

Abstract
Abnormal levels of inflammation are associated with cardiovascular disease and mortality in human immunodeficiency virus (HIV)–infected patients. Microbial translocation, which may cause inflammation, is decreased by sevelamer in patients undergoing hemodialysis. In this single-arm study, we evaluated the effects of 8 weeks of sevelamer therapy on 36 HIV-infected subjects who were not receiving antiretroviral therapy. Sevelamer did not significantly change markers of microbial translocation, inflammation, or T-cell activation. During sevelamer treatment, however, levels of soluble tissue factor, low-density lipoprotein (LDL) cholesterol, and oxidized LDL cholesterol decreased significantly, whereas D-dimer levels increased. Thus, in this study population, sevelamer did not reduce microbial translocation but may have yielded cardiovascular benefits.
Clinical Trials Registration. NCT 01543958.
Keywords: HIV, sevelamer, microbial translocation, LPS, sCD14, soluble tissue factor, LDL, oxLDL
AIDS-related complications and mortality have decreased dramatically in the combination antiretroviral therapy (cART) era, but the risk of cardiovascular disease and other morbidities linked to chronic immune dysfunction and inflammation remains high [1]. Increased CD8 T-cell activation and inflammatory markers are associated with impaired CD4 T-cell recovery and excess mortality in ART recipients [1, 2].
Systemic translocation of microbial products such as lipopolysaccharide (LPS) across a permeable gut mucosa may contribute to persistent inflammation [2]. Individuals with chronic human immunodeficiency virus (HIV) infection have increased circulating levels of LPS, LPS-binding protein (LBP), and soluble CD14 (sCD14), an LPS coreceptor [2]. ART decreases but does not normalize these levels [2]. Thus, an agent that decreases levels of LPS and its inflammatory consequences may improve clinical outcomes in HIV infection.
Sevelamer carbonate, a phosphate-lowering drug, decreases circulating LPS levels in patients with renal insufficiency, possibly by binding chylomicron-LPS complexes and preventing their reabsorption; in this population, sevelamer also reduces levels of sCD14, interleukin 6 (IL-6), C-reactive protein (CRP), and total and low-density lipoprotein (LDL) cholesterol [3–6]. These effects may contribute to the decreased mortality risk observed in sevelamer-treated patients undergoing dialysis [7].
Given the possible role of microbial translocation in HIV-associated inflammation and the potential for its reversal by sevelamer, we performed a single-arm, open-label clinical trial (AIDS Clinical Trials Group [ACTG] A5296), to test the hypothesis that 8 weeks of sevelamer treatment would decrease plasma LPS and sCD14 levels. We studied its effects in subjects who were not receiving cART because (1) sevelamer-cART interactions are unknown, (2) sevelamer might exacerbate tenofovir's phosphate-lowering effect, and (3) microbial translocation is more consistently elevated in untreated disease, increasing the likelihood of seeing an effect.
METHODS
Institutional review boards at each site approved the study. All subjects provided written informed consent.
Study Subjects
Eligibility criteria included ≥18 years of age, chronic HIV infection, no cART for ≥24 weeks, a plasma HIV RNA level of >50 copies/mL, and a CD4 T-cell count of ≥400 cells/mm3. Major exclusion criteria were recent severe illness, liver or kidney disease, pregnancy or breast-feeding, and use of immunosuppressive medications within 24 weeks of study entry.
Study Design
ACTG A5296 was a phase 2, single-arm trial. Subjects received 1600 mg of sevelamer carbonate orally 3 times daily for 8 weeks; each dose was administered 1 hour after meals to minimize dietary phosphate binding. Subjects were followed for 8 weeks after treatment was stopped. Two blood samples were collected 2–4 weeks apart before initiation of sevelamer treatment; the average of test results for both specimens, if available, were used for the baseline measurement. Blood specimens for the baseline measurements and at weeks 4, 8 and 6 were obtained after participants had fasted for ≥8 hours. Subjects underwent clinical evaluations, safety laboratory testing and blood sample collection and storage at these visits and at weeks 1, 2, 6 and 12. Serum specimens, EDTA-treated and citrated-treated plasma specimens, and peripheral blood mononuclear cells (PBMCs) were cryopreserved for batched assays.
