section in the Methods).
Star plot visualization of individual patient pathways
Star plot visualization of individual patient pathways (figure 6, table 2, and online supplementary figure S3), also known as a spider or radar plot, was performed in R using the stars function in the default graphics package (figure 6C, and online supplementary figure S3C). Each patient had an individual star plot where each edge represented a particular GO-BP z score value (see the ‘N-of-1-pathways framework’ section in the Methods). In order to obtain a relevant representation of the star plot surface, the GO-BP terms were ordered according to their GO-ITS similarity and clustered using the hierarchical clustering hclust method in R. In figure 6A, each hierarchical cluster was manually curated by a biologist to a representative GO-BP category as shown in figure 6B and table 2.
Table 2.
GO-BP terms deregulated between diametric extreme survival phenotypes in the exploration dataset
| Curated classes | GO ID | GO description |
|---|---|---|
| Immune response – antigen presentation | GO:0002479 | Antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent |
| GO:0042590 | Antigen processing and presentation of exogenous peptide antigen via MHC class I | |
| GO:0072474 | Signal transduction involved in mitotic cell cycle G1/S checkpoint | |
| Immune response to type I interferon | GO:0034340 | Response to type I interferon |
| GO:0071357 | Cellular response to type I interferon | |
| GO:0060337 | Type I interferon-mediated signaling pathway | |
| Immune response | GO:0009615 | Response to virus |
| Cell division/cell cycle | GO:0000082 | G1/S transition of mitotic cell cycle |
| GO:0031329 | Regulation of cellular catabolic process | |
| GO:0051325 | Interphase | |
| GO:0000087 | M phase of mitotic cell cycle | |
| GO:0007050 | Cell cycle arrest | |
| GO:0010564 | Regulation of cell cycle process | |
| DNA repair and recombination | GO:0006281 | DNA repair |
| GO:0006310 | DNA recombination |
This table lists the GO-BP terms presented in figure 6 along with their curated classes and complete GO description.
GO-BP, Gene Ontology annotations of Biological Process.
Results
N-of-1-pathways successfully identifies deregulated pathways in a synthetic simulation
The N-of-1-pathways statistical analysis component is the validation of a non-parametric statistical based method, applied to single patient paired samples, rather than a larger multi-sample based patient cohort. It assumes that the RNA of a single patient is the population and the patient is the mathematical universe. Cross-patient studies tend to generalize commonly found pathways, whereas our unconventional approach straightforwardly delineates unique pathways in an individual patient. Figure 1 confirms the obvious: genesets containing fewer genes (n) require a larger proportion of deregulated genes (r) to be recognized as statistically significant by N-of-1-pathways. Moreover, N-of-1-pathways takes into account the concordance of deregulation of the genes in the studied pathway, and thus the results are robust given different fold changes. Of note, we compared N-of-1-pathways with a non-classic alternative: Fisher's exact test (FET) enrichment of genes at certain fold changes (see online supplementary figure S1). Results show that N-of-1-pathways obtains similar or better results, without requiring to specify a threshold for the fold change, while it is needed for the alternative.
Internal validation: N-of-1-pathways unveiled deregulated pathways comparable to those of conventional geneset-level methods
In order to assess the relevance of the genesets uncovered by N-of-1-pathways, we compared its results against those computed with conventional enrichment methods: GSEA and DEG enrichment within the exploratory datasets comparing uninvolved to tumoral lung tissues. We also compared the results with the ssGSEA method,21 as a single-sample alternative method (see ‘ssGSEA’ in the online supplementary methods). Because N-of-1-pathways and ssGSEA identify deregulated genesets for each patient individually, instead of a global estimation over a cohort, we grouped the results of the 55 patients together in order to make the comparison. We compared the union of the deregulated genesets over the 55 patients derived from N-of-1-pathways (905 for Bonferroni 1% cutoff and 2662 for FDR 5% cutoff) and ssGSEA (78 for FDR 5% cutoff) to the genesets of GSEA30 and DEG enrichment. Throughout the study, we used GO Biological Processes (GO-BP) terms as genes annotation to genesets (also referred as pathways). We found 1659 differentially expressed genes in the exploration set (480 down-regulated and 1179 up-regulated) that we further enriched into 65 GO-BP associated pathways using a FET (see ‘DEG enrichment’ in the online supplementary methods) at FDR ≤5%. Second, we identified a group of 725 GO-BP terms at FDR ≤5% using GSEA (see ‘GSEA’ in the online supplementary methods). In order to evaluate the effectiveness of the N-of-1-pathways statistical analysis component, we compared the pathways uncovered at the single patient level to those found by DEG enrichment using GSEA as a proxy GS (figure 2). We also used an information theory semantic similarity technique (see the ‘Information theoretic similarity’ section in the Methods) to relate the highly predicted GO-BP terms to those of the GS, using a cutoff we have previously derived as significant (GO-ITS ≥0.7; see the ‘Information theoretic similarity’ section in the Methods).
The results shown in figure 2 reveal that N-of-1-pathways outperforms ssGSEA in both recall and precision regardless of the chosen GS. Different ssGSEA cutoffs (FDR 25% and FDR 50%) are concordant (data not shown; N-of-1-pathways outperforms ssGSEA). The low accuracy of ssGSEA is unsurprising in these conditions as it was not designed for paired samples or for fold change as inputs (see ‘ssGSEA’ in the online supplementary methods). Results also show that when using GSEA as a proxy GS (figure 2A, B), N-of-1-pathways obtains more similar pathways to the proxy GS (see online supplementary table S4) than when using ssGSEA or the DEG enrichment method. When DEG enrichment serves as a proxy GS, N-of-1-pathways shows better precision and recall than either ssGSEA or GSEA (see online supplementary figure S2 and table S4; maximum precision ∼35%). The lower precision observed for both GSEA and N-of-1-pathways when DEG enrichment is used as a proxy GS is likely related to the lower number of GO-BP terms of the latter (over-conservative GS).
External validation: biological relevance of the deregulated mechanisms found in the context of lung adenocarcinoma
In order to further assess the accuracy of the pathways we found deregulated in the exploration dataset, we used the three independent lung adenocarcinoma studies described in table 1 to derive GSs (see the ‘Proxy gold standard for the internal and external validations’ section in the Methods). In figure 3, the GO-BP genesets found deregulated across patients in the exploration set were compared to those found deregulated in each of the three independent validation studies. We used a GO-ITS >0.7 semantic similarity threshold for these curves. We conducted GSEA and DEG enrichment methods on the exploration dataset using the GO-BP terms published in study I (figure 3B) and GO-BP terms enriched with both DEG enrichment and GSEA in studies II and III (figure 3C–F, respectively). When selecting GSEA results as the proxy GS, N-of-1-pathways (Bonferroni ≤1% and FDR ≤5%) predictions from the exploration set are comparable or even more similar to the proxy GS than those of GSEA and DEG enrichment. When DEG enrichment results are chosen as the proxy GS, N-of-1-pathways generated comparable precision and recall to those of GSEA. Further exploration shows that GSEA obtains higher precision and recall than those of DEG enrichment when the GS is derived from GSEA and vice versa when the GS is derived from DEG enrichment. Thus, it appears that pooled N-of-1-pathways predictions across patients are more related to those of GSEA.
In figure 3A, a modest GO-BP pathway overlap is shown between all three validation studies (see the Venn diagram in the online supplementary methods). When the overlap of two studies is compared, the commonality is modest to moderate. This observation concurs with the reports that lung adenocarcinoma is a very complex and heterogeneous disease.23 Further, these results also suggest that high heterogeneity and convergence of molecular mechanisms underlie both disease development and chemoresistance.32 These results support our initial hypothesis that patient-specific interpretations of deregulated pathways are required. However, the stronger overlap found between studies II and III suggests a confounded artifact: an increased overlap due to the enrichment method used. We confirmed this by comparing GSEA with DEG enrichment on the same datasets and across datasets (data not shown).