Biomarker Analysis
Serum LPS levels were measured independently in the laboratory of one author (D. C. D.) for the primary analysis and in the laboratory of another author (M. M. L.) for confirmation, using the Limulus Amebocyte Lysate Kit (Lonza, Walkersville, MD) on serum diluted 10-fold in water (Sigma, St. Louis, MO). The absorbance of diluted serum was subtracted from the absorbances at assay completion. Levels of the following markers were measured by ELISA in accordance with the manufacturer's instructions: plasma sCD14, IL-6, CRP, interferon γ–inducible protein 10 (IP-10), interleukin 1β (IL-1β), soluble CD163 (sCD163), fetuin A, and soluble tissue factor (sTF; R&D Systems, Minneapolis, MN), LBP, endotoxin core immunoglobulin G antibody (EndoCAb), (Hycult Biotech, Plymouth Meeting, PA); D-dimer (Diagnostica Stago, Parsippany, NJ); and oxidized LDL (oxLDL) cholesterol (Mercodia, Uppsala, Sweden).
T-cell Activation
PBMCs were thawed (median viability, 89.4%; interquartile range [IQR], 84.2%–92.8%); stained with Live/Dead Aqua viability dye, anti-CD4 PE-Texas Red (Invitrogen, Grand Island, NY), anti-CD3 V450, anti-CD8 APC-H7, and either anti-HLA-DR PE and anti-CD38 APC or, after permeabilization, anti-Ki67 PE (BD Biosciences, San Jose, CA); and acquired on an LSRII flow cytometer (BD Biosciences, San Jose, CA), using FacsDiva software. Frequencies of activated (CD38+HLA-DR+) and cycling (Ki67+) CD4 and CD8 T cells were determined using FlowJo software (Tree Star, Ashland, OR).
Statistical Analysis
The primary end points were the changes in LPS and sCD14 levels from baseline to week 8. Secondary end points included changes from week 8 to 16 in sCD14 and LPS levels and changes from baseline to week 8 and from week 8 to 16 in activated and cycling T-cell frequencies, biomarkers of systemic inflammation and coagulation, plasma HIV-1 RNA levels, CD4 T-cell counts, serum phosphate levels, and fasting lipid and glucose levels. Signs and symptoms with a severity grade of ≥2 were analyzed.
Primary and secondary as-treated analyses were limited to subjects who had data from baseline and week 8 and who received sevelamer therapy through week 8, to examine the biologic effects of sevelamer.
Median values were reported for baseline characteristics and primary and secondary end points. Frequencies and percentages were reported for categorical variables. Changes in primary and secondary end points were tested using the sign test. Results obtained using paired t tests were similar (not shown). Relationships among variables were assessed with Spearman correlations.
RESULTS
Forty subjects enrolled at 15 US sites between December 2011 and August 2012. Baseline characteristics are described in Supplementary Table 1, and reasons for the ineligibility of 16 additional patients are described in Supplementary Table 2. Four subjects were excluded from analysis: 1 initiated cART at week 7, and 3 missed key study visits. The baseline characteristics of included subjects did not differ markedly from those of excluded subjects. The median time since the first HIV-positive test result was 1084 days (IQR, 463–2737 days). Among 24 subjects for whom data were available, 5 previously received cART but stopped treatment ≥2 years before enrollment. Self-reported adherence to sevelamer therapy was high: at study weeks 4 and 8, 81% and 92% of subjects, respectively, reported having not missed >1 dose in the previous 4 days.
To test the hypothesis that sevelamer would decrease microbial translocation, we measured serum LPS and plasma sCD14 levels. LPS levels did not change significantly during sevelamer treatment or after its discontinuation (Figure 1A and Table 1). Results from the laboratories of 2 authors (D. C. D. and M. M. L.) were similar (data not shown). Plasma sCD14, LBP, and EndoCAb levels did not change significantly either during sevelamer treatment or after its discontinuation (Figure 1B and Table 1). Thus, we did not find evidence that sevelamer treatment for 8 weeks decreased microbial translocation in HIV-infected subjects who were not receiving cART.