Paired sample analysis unveils individual and shared patient associated pathways
We assessed the biological relevance of the pathways uncovered by N-of-1-pathways to underline unique and shared deregulated pathways among patients, and relate these pathways to survival outcomes. Using the z scores of each of the 905 GO-BP terms found deregulated in at least one of the 55 patients, we conducted a hierarchical clustering of patients and mechanisms (figure 4A; see the ‘Heatmap’ section in the Methods). In addition, three additional analyses are presented in figure 4A: (i) the number of patients sharing any given pathway (patient count), (ii) the distribution of the GO-BP terms grouped in curated classes, and (iii) the similarity of predicted GO-BP terms to the GS based on information theoretic similarity (see the ‘Information theoretic similarity’ section in the Methods). Further, clusters of GO-BP terms are highlighted by a background color that underlines patterns that correlate patient counts to GO-BP classes. The most striking cluster (figure 4A, ‘Patient count’ and ‘GO-BP classes,’ gray, rightmost) contains nearly half of the deregulated GO-BP genesets, each shared by less than 10 patients (20%). This cluster comprises the majority of the GO-BP terms for metabolic process/transport signaling/molecular pathway, tissue/organ development, and immune response. These GO-BP classes can be attributed to the results of the inherent property of individuals or response to therapy. In contrast, the leftmost clusters of GO-BP terms comprise 25–49 patients each and pertain almost exclusively to DNA repair chromatin assembly, cell division/cell cycle, and RNA processes (figure 4A, ‘Patient count’ and ‘GO-BP classes,’ pink, yellow, and green). This cluster relates to GO-BP classes that are commonly found in cancer gene-level and pathway-level expression profiles. Further, these GO-BP classes are almost absent from the gray cluster. However, the gray and blue clusters share nearly all the immune response GO-BP terms and may infer a patient's response to therapy or tumor progression.
GO-ITS study provides comprehensive information on GO-BP terms that are overlapped by the N-of-1-pathways method and the union of the three external validation studies used as GS (figure 4A, ‘GO-ITS analysis’). Here are few examples of related GO-BP terms at different GO-ITS thresholds: with perfect overlap (GO-ITS=1), GO:0000087, M phase of mitotic cell cycle shared by 49 patients; highly related (GO-ITS=0.85), GO:0045619, regulation of lymphocyte differentiation shared by two patients; somewhat related (GO-ITS=0.7), GO:0031667, response to nutrient levels; unrelated (GO-ITS=0.25), GO:0032259, methylation.
In order to evaluate whether the individual mechanisms uncovered by N-of-1-pathways were related to disease outcome, we performed a survival analysis (see ‘The Kaplan–Meier survival curve’ section in the Methods). Patients were separated into three groups using the PAM clustering method on the individual deregulated mechanism signatures uncovered by N-of-1-pathways (figure 4B). The results show an overall trend predicting survival prognosis using individual deregulated GO-BP terms (figure 4). Given these preliminary survival results, we curated two diametric extreme survival phenotypes: ‘death of disease in less than a year’ and ‘disease-free survival >5 years’, and then performed a PCA (see the ‘Principal component analysis of individual GO-BP terms’ section in the Methods). The component plot shows a clear clustering of the two diametric extreme prognosis phenotypes (Wilcoxon test, p<0.05) (figure 4C). This result suggests that N-of-1-pathways is able to uncover deregulated GO-BP terms terms that may predict tumor severity, and of note, these GO-BP terms are individually predicted. In addition, we show in figure 4D that the second principal component of the PCA allows the best outcome phenotype ‘disease-free survival >5 years’ to be distinguished from more heterogeneous phenotype groups (t test, p<0.05).
Early cancer death associated to higher heterogeneity of deregulated pathways
The main purpose of the N-of-1-pathways framework is to unveil individual deregulated mechanisms in order to obtain a patient-oriented representation of a disease, here applied to lung adenocarcinoma. We showed (figure 4C, D) that the z scores computed from N-of-1-pathways were able to dichotomize two diametric extreme prognosis phenotypes. Such difference could be predictable and visualized in two ways: (i) by comparing the patient deregulated mechanisms to a GS built from external reference studies (figure 5) and (ii) by plotting each patient's own z scores of related deregulated mechanisms (figure 6).
Figure 5A shows that 92% of the GO-BP terms found by the external GS (75% overlap with GO-ITS=1 and 17% highly related with 0.7<GO-ITS<1) are predicted by N-of-1-pathways. This represents common deregulated mechanisms of the disease that are traditionally found by cohort-based methods. However, N-of-1-pathways also reveals deregulated mechanisms in smaller proportions of individuals and even unique to an individual of this cohort. Hence, only 61% of the GO-BP terms found across 55 patients are common or highly related to the GS (GO-ITS=1 and 0.7<GO-ITS<1), while 29% and 10% are found less related (0.3<GO-ITS<0.7) and unrelated to the GS, respectively (0.3<GO-ITS). Figure 5B shows each patient's ratio of shared mechanisms relative to the GS. The results clearly demonstrate that disease-free survival is increased for patients with a larger proportion of deregulated phenotypes similar to the external GS. In other words, an average-looking patient survives better than those very different from the GS. As these therapies are developed for the ‘average,’ this suggests that the N-of-1-pathways may provide insight into resistance mechanisms. Figure 6 may provide some understanding of those mechanisms as it illustrates the 15 most significant deregulated GO-BP terms (table 2) between the diametric extreme survival patients identified in figure 4C. The best-survival-outcome (BSO) patients tend to have cell cycle-related mechanisms less up-regulated, or non-deregulated, compared to the worst-survival-outcome (WSO) patients. Moreover, BSO patients show that the immune response-related mechanisms are generally down-regulated when compared to their significant up-regulation in WSO patients. Such specific individual patient representation allows an excellent visual of each patient's profile of deregulated mechanisms. We also computed the same analysis using FAIME scores15 instead of N-of-1-pathways z scores (see online supplementary figure S3), which shows that this type of representation is scalable to any other scoring method at the level of the patient.
Discussion
Single patient DNA-Seq analyses of tumoral versus uninvolved tissues can readily identify somatic mutations, which can thereafter be enriched into pathways.33 However, a simple measure of difference or fold change between gene expressions of paired RNA-Seq samples of a single patient cannot be interpreted with a theoretical statistical model and have conventionally been unreliable for identifying biological significance. On the other hand, pooling many genes together in paired studies according to an information model may yield significant enrichments and further model the dynamic range of individual biological mechanisms involved in disease pathophysiology. In the present study, we envisioned bridging such a gap and have shown that N-of-1-pathways is accurate (figures 2 and 3) and provides individual patient interpretations for which conventional enrichment approaches were not designed. N-of-1-pathways is a method intended for precision medicine, requiring paired samples from a single patient. Our method predicts patient-specific deregulated pathways overlooked by traditional methodologies, as they focus on identifying common genes or pathways across patients. Combining N-of-1 studies together is not novel, as this has been done using meta-analyses or Bayesian-mixed model approaches for generalizing the value of therapeutic interventions to a population.34–36 However, the latter were not designed to address two samples per patient or the overwhelming number of multiple comparisons arising from genomic datasets. In contrast, N-of-1-pathways can also be applied to different types of 'omics datasets (eg, transcriptomes, epigenomes, methylomes, etc). Specifically, the principle of generating the statistical power from multiple genes (or molecules of life)—in each geneset of a pair of samples—can be extended to methylations, or related modifications of histone or DNA binding proteins, etc. These could allow for the inclusion of novel patient-centered ’omics reports in electronic medical records for clinical interpretation (figure 6).