Figure 1.
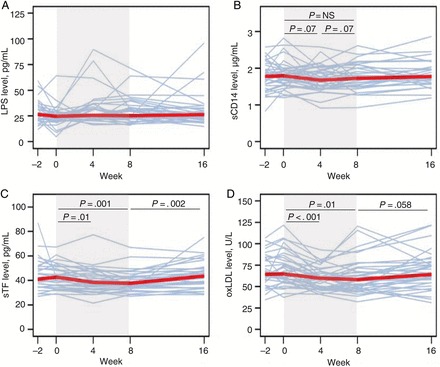
Sevelamer does not significantly change circulating markers of microbial translocation. A, Serum lipopolysaccharide (LPS) levels measured 2–4 weeks before initiation of sevelamer treatment, on the day of initiating treatment, after 4 weeks of treatment, after 8 weeks of treatment, and 8 weeks after cessation of treatment (week 16). B, Plasma soluble CD14 (sCD14) levels measured 2–4 weeks before initiation of sevelamer treatment, on the day of initiating treatment, after 4 weeks of treatment, after 8 weeks of treatment, and 8 weeks after cessation of treatment (week 16). C, Sevelamer decreases plasma soluble tissue factor (sTF) levels, which rebound after treatment cessation. Levels were measured 2–4 weeks before initiation of sevelamer treatment, on the day of initiating treatment, after 4 weeks of treatment, after 8 weeks of treatment, and 8 weeks after cessation of treatment (week 16). D, Sevelamer reduces oxidized low-density lipoprotein cholesterol (oxLDL) levels. oxLDL levels were measured 2–4 weeks before initiation of sevelamer treatment, on the day of initiating treatment, after 4 weeks of treatment, after 8 weeks of treatment, and 8 weeks after cessation of treatment (week 16). Shading indicates treatment period. Thin lines represent individual subjects over time. Thick red lines represents the median values at each study week. P values were calculated using the sign test. Abbreviation: NS, not significant.
Table 1.
Biomarkers of Microbial Translocation, Inflammation, Coagulation, and Immune Activation
| Biomarker | Baseline, Median (IQR) | Week 8,a Median (IQR) | Week 16,b Median (IQR) | Week 8 vs Baseline | Week 16 vs Week 8 | ||
|---|---|---|---|---|---|---|---|
| Median Change (IQR) | P value | Median Change (IQR) | P value | ||||
| Microbial translocation | |||||||
| LPS level, pg/mL | 25.2 (21.3–29.8) | 25.3 (22.3–32.9) | 26.4 (21.4–32.0) | +0.2 (−4.1 to 5.8) | .87 | −2.4 (−8.0 to 2.7) | .30 |
| sCD14 level, μg/mL | 1.81 (1.57–2.00) | 1.73 (1.53–2.05) | 1.78 (1.61–1.98) | −0.02 (−0.18 to 0.06) | .62 | +0.06 (−0.03 to 0.24) | .23 |
| LBP level, μg/mL | 13.5 (12.0–17.4) | 14.1 (11.3–17.8) | … | +0.5 (−1.6 to 2.5) | .24 | … | |
| EndoCAb level, GMU/mL | 134.9 (62.9–250.0) | 117.0 (61.2–212.7) | … | −5.8 (−21.8 to 12.9) | .13 | … | |
| T-cell activation | |||||||
| CD38+HLA-DR+ CD4 T cells, % | 10.3 (7.0–13.0) | 11.2 (6.8–13.4) | 10.2 (6.9–13.9) | +0.1 (−0.7 to 2.6) | 1.00 | +0.5 (−0.8 to 1.6) | .20 |
| CD38+HLA-DR+ CD8 T cells, % | 36.0 (25.6–51.4) | 36.1 (24.0–53.6) | 34.3 (24.2–51.8) | −0.5 (−1.8 to 2.0) | .73 | −0.2 (−1.9 to 6.0) | 1.00 |