We and others have previously developed a single sample scoring method based on genesets that outperformed gene expression classification.14–17 However, these scoring systems are designed for interpretation of a single patient against a reference standard or a geneset-level classifier previously calculated from a cohort. In contrast, the framework of N-of-1-pathways is applicable to clinical and experimental designs with as few as two samples from the same subject (eg, treated vs non-treated cell lines). We completed a series of shRNA conditional knock-down experiments of an alternative splicing protein, PTBP1, with paired samples of ovarian or breast cancer cell lines and confirmed the biological significance of N-of-1-pathways predictions.37 In this study, we also compared N-of-1-pathways with ssGSEA, an alternative single sample method, and showed that N-of-1-pathways results were more proximate with those of GSEA and DEG enrichment (figure 3) when all individual scores were pooled together than with ssGSEA as single level results (figure 2). This supports the ability of N-of-1-pathways to recapitulate common signatures of conventional geneset methods although designed for paired data. Moreover, N-of-1-pathways predictions can be scaled down to a single geneset, which provides opportunities to target limited number of genes of a given deregulated pathway for a qPCR analysis, while ssGSEA requires the whole background of genes for the same evaluation.
Although N-of-1-pathways performed similarly to GSEA and DEG enrichment (figure 3), we are planning to conduct a number of studies to quantify the gain of accuracy of this new method. We are considering a resampling by decreasing subsets of patients to estimate the threshold for which N-of-1-pathways becomes no more accurate than alternate methods when conducted over the full dataset of 55 patients. We have already undertaken empirical studies (10 000 permutations, data not shown) to calculate the p value associated to a GO-BP pathway identified across patients, using the N-of-1-pathways statistical analysis component. This approach should yield better ranking of GO terms than the simple count of number of patients used in this current study. We also plan to incorporate FAIME scores or fold change calculation to provide a more accurate measurement of deregulated pathways. Moreover, we are considering using uncurated biomodules to generate genesets required for N-of-1-pathways from co-expression patterns or yeast two-hybrid protein interactions. We have previously shown improvement of geneset scoring methods with unbiased biomodules (eg, co-expression networks) rather than curated genesets.16 The N-of-1-pathways framework is designed to utilize the most appropriate statistics to compare paired samples. In the current application, use of the Wilcoxon signed-rank test identified deregulated pathways from genesets where genes were preferentially deregulated in one direction (‘up vs down’ or ‘down vs up’). This type of statistics may produce false negatives on genesets containing genes with opposite expression patterns. Of note, the enriched GO terms of the proxy GSs were designed with up-regulated genes separately from the down-regulated genes. In future studies, we will develop statistics that enable the identification of genes deregulated in both directions and attribute a gene weight to score significant pathways. The latter may provide insight on biological mechanisms at play in response to therapy and survival.
Finally, with the abundance of microarray datasets, an analysis of the accuracy might be valuable for obtaining a signal in microarrays by re-analyzing well-understood legacy datasets with the intention of unveiling individual differences overlooked by cross-patient studies. TCGA also contains paired DNA-sequencing of uninvolved and tumoral lung tissues in the context of lung adenocarcinoma disease for the same patients than the RNA-Seq exploration dataset, from which somatic mutations can readily be extracted. We plan to improve analytical techniques for N-of-1 genomic studies through the use of additional sources of external knowledge (eg, eQTL) and paired samples from other genomic scales for predicting individualized response to therapy. Indeed, the new pathologic classification of lung cancer identifies key mutations relevant to chemoresistance and disease progression such as EGFR, ALK, TTF-1, p53, and KRAS.23 38 39
Conclusion
In this study, we validated a novel framework, N-of-1-pathways, for unveiling deregulated pathways from only two paired samples. In classic comparative study analyses, many samples of different categories are required for achieving sufficient statistical power to draw conclusions at the level of the studied population. Here the power is available for a single patient with as few as two samples, yet population-based generalizations can be conducted in a deceptively simple way: by adding significant patient results together.
The N-of-1-pathways framework relies on three main components: single-patient pathway statistics, biological modules, and information theory similarity. First, the statistical universe is a single patient or a set of paired samples, providing statistical evidence where other methods were not designed to operate. Second, mechanisms unveiled within paired samples can be measured from genesets. Third, the ‘naive’ exact overlap of the mechanism's coded terms is insufficient to comprehensively assess commonality or differences between patients, and here we validate an information theoretic similarity approach.
We compared the results of N-of-1-pathways with two well-known methods: GSEA and DEG enrichment, which are current state-of-the-art techniques for identifying deregulated pathways from multiple samples. The results show that N-of-1-pathways could be effectively used to identify deregulated pathways at the patient level.
Our results show that about 20% of pathways deregulated in individual patients were overlooked in three previous well-powered studies; yet, some were shared by a number of patients when measured individually. This highlights the variability of individual patients who can be further stratified into subgroups at the molecular level, a missed opportunity of studies relying on mechanisms ‘common’ to a large proportion of patients. In comparison, the N-of-1-pathways approach unveils a significant number of mechanisms shared by fewer patients and provides the opportunity to tailor precision and molecular therapies to individual deregulation. Indeed, precision medicine is a nascent field lacking in robust quantitative and qualitative methodologies, particularly at the level of individualized transcriptome interpretation for predicting personal response to therapy.
Supplementary Material
Acknowledgments
We thank Dr Liqiang Xi for providing the datasets of validation study II. We thank Ms Colleen Kenost for help with proofreading. We thank Drs Terry Speed, Walter W Piegorsch, and Dean Billheimer for their statistical insights.
Footnotes
Contributors: YAL, VG: conceived the experiments; VG, JL, GP, MMC: conducted the computational biology and high-throughput experiments; VG, IA: provided the figures and tables; IA: provided the knowledge bases; VG, IA, LP, RW, JGNG, YAL: analyzed the results; VG, IA, MMC, HL, JGNG, RW, IF, NB, YAL: wrote and revised the manuscript.
Funding: The study was supported in part by NIH grants UL1TR000050 (University of Illinois CTSA), 1S10RR029030-01 (BEAGLE Cray Supercomputer), and K22LM008308, and the University of Illinois Cancer Center.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: All software/programs are provided at http://Lussierlab.org/publications/N-of-1-pathways.
References
- 1.Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet 2010;375:1525–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009;106:9362–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y, Li H, Li J, et al. Network models of genome-wide association studies uncover the topological centrality of protein interactions in complex diseases. J Am Med Inform Assoc 2013;20:619–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee Y, Gamazon ER, Rebman E, et al. Variants affecting exon skipping contribute to complex traits. PLoS Genet 2012;8:e1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee Y, Yang X, Huang Y, et al. Network modeling identifies molecular functions targeted by miR-204 to suppress head and neck tumor metastasis. PLoS Comput Biol 2010;6:e1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen JL, Li J, Stadler WM, et al. Protein-network modeling of prostate cancer gene signatures reveals essential pathways in disease recurrence. J Am Med Inform Assoc 2011;18:392–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Lee Y, Chen JL, et al. Complex-disease networks of trait-associated single-nucleotide polymorphisms (SNPs) unveiled by information theory. J Am Med Inform Assoc 2012;19:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kern SE. Why your new cancer biomarker may never work: recurrent patterns and remarkable diversity in biomarker failures. Cancer Res 2012;72:6097–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher RA. The design of experiments. 1st edn. Edinburgh: Oliver & Boyds, 1935 [Google Scholar]
- 10.Janosky JE, Leininger SL, Hoerger MP, et al. Single subject designs in biomedicine. USA: Springer, 2009 [Google Scholar]