| Ki67+ CD4 T cells, % | 3.45 (2.75–4.78) | 3.30 (2.45–5.00) | 3.90 (3.00–4.90) | +0.03 (−0.58 to 0.40) | 1.00 | +0.20 (−0.30 to 0.80) | .14 |
| Ki67+ CD8 T cells, % | 4.18 (2.98–5.68) | 4.60 (2.55–5.50) | 5.00 (3.20–5.70) | +0.10 (−0.43 to 0.78) | 1.00 | 0.30 (−0.50 to 0.90) | .44 |
| Inflammation and cardiovascular disease | |||||||
| IL-6 level, pg/mL | 1.65 (1.23–2.20) | 1.71 (1.27–2.31) | 1.68 (1.20–3.08) | −0.02 (−0.58 to 0.63) | .87 | 0.19 (−0.43 to 1.07) | .39 |
| CRP level, ng/mL | 1768 (883–4029) | 2199 (708–4249) | 2353 (834–4077) | +46 (−653 to 1540) | .62 | −34 (−493 to 527) | .39 |
| IL-1β level, pg/mL | 0.23 (0.14–0.32) | 0.26 (0.12–0.35) | 0.24 (0.08–0.33) | +0.03 (−0.09 to 0.10) | .38 | −0.01 (−0.07 to 0.06) | .70 |
| IP-10 level, pg/mL | 278 (198–463) | 287 (168–412) | 343 (201–4356) | −16 (−60 to 24) | .24 | +19 (−10 to 83) | .06 |
| sCD163 level, ng/mL | 1006 (690–1352) | 968 (735–1236) | 991 (638–1398) | +8 (−117 to 98) | .87 | +29 (−72 to 119) | .86 |
| Fetuin A level, mg/mL | 0.88 (0.46–1.34) | 0.55 (0.22–0.98) | 0.66 (0.30–1.16) | −0.34 (−0.70 to 0.22) | .41 | +0.05 (−0.23 to 0.70) | .61 |
| Coagulation | |||||||
| D-dimer level, ng/mL | 184 (122–313) | 201 (143–335) | 199 (113–277) | +20 (−16 to 75) | .03 | −17 (−66 to 26) | .39 |
| sTF level, pg/mL | 41.8 (36.0–50.0) | 37.7 (35.3–43.3) | 43.6 (37.3–49.4) | −2.0 (−6.7 to 0.1) | .001 | +3.8 (0.0 to 8.0) | .002 |
| Metabolic index | |||||||
| Total cholesterol level, mg/dL | 172 (154–198) | 155 (141–191) | 169 (154–200) | −13 (−29 to 6) | .18 | +9 (−4 to 25) | .01 |
| HDL cholesterol level, mg/dL | 42 (35–53) | 42 (35–51) | 41 (36–54) | +1 (−6 to 3) | .61 | +1 (−3 to 6) | .59 |
| Non-HDL cholesterol level, mg/dL | 127 (105–158) | 117 (92–140) | 125 (110–140) | −12 (−25 to 2) | .02 | +4 (−2 to 24) | .04 |
| Triglycerides level, mg/dL | 121 (93–178) | 123 (101–193) | 130 (74–154) | −4 (−13 to 41) | .50 | −3 (−56 to 7) | .16 |
| LDL cholesterol level, mg/dL | 101 (83–122) | 81 (68–101) | 94 (85–116) | −18 (−32 to 2) | .009 | +14 (−1 to 33) | .01 |
| oxLDL cholesterol level, U/L | 64.4 (60.6–81.7) | 58.2 (48.3–72.3) | 64.2 (54.8–77.8) | −7.3 (−16.8 to 1.7) | .01 | +5.3 (−5.1 to 16.2) | .06 |
| Glucose level, mg/dL | 88 (80–93) | 88 (76–95) | 89 (80–99) | −2 (−12 to 2) | .24 | +2 (−3 to 12) | .22 |
| HIV disease parameter | |||||||
| HIV-1 RNA load, log10 copies/mL | 3.58 (2.95–4.02) | 3.44 (2.96–4.04) | 3.73 (3.12–4.24) | −0.02 (−0.12 to 0.10) | .38 | 0.16 (−0.11 to 0.31) | .01 |
| CD4 T-cell count, cells/mm3 | 585 (494–716) | 630 (505–738) | 573 (477–701) | −14 (−70 to 45) | .29 | −1 (−93 to 69) | .61 |
P values of <.05 are considered statistically significant.