- 11.Fu X, Fu N, Guo S, et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lillie EO, Patay B, Diamant J, et al. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Personalized Med 2011;8:161–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chuang HY, Lee E, Liu YT, et al. Network-based classification of breast cancer metastasis. Mol Syst Biol 2007;3:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, Regan K, Huang Y, et al. Single sample expression-anchored mechanisms predict survival in head and neck cancer. PLoS Comput Biol 2012;8:e1002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen JL, Hsu A, Yang X, et al. Curation-free biomodules mechanisms in prostate cancer predict recurrent disease. BMC Med Genomics 2013;6(Suppl 2):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Li H, Regan K, et al. Towards mechanism classifiers: expression-anchored Gene Ontology signature predicts clinical outcome in lung adenocarcinoma patients. AMIA Annu Symp Proc 2012;2012:1040–9 [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Rathke A, Li H, Lussier YA. Interpreting personal transcriptomes: personalized mechanism-scale profiling of RNA-seq data. Pac Symp Biocomput 2013:159–70 [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham G, Kowalczyk A, Loi S, et al. Prediction of breast cancer prognosis using gene set statistics provides signature stability and biological context. BMC Bioinformatics 2010;11:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Wang L, Ishwaran H. An integrative pathway-based clinical-genomic model for cancer survival prediction. Statistics Probability Lett 2010;80:1313–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travis WD, Brambilla E, Riely GJ. New pathologic classification of lung cancer: relevance for clinical practice and clinical trials. J Clin Oncol 2013;31:992–1001 [DOI] [PubMed] [Google Scholar]
- 24.Lu J, Kerns RT, Peddada SD, et al. Principal component analysis-based filtering improves detection for Affymetrix gene expression arrays. Nucleic Acids Res 2011;39:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yap YL, Lam DC, Luc G, et al. Conserved transcription factor binding sites of cancer markers derived from primary lung adenocarcinoma microarrays. Nucleic Acids Res 2005;33:409–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xi L, Feber A, Gupta V, et al. Whole genome exon arrays identify differential expression of alternatively spliced, cancer-related genes in lung cancer. Nucleic Acids Res 2008;36:6535–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim IJ, Quigley D, To MD, et al. Rewiring of human lung cell lineage and mitotic networks in lung adenocarcinomas. Nat Commun 2013;4:1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gene Ontology Consortium. The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res 2010;38(Database issue):D331–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha V, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang J, Conrath D. Multi-word complex concept retrieval via lexical semantic similarity. Information Intelligence and Systems, International Conference on 03/1999; 1999:407–14 [Google Scholar]
- 32.Sangodkar J, Katz S, Melville H, et al. Lung adenocarcinoma: lessons in translation from bench to bedside. Mt Sinai J Med 2010;77:597–605 [DOI] [PubMed] [Google Scholar]
- 33.Chittenden TW, Howe EA, Culhane AC, et al. Functional classification analysis of somatically mutated genes in human breast and colorectal cancers. Genomics 2008;91:508–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zucker DR, Schmid CH, McIntosh MW, et al. Combining single patient (N-of-1) trials to estimate population treatment effects and to evaluate individual patient responses to treatment. J Clin Epidemiol 1997;50:401–10 [DOI] [PubMed] [Google Scholar]
- 35.Huber AM, Tomlinson GA, Koren G, et al. Amitriptyline to relieve pain in juvenile idiopathic arthritis: a pilot study using Bayesian metaanalysis of multiple N-of-1 clinical trials. J Rheumatol 2007;34:1125–32 [PubMed] [Google Scholar]
- 36.Zucker DR, Ruthazer R, Schmid CH. Individual (N-of-1) trials can be combined to give population comparative treatment effect estimates: methodologic considerations. J Clin Epidemiol 2010;63:1312–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardeux V, Arslan AD, Achour I, et al. Concordance of deregulated mechanisms unveiled in underpowered experiments: PTBP1 knockdown case study. BMC Med Genomics 2014;7(Suppl 1):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Travis WD, Brambilla E, Noguchi M, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society: international multidisciplinary classification of lung adenocarcinoma: executive summary. Proc Am Thorac Soc 2011;8:381–5 [DOI] [PubMed] [Google Scholar]
- 39.Kalari KR, Rossell D, Necela BM, et al. Deep sequence analysis of non-small cell lung cancer: integrated analysis of gene expression, alternative splicing, and single nucleotide variations in lung adenocarcinomas with and without oncogenic KRAS mutations. Front Oncol 2012;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.