Abbreviations: CRP, C-reactive protein; EndoCAb, endotoxin core antibody; GMU, immunoglobulin G median units; HDL, high-density lipoprotein; IL-1β, interleukin 1β; IL-6, interleukin 6; IP-10, interferon γ–inducible protein 10; IQR, interquartile range; LBP, lipopolysaccharide-binding protein; LDL, low-density lipoprotein; LPS, lipopolysaccharide; oxLDL, oxidized low-density lipoprotein; sCD14, soluble CD14; sCD163, soluble CD163; sTF, soluble tissue factor.
a During sevelamer treatment.
b Eight weeks after cessation of sevelamer treatment.
Because sevelamer decreases other markers of immune activation and inflammation in patients undergoing hemodialysis [4, 5], we evaluated its effects in our population. The frequencies of activated (HLA-DR+CD38+) and cycling (Ki67+) CD4 and CD8 T cells did not change significantly during or after discontinuation of treatment. Similarly, levels of markers associated with cardiovascular events, poor recovery of the CD4 T-cell count, and/or mortality, including plasma IL-6, CRP, IL-1β, IP-10, sCD163, and fetuin A levels, did not change significantly during sevelamer treatment or after its discontinuation (Table 1).
To assess sevelamer's effect on coagulation, we measured sTF and D-dimer levels. The median sTF level decreased during sevelamer treatment, from 41.8 pg/mL to 37.7 pg/mL (P = .001), and rebounded after sevelamer therapy was stopped (Figure 1C and Table 1). In contrast, the median D-dimer level increased during sevelamer treatment, from 184 ng/mL to 201 ng/mL (P = .03), and remained elevated after treatment cessation (Table 1).
Evaluations of the associations among biomarkers revealed that LPS, sCD14, and EndoCAb levels neither correlated with each other nor consistently with other biomarkers. However, levels of LBP, IL-6, and CRP, all acute phase reactants, correlated significantly with each other at all time points (data not shown). sTF and D-dimer levels were also associated at baseline (r = 0.40; P = .01), week 4 (r = 0.37; P = .02), and week 16 (r = 0.37; P = .03) but not at week 8 (r = 0.19; P = .26).
Because of sevelamer's lipid-lowering action in patients undergoing hemodialysis [4, 6, 7], we examined its effect on fasting lipid levels in our subjects. The total cholesterol level decreased, but not significantly, during treatment and increased significantly after treatment cessation. LDL, non–high-density lipoprotein (HDL), and oxLDL cholesterol levels decreased during treatment (median change, −18 mg/dL, −12 mg/dL, and −7.3 U/L, respectively) and increased with treatment cessation (Table 1 and Figure 1D). The ratio of the oxLDL cholesterol level to the LDL cholesterol level did not change significantly during or after treatment (data not shown), suggesting no change in oxidative stress [8]. The HDL cholesterol level, the ratio of the total cholesterol level to the HDL cholesterol level, the triglycerides level, and the glucose level did not change significantly.
CD4 T-cell counts did not change significantly during or after discontinuation of sevelamer treatment (Table 1). Plasma HIV RNA levels did not change during treatment but increased significantly after stopping sevelamer therapy (P = .01). The slopes of the changes in plasma HIV RNA load during and after treatment were not statistically significantly different (P = .60).
There were no study discontinuations due to adverse events. Adverse events included hypophosphatemia (in 10 subjects), constipation (in 2), and diarrhea (in 1). Seven subjects received phosphate supplementation for levels of <2.5 mg/dL. One had a 2-week reduction in the sevelamer dose, for a phosphate level of 1.9 mg/dL at week 2. Three subjects had 4 episodes of grade 3 hypophosphatemia at study weeks 2, 8, 12, and 16. Serum phosphate levels did not change significantly between baseline and week 8 (median change, −0.10 mg/dL; P = .95) or between weeks 8 and 16 (median change, 0.10 mg/dL; P = .63).
DISCUSSION
Persistent microbial translocation has been postulated to contribute to inflammation, morbidity, and mortality in HIV disease. We performed a prospective clinical trial to measure the effects of sevelamer, which binds LPS in the intestine, on markers of microbial translocation, inflammation, coagulation, and lipids. In these untreated individuals with early stage HIV disease, we observed no effect on LPS level, sCD14 level, CD4 T-cell count, or viremia. Despite increasing D-dimer levels, sevelamer decreased LDL cholesterol, oxLDL cholesterol, and sTF levels, suggesting it may have net beneficial cardiovascular effects.
It is unclear why sevelamer decreased microbial translocation and inflammation in HIV-uninfected patients undergoing hemodialysis but not in our subjects. In patients undergoing hemodialysis, microbial translocation may result from intestinal edema and ischemia, pathogen overgrowth, and tight junction degradation [9]; sevelamer may bind enough LPS in the intestine to prevent microbial translocation. In HIV-infected patients, additional contributors to microbial translocation may include mucosal CD4 T-cell depletion, decreased mucosal immunoglobulin A production, and impaired clearance of microbial products [2], mechanisms that may be unaffected by sevelamer. Other potential explanations for sevelamer's failure to affect LPS levels and inflammation here include (1) ongoing HIV replication causing persistent intestinal damage; (2) minimally increased LPS levels in this relatively healthy population [10]; (3) sevelamer administration without rather than with food; (4) proinflammatory effects of other microbial products, HIV, and coinfecting pathogens; (5) absence of underlying hyperphosphatemia, a driver of inflammation in renal insufficiency [5]; and (6) type II error. Baseline LPS and sCD14 levels here were lower than we have previously reported in individuals with untreated HIV infection [11]. Whether sevelamer treatment would decrease microbial translocation and inflammation in cART recipients is unknown. Regardless, sevelamer may have beneficial cardiovascular effects, although interactions or overlapping toxicities with tenofovir and other antiretroviral drugs might limit its utility.
Sevelamer significantly decreased LDL and non-HDL cholesterol levels here, consistent with effects reported elsewhere [4, 6, 7]. The 20% decrease in LDL cholesterol levels here is comparable to findings in subjects undergoing hemodialysis and to the effects of statins in HIV-infected patients [12]. Sevelamer, like statins [8], also decreased levels of oxLDL cholesterol and sTF. oxLDL cholesterol promotes the development of atherosclerotic lesions by inducing foam cell formation [8], proinflammatory cell recruitment [13], and TF expression on monocytes and other cells [8]. TF expression also increases on circulating monocytes in HIV infection and in acute coronary syndrome [14]. Thus, by decreasing levels of oxLDL cholesterol and sTF, sevelamer may reduce coagulation-associated cardiovascular disease. Decreasing the oxLDL cholesterol level should also lower the plasminogen activator inhibitor 1 level, thereby increasing fibrinolysis [15]. Thus, the increases in D-dimer level observed here, despite a reduced sTF level, may reflect sevelamer's impact on fibrinolysis. This hypothesis will be explored in future studies.
This study has several limitations. A modest treatment effect might have been missed because of the small sample size. The bioassay for LPS is technically challenging; serum proteins may inhibit accurate measurement, and alterations in serum lipids may interfere with interpretation of the results and obscure changes [2]. Nonetheless, we evaluated other markers of endotoxemia, and none reflected altered microbial translocation.
In summary, receipt of sevelamer therapy for 8 weeks did not decrease microbial translocation, inflammation, or immune activation in untreated individuals with early stage HIV disease. Sevelamer did, however, lower sTF and oxLDL cholesterol levels, which may have beneficial cardiovascular effects. Additional studies of the mechanism by which sevelamer affects coagulation and lipids are warranted.
ACTG A5296 STUDY TEAM MEMBERS AND SITES
In addition to the coauthors, members of the study team are as follows: Jennifer Tiu (clinical trials specialist), Jennifer Janik (data manager), Ruth Ebiasah and Thucuma Sise (study pharmacists), Gene Morse (pharmacologist), George Bishopric (community scientific subcommittee representative), Michael Klebert (field representative), LuAnn Borowski and Paul Harding (laboratory technologists), Amy Gonzalez (laboratory data coordinator), Russell Tracy (consultant), and Brian Claggett (Case Western Reserve University Immunology Laboratory). Enrolling research sites were as follows: Alabama Therapeutics (Alicarmen Alvarez, RN, and Tamara James), Case Western Reserve University (Patricia Walton, RN, and Kristen Allen, RN), Harbor-UCLA Medical Center (Eric Daar, MD, and Ruben Lopez, MD), Hospital of the University of Pennsylvania (Pablo Tebas, MD, and Aleshia Thomas, RN, BSN), Massachusetts General Hospital (Amy Sbrolla, RN, and Teri Flynn, ANP), MetroHealth (Kim Whitely, RN, and Traci Davis, RN), Northwestern University (Donna McGregor, MSN, NP, and Babafemi Taiwo, MBBS, MD), Trillium Health (Christine Hurley, RN, BSN), University of California–Los Angeles (UCLA; Jordan Lake, MD), University of California–San Francisco (Annie Luetkemeyer, MD, and Anna Smith, RN), University of Cincinnati (Carl J. Fichtenbaum, MD, and Jan M. Stockton, RN, MSN, APRN, AACRN), University of Colorado (Kristine Griesmer, BSN, and Graham Ray, RN, MSN), University of Miami (Hector H. Bolivar, MD, and Margaret A. Fischl, MD), University of Rochester (Mary Adams, RN, MPH), and Washington University School of Medicine (Michael K. Klebert, PhD, RN, ANP-BC, and Michael Royal, RPh).
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the patients, for volunteering to participate; Irini Sereti, Alvin Schmaier, Nehal Mehta, and Stephen Smiley, for sharing their expertise on tissue factor, coagulation, and lipids; Peter Hunt, for his expertise on microbial translocation, lipids, and inflammation and for his helpful review of the manuscript.
This article is dedicated to Dr Andy I. Choi, who proposed this study but passed away on 14 August 2010, before the study could be completed.
Financial support. This work was supported by the National Institutes of Health (grant U01 AI-68636 to the ACTG, grants to the Statistical and Data Analysis Center [AI-68634] and participating clinical trials units [2UM1AI069452-08 to Alabama Therapeutics, AI069501 to Case Western Reserve University, UM1-A1069424 to Harbor-UCLA, UM1-AI069534-08 to the Hospital of the University of Pennsylvania, 2UM1AI069412-08 to Massachusetts General Hospital, AI069471 to Northwestern University, 2UMIAI069511-08 to Trillium Health, UM1 AI069424 to the UCLA CARE Center, 5UO1 AI069502-07 to the University of California–San Francisco, A1-069513 to the University of Cincinnati, 2UM1AI069432 and UL1 TR001082 to University of Colorado Health, AI069477 to the University of Miami, 2UMIAI069511-08 to the University of Rochester, and UM1AI069495 to Washington University)], grant UL1TR000124 to the UCLA Clinical and Translational Science Institute, grants to the Center for AIDS Research at the Hospital of the University of Pennsylvania [5-P30-AI-045008-15] and affiliated clinical research centers [UL1 RR024160 to Trillium Health and UL1 RR024160 to the University of Rochester], grants UM1-AI068636 and P30-AI027757 to R. W. C., and grant AI-076174 to the Cleveland Immunopathogenesis Consortium).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Contributor Information
Collaborators: Jennifer Tiu, Jennifer Janik, Ruth Ebiasah, Thucuma Sise, Gene Morse, George Bishopric, Michael Klebert, LuAnn Borowski, Paul Harding, Amy Gonzalez, Russell Tracy, Brian Claggett, Alicarmen Alvarez, Tamara James, Patricia Walton, Kristen Allen, Eric Daar, Ruben Lopez, Pablo Tebas, Aleshia Thomas, Amy Sbrolla, Teri Flynn, Kim Whitely, Traci Davis, Donna McGregor, Babafemi Taiwo, Christine Hurley, Jordan Lake, Annie Luetkemeyer, Anna Smith, Carl J. Fichtenbaum, Jan M. Stockton, Kristine Griesmer, Graham Ray, Hector H. Bolivar, Margaret A. Fischl, Mary Adams, Michael K. Klebert, and Michael Royal
References
- 1.Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol. 2013;119:51–83. doi: 10.1016/B978-0-12-407707-2.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10:655–66. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 3.Sun PP, Perianayagam MC, Jaber BL. Sevelamer hydrochloride use and circulating endotoxin in hemodialysis patients: a pilot cross-sectional study. J Ren Nutr. 2009;19:432–8. doi: 10.1053/j.jrn.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Stinghen AE, Goncalves SM, Bucharles S, et al. Sevelamer decreases systemic inflammation in parallel to a reduction in endotoxemia. Blood Purif. 2010;29:352–6. doi: 10.1159/000302723. [DOI] [PubMed] [Google Scholar]
- 5.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Donate-Correa J, Cazana-Perez V, Garcia-Perez J. Effect of phosphate binders on serum inflammatory profile, soluble CD14, and endotoxin levels in hemodialysis patients. Clin J Am Soc Nephrol. 2011;6:2272–9. doi: 10.2215/CJN.01650211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferramosca E, Burke S, Chasan-Taber S, Ratti C, Chertow GM, Raggi P. Potential antiatherogenic and anti-inflammatory properties of sevelamer in maintenance hemodialysis patients. Am Heart J. 2005;149:820–5. doi: 10.1016/j.ahj.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 7.Iimori S, Mori Y, Akita W, et al. Effects of sevelamer hydrochloride on mortality, lipid abnormality and arterial stiffness in hemodialyzed patients: a propensity-matched observational study. Clin Exp Nephrol. 2012;16:930–7. doi: 10.1007/s10157-012-0640-4. [DOI] [PubMed] [Google Scholar]
- 8.Owens AP, III, Mackman N. Sources of tissue factor that contribute to thrombosis after rupture of an atherosclerotic plaque. Thromb Res. 2012;129(suppl 2):S30–3. doi: 10.1016/j.thromres.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anders HJ, Andersen K, Stecher B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013;83:1010–6. doi: 10.1038/ki.2012.440. [DOI] [PubMed] [Google Scholar]
- 10.Manner IW, Troseid M, Oektedalen O, Baekken M, Os I. Low nadir CD4 cell count predicts sustained hypertension in HIV-infected individuals. J Clin Hypertens (Greenwich) 2013;15:101–6. doi: 10.1111/jch.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandler NG, Wand H, Roque A, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. 2011;203:780–90. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silverberg MJ, Leyden W, Hurley L, et al. Response to newly prescribed lipid-lowering therapy in patients with and without HIV infection. Ann Intern Med. 2009;150:301–13. doi: 10.7326/0003-4819-150-5-200903030-00006. [DOI] [PubMed] [Google Scholar]
- 13.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–12. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 14.Funderburg NT, Zidar DA, Shive C, et al. Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndrome. Blood. 2012;120:4599–608. doi: 10.1182/blood-2012-05-433946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dichtl W, Stiko A, Eriksson P, et al. Oxidized LDL and lysophosphatidylcholine stimulate plasminogen activator inhibitor-1 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:3025–32. doi: 10.1161/01.atv.19.12.3025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